

INTRODUCTION
Arthritis is a common and disabling problem in our population. Joint inflammation, arthritis, is associated with behavioral changes such as limping, guarding, decreased range of motion, and an increased response to peripheral noxious stimuli (hyperalgesia). For these reasons, experimental animal models have been designed to study various arthritic conditions. The injection of a mixture of kaolin and carrageenan into the knee joint is a model designed to study the acute inflammatory response. These irritants produce a localized joint inflammation and limping and guarding of the limb (Coggeshall et al. 1983; Sluka and Westlund 1992). This arthritis model has been studied in the anesthetized cat (Schaible et al. 1987; Neugebauer and Schaible 1990), anesthetized monkey (Dougherty et al. 1992b; Sluka et al. 1992; Sorkin et al. 1992; Westlund et al. 1992), and awake rat (Sluka and Westlund 1992, 1993a,b,c,d). Neural changes have been observed in both the peripheral and central nervous systems. Sensitization of articular primary afferent fibers (Coggeshall et al. 1983) and dorsal horn neurons (Dougherty et al. 1992b; Neugebauer and Schaible 1990; Schaible et al. 1987) to cutaneous and joint stimuli occurs after induction of arthritis.
From the symptomatic point of view, arthritis can be divided into two components: (1) the inflammation itself (redness, swelling, increased temperature) in and around the joint, and (2) the pain that accompanies this inflammation. Blood-borne or local factors have been considered the sole players in the development of inflammation (Hurley 1983) until relatively recently. There is clear evidence that both the peripheral somatic and sympathetic nervous systems are involved in a variety of inflammatory conditions (see Levine et al. 1988). For example, primary afferent fibers are known to release the inflammatory neuropeptides, substance P (SP) and calcitonin gene-related peptide (CGRP), into the knee joint (Yaksh et al. 1988; Larsson et al. 1991). These neuropeptides have been found in the exudate in both human arthritic conditions and in animal models of arthritis (Applegren et al. 1991; Larsson et al. 1991; Marshall et al. 1990). In adjuvant arthritis, a chronic inflammatory model, chemical sympathectomies decrease the severity of the arthritis (Levine et al. 1986).
We used the kaolin and carrageenan model of inflammation in our experiments to investigate the involvement of:
spinal excitatory amino acid (EAA) receptors of the non-N-methyl-D-aspartate (non-NMDA) (n = 8) and NMDA (n = 8) types,
spinal gamma-aminobutyric acidA (GABAA) receptors (n = 8),
primary afferent fibers (unilateral dorsal rhizotomy) (n = 6), and
the sympathetic nervous system (surgical and/or chemical sympathectomy) (n = 6)
In rats, the knee joint is injected with a mixture of 0.1 cc of 3% kaolin and 3% carrageenan while animals are briefly anesthetized (Sluka and Westlund 1992). Following induction of arthritis, we assessed pain-related behaviors using paw withdrawal latency (PWL) to radiant heat and a subjective pain rating scale. The degree of inflammation was determined by measuring joint circumference and with thermography. Additionally, we assessed the effects of non-NMDA and NMDA receptors on excitatory amino acid (EAA) release in the dorsal horn. Antagonists were infused into the spinal cord through a microdialysis fiber implanted into the dorsal horn (lamina III–IV of the lumbar [L3–L6] spinal cord). As a control for the localization of the effects of receptor antagonists, a microdialysis fiber was placed into the sacral spinal cord. Untreated arthritic (n = 10) and saline-injected control animals (n = 4) were used for comparison. All experiments were approved by the Animal Care and Use Committee at our institution.
EXCITATORY AMINO ACID RELEASE
The release of the EAAs, aspartate (ASP) and glutamate (GLU), in the deep dorsal horn of the spinal cord was studied by measuring changes in their concentrations in extracellular fluid collected via a microdialysis fiber (Skilling et al. 1988; Sluka and Westlund. 1992; Sorkin et al. 1988) in awake rats during the development of the inflammation. An initial increase in ASP and GLU concentration was observed upon injection of the irritants (Fig. 1). Subsequently, a prolonged release of ASP and GLU in the deep dorsal horn began after three hours and persisted throughout the collection period (eight hours). If animals were pretreated either with the non-NMDA receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 1.1 mM), or the NMDA receptor antagonist, DL-2-amino-7-phosphonoheptanoic acid (AP7; 2 mM), the increased release of ASP and GLU was blocked both at the time of injection and in the prolonged release phase. In fact, ASP concentrations fell significantly below baseline in the CNQX-treated arthritic animals. The antagonists—CNQX and AP7—had no effect on baseline levels of ASP or GLU (Fig. 1).
Fig. 1.
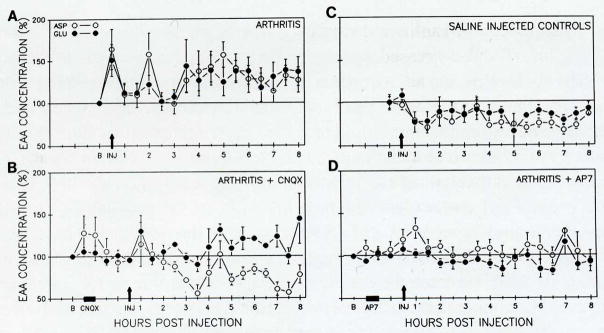
The release of ASP and GLU in the dorsal horn of the spinal cord before and eight hours after induction of arthritis is represented in panel A. At the time of injection (arrow) there was a significant increase in the release of ASP (ANOVA; P = 0.04) and GLU (P = 0.04). A transient increase in ASP at two hours is followed by a prolonged increase in release of ASP and GLU beginning at 3.5 hours and continuing for at least eight hours. The increase in the prolonged release phase (3.5–8 hours) was significant for ASP (P = 0.01) and GLU (P = 0.02). In saline-injected control animals (C), no change occurred at the time of injection for either ASP or GLU. Interestingly, there was a significant decrease in release of ASP (P = 0.0008) in the prolonged-release phase. Animals were pretreated for one hour with either CNQX (2 mM; dissolved in artificial CSF and filtered; Research Biochemicals), a non-NMDA receptor antagonist, or AP7 (2 mM; dissolved in ACSF; Sigma), an NMDA receptor antagonist, followed by one hour of washout for CNQX or 30 minutes for AP7 before injection of the knee joint with 0.1 cc 3% kaolin and 3% carrageenan, (B and D, respectively). In the group of animals pretreated with CNQX (B; thick bar), no release at the time of injection (arrow) occurred. In the prolonged-release phase, delayed and attenuated release of GLU (P = 0.02) occurs with a significant decrease in the release of ASP (P = 0.04). If the animals were pretreated with AP7 (D; thick bar), no increase in ASP or GLU occurs at the time of injection (arrow), although there is a delayed peak in ASP between 30 minutes and one hour. No change in ASP or GLU concentration occurred in response to delivery of either CNQX or AP7.
BEHAVIORAL CHANGES ASSOCIATED WITH JOINT INFLAMMATION
Arthritic animals treated with a non-NMDA, an NMDA, or a GABAA receptor antagonist did not display the increased sensitivity to radiant heat applied to the paw (heat hyperalgesia) that is characteristic of arthritic rats (cf. Hargreaves et al. 1988). In untreated arthritic rats, a faster withdrawal to radiant heat (1–2 seconds) occurs by four hours and is maintained through 24 hours (Sluka and Westlund 1993d) (Fig. 2). This hyperalgesia represents a central neuronal sensitization because the paw itself was not inflamed, but is now responsive to stimuli that were previously innocuous. The blockade of hyperalgesia by CNQX, AP7, or bicuculline implies that polysynaptic mechanisms are involved in the development of heat hyperalgesia. In contrast, arthritic animals sympathectomized by surgery and/or by phentolamine (1 mg/kg) were still hyperalgesic similar to the untreated arthritic group.
Fig. 2.
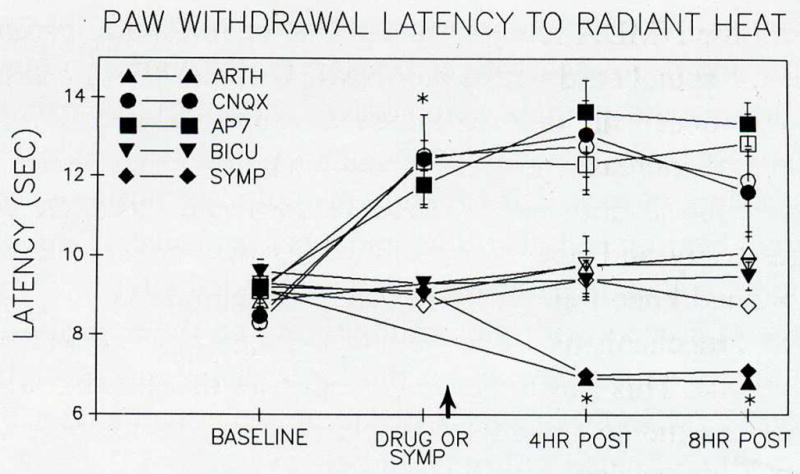
This graph shows paw withdrawal latency (PWL) to radiant heat for animals that were untreated (triangles), CNQX-treated (circles), AP7-treated (squares), bicuculline-treated (inverse triangles), and sympathectomized (diamonds) arthritic animals. The response latency of the hindpaw ipsilateral to the inflamed knee joint is represented by filled symbols whereas the contralateral side is represented by unfilled symbols. Before the induction of arthritis or infusion of the antagonists, the withdrawal latencies averaged between 8 and 10 seconds. The receptor antagonists were infused for the duration of the experiment to account for differences in length of effectiveness of the drugs. After one hour infusion of either CNQX (non-NMDA) or AP7 (NMDA) through the dialysis fiber, the PWL increased significantly for both the ipsilateral and the contralateral sides when compared to baseline (paired t test; P ≤ 0.05), suggesting the animals were hypoalgesic. Following the induction of arthritis, untreated and sympathectomized arthritic animals showed a 1–2 second decrease in PWL to radiant heat when compared to baseline at four and eight hours after injection of kaolin and carrageenan. In the animals treated with either CNQX or AP7, there was no further decrease in withdrawal times of either the ipsilateral or contralateral paws after induction of arthritis when compared to the withdrawal times induced by the application of the antagonists. Therefore, the heat hyperalgesia seen in untreated arthritic animals is blocked by the treatment of the spinal cord with either a non-NMDA (CNQX), an NMDA (AP7), or a GABAA (bicuculline) receptor antagonist.
In another model, the heat hyperalgesia associated with inflammation of the paw by carrageenan appears to be reversed by posttreatment with intrathecal injection of NMDA receptor antagonists and not of non-NMDA antagonists (Ren et al. 1992). This may reflect the time when the antagonists were delivered, i.e., pretreatment vs. posttreatment. We pretreated animals with the antagonists. If the animal is already hyperalgesic (as in the study by Ren and colleagues), the further activation of the non-NMDA receptor may not be a requirement for continued sensitization of central neurons. NMDA receptor activation is necessary for wind-up (Davies and Lodge 1987) and also is involved in reducing the hyperexcitability of already sensitized dorsal horn neurons (Schaible et al. 1991). Therefore, the differences between the two studies suggest a critical time of non-NMDA receptor activation during the initial stages of the development of the inflammation, while the secondary hyperalgesia, once developed, is maintained by activation of the NMDA receptor. Another likely explanation concerns methodological differences between the delivery of the drugs to the superficial dorsal horn by intrathecal administration versus the deeper dorsal horn by microdialysis.
The injection of kaolin and carrageenan into the knee joint cavity results in guarding of and decreased weight bearing on the limb. To quantify these behavioral changes, the animals were graded by a subjective pain rating scale (0–5) modified from Guilbaud and colleagues (Attal et al. 1990) where: 0 is normal; 1 is curling of toes; 2 is eversion of foot; 3 is partial weight bearing; 4 is non–weight bearing and guarding; and 5 is avoidance of any contact with the limb. The untreated arthritic animals were graded 4.6 ± 0.15; they exhibited avoidance of contact with the limb by lying on the opposite flank in the cage and/or they would not walk on the limb at the end of eight hours. In contrast, scores of CNQX-treated arthritic animals averaged 1.5 ± 0.5 and those of bicuculline-treated arthritic animals averaged 1.2 ± 0.2. These animals all exhibited curling of the toes and some exhibited eversion of the foot. None of the animals in either group limped or guarded the limb. Similar to the untreated arthritic animals, subjective pain ratings for AP7-treated arthritic animals averaged 4.25 ± 0.4 and the sympathectomized animals averaged 4.3 ± 0.2. All of these animals demonstrated guarding of the limb and no weight bearing during ambulation of the limb. Therefore, the behavioral manifestations were greatly decreased only by treatment with the non-NMDA and the GABAA receptor antagonists and not by treatment with the NMDA receptor antagonist or by sympathectomy, which is consistent with a decrease in the degree of peripheral inflammation.
CENTRAL CONTROL OF JOINT INFLAMMATION
One of the most surprising findings was that spinal cord administration of either a non-NMDA (CNQX; 1.1 mM) or a GABAA (bicuculline; 0.01 mM) receptor antagonist prior to induction of arthritis significantly reduced the extent of joint inflammation compared to untreated arthritic animals (Fig. 3). Measurement of knee joint circumference revealed that the joint circumferences in animals pretreated with a non-NMDA or a GABAA receptor antagonist were significantly reduced compared to untreated arthritic animals. Similarly, following unilateral dorsal rhizotomy (L2–L6, 2–3 days) the joint circumferences of inflamed knees were significantly less than those in untreated arthritic animals (Fig. 3). In contrast, treatment of the spinal cord with an NMDA receptor antagonist did not reduce the degree of peripheral joint inflammation when compared to untreated arthritic animals. Nor did a combination of surgical and chemical (phentolamine, 1 mg/kg) sympathectomies reduce the degree of inflammation.
Fig. 3.

The circumference of the knee joint was measured before and eight hours after induction of arthritis for the inflamed and the contralateral knee joints. The inflamed knee joint significantly (paired t test; P < 0.05) increased in girth when compared to its own size before induction of arthritis. The sympathectomized arthritic animals and those treated with AP7, an NMDA receptor antagonist, were significantly increased from baseline for the inflamed knee and were not significantly different from the untreated arthritic group. In animals treated with the non-NMDA (CNQX) or the GABAA (bicuculline) receptor antagonist, the increase in girth was approximately half of the untreated arthritic animals and was significantly less than that of untreated arthritic animals. Difference from untreated arthritic animals (Scheffé test) is represented by an asterisk (*).
As a control for systemic effects, the microdialysis fiber was placed into the sacral spinal cord and the antagonist (CNQX or bicuculline) administered in the same manner as in all other experiments. In these animals the degree of joint inflammation and the behavioral changes were similar to those of untreated arthritic animals. Therefore, the application of CNQX and bicuculline must be into the levels of the spinal cord receiving articular inputs to interfere with the progression of the inflammation and the resultant behavioral changes. Further, this finding demonstrated that application to the spinal cord through the microdialysis fiber remained confined regionally. Additionally, in animals (n = 3) in which methylene blue dye was passed through the fiber for the duration of infusion of the antagonist, the spread of the dye did not extend beyond one spinal segment or ventrally into the motor neuron pool.
Thermographic readings taken before and after induction of arthritis revealed that a major temperature increase occurs in the knee joint with inflammation. Similar changes were evident for arthritic animals after receiving treatment with the NMDA receptor antagonist or following sympathectomy. Treatment of the spinal cord with a non-NMDA or a GABAA receptor antagonist, however, reduced the thermal increases typical in knee joint inflammation. The thermal increases also were reduced following dorsal rhizotomy. The reduction in the severity of the inflammatory events by CNQX, bicuculline, or dorsal rhizotomy and not by AP7 or sympathectomy, as determined by thermographic and knee joint circumference measurements, suggest that differential control mechanisms are involved for the peripheral inflammatory and nociceptive events. This finding is consistent with the decrease in the degree of inflammatory exudate present in the knee joints of animals treated with CNQX, bicuculline, or dorsal rhizotomy.
DISCUSSION
These data support a contribution by central pathways to the development of peripheral joint inflammation. The central pathway(s) involves spinal non-NMDA (Sluka and Westlund 1993b) and GABAA (Sluka et al. 1993) receptors as well as the central terminals of primary afferent fibers. These data demonstrate that a central neuronal pathway exists that when blocked at the spinal cord level, not only reduces the transmission of nociceptive information but also influences the development of peripheral inflammation. Consequent behavioral manifestations of the arthritis also are decreased. Peripheral joint inflammation is reduced by dorsal rhizotomy, spinal application of CNQX or bicuculline, but not by AP7 or sympathectomy. Efferent activity was recorded in knee joint afferents in response to mechanical stimulation of the hindlimb following induction of acute arthritis (Rees et al. 1994). Thus, we hypothesize that the central pathway for increasing peripheral inflammation involves primary afferent depolarization of terminals in the dorsal horn and the consequent dorsal root reflexes (DRRs). The central terminals of primary afferent fibers must be intact for the joint inflammation to develop fully. The peripheral terminals of these afferents may directly release neuropeptides into the joint when invaded by antidromic nerve impulses (DRRs) triggered by primary afferent depolarization (PAD).
There is substantial evidence for the involvement of the peripheral nervous system in the inflammatory process. Peripherally, changes in content of fibers labeled for SP and CGRP occur in both human inflammatory conditions and animal models of inflammation (Mapp et al. 1990; Pereira da Silva and Foneseca 1990; Weihe et al. 1988). A peripheral release of SP and CGRP occurs in the inflammatory exudate of the joint (Appelgren et al. 1991; Larsson et al. 1991; Marshall et al. 1990) and these peptides increase the inflammatory events in the periphery (Levine et al. 1984; Brain and Williams 1985). This indicates that primary afferent neurons are involved in tissue extravasation during arthritis. Additionally, denervation of the knee joint or intra-articular injection of capsaicin, which depletes SP and CGRP, reduces peripheral joint inflammation (Lam and Ferrell 1989a,b).
Centrally, an early depletion of SP followed by a later increase of SP and CGRP are observed after induction of arthritis (Sluka et al. 1992; Sluka and Westlund 1993d). These dorsal horn changes occur across the entire superficial dorsal horn (Sluka et al. 1992; Sluka and Westlund 1993d). This includes not only the termination sites of the knee joint afferents (Craig et al. 1988), but also areas receiving input from cutaneous, muscle, and other joint primary afferent fibers. Dorsal horn CGRP is solely of primary afferent origin (Chung et al. 1988), so widespread release suggests long-term facilitatory events are occurring that affect a large number of primary afferent fibers. The increased degree of inflammation mediated through non-NMDA and GABAA mechanisms in the spinal cord and the widespread changes in GLU, SP, and CGRP (Sluka et al. 1992; Sluka and Westlund 1993d) that occur following induction of arthritis may be the result of PAD, which occurs through GABAA synapses on the central terminals of primary afferent fibers (Eccles et al. 1963; Jiminez et al. 1987). This is consistent with the reduction in the peripheral inflammation by spinal application of a GABAA receptor antagonist, bicuculline. Increased extracellular potassium concentration also results in PAD (Svobada et al. 1988), and a small increase [K+] has been shown in this arthritis model (Heinemann et al. 1990). Additionally, activation of descending pathways causes a depolarization of primary afferent fibers (Carpenter et al. 1963; Martin et al. 1979). This central pathway influencing joint inflammation is further supported by recent studies that demonstrated: (1) an increased release of neuropeptides into the knee joint on the side contralateral to the inflammation (Bileviciute et al. 1993), and (2) a blockade of the spread of inflammation to the contralateral limb following capsaicin pretreatment of either sciatic nerve (Donaldson et al. 1993).
Another alternative is increased activity in muscle efferents, which would result in behavioral signs similar to those observed in this model, i.e., limping, and guarding of the limb. Indeed, an increase in activity in muscle efferents has been observed in a variety of nociceptive conditions (Woolf 1983; Woolf and Wall 1986; Woolf and Thompson 1991). However, though many mechanisms may be responsible for the central control of peripheral joint inflammation, we believe that DRRs triggered by PAD are likely to play a major role.
The increased central EAA release and content of GLU, SP, and CGRP (Schaible et al. 1990; Sluka and Westlund 1992, 1993d; Sorkin et al. 1992) may contribute to the sensitization of dorsal horn neurons. In fact, the release and increased content of GLU is blocked by spinal application of either a non-NMDA or an NMDA receptor antagonist, while the increase in SP is blocked only by non-NMDA receptor antagonists (Sluka and Westlund 1993c). In addition, the changes in PWL have been correlated with the changes in GLU immunoreactivity in the superficial dorsal horn (Sluka and Westlund 1993d), further implicating GLU in the development of heat hyperalgesia. Lastly, central sensitization can be reduced by iontophoretic application of an NMDA receptor antagonist (Schaible et al. 1991). Increased responsiveness of nociceptive neurons due to sensitization would lead to an increased firing of the neurons and therefore an increased transmission of nociceptive information to higher centers.
The heat hyperalgesia of the paw that develops in these arthritic animals reflects a central neuronal sensitization because the inflammation itself is localized to the knee joint and does not include the sensitized paw. The sensitization of central neurons (Dougherty et al. 1992b) could be a result of the increased spinal release and content of EAAs (Sorkin et al. 1992; Sluka et al. 1992; Sluka and Westlund 1992, 1993a,c) and of neuropeptides (Schaible et al. 1990; Sluka et al. 1992; Sluka and Westlund 1993d). The heat hyperalgesia of the paw does not occur if EAA release and content are blocked by both non-NMDA and NMDA receptor antagonists (Sluka and Westlund 1993a,b,c). Previous studies have demonstrated involvement of NMDA (Aanonsen and Wilcox 1987; Ault and Hildebrand 1993; Ren et al. 1992; Schaible et al. 1991; Nasstrom and Karlsson 1992) and non-NMDA (Ault and Hildebrand 1993; Dougherty et al. 1992a,b; Näsström et al. 1992; Sluka and Westlund 1993a) receptors in a variety of nociceptive events.
In summary, these studies indicate that there are central neuronal networks not only for processing nociceptive information but also for activating events resulting in peripheral inflammation. Transmission of the nociceptive information involves the release of the EAAs, ASP, and GLU, as well the activation of both non-NMDA and NMDA receptors. In contrast, the development of the peripheral inflammation and the behavioral manifestations (guarding and limping) are affected differentially by the two EAA receptor types. The joint inflammation is also reduced by spinal administration of a GABAA receptor antagonist and dorsal rhizotomy. The central pathway initially involves the activation of non-NMDA receptors in the spinal cord that in turn activate GABAA synapses on the central terminals of primary afferent fibers. Activation of these terminals would cause PAD and dorsal root reflexes that would produce a peripheral release of inflammatory neuropeptides into the joint. Clinically, these data seem to suggest that recovery time following surgical trauma and joint inflammation could be significantly reduced with preemptive treatment of the anticipated pain and inflammatory events.
Acknowledgments
We thank Heidi Jordan for surgical assistance with the microdialysis experiments. This was supported by NIH grants NS11255 and NS01445, RO1 NS28064, and an unrestricted grant from the Bristol-Myers Squibb Company.
References
- Aanonsen LM, Wilcox GL. Nociceptive action of excitatory amino acids in the mouse: effects of spinally administered opioids, phencyclidine and sigma agonists. J Pharmacol Exp Ther. 1987;243:9–19. [PubMed] [Google Scholar]
- Appelgren A, Appelgren B, Eriksson S, Kopp S, Lundeberg T, Nylander M, Theodorsson E. Neuropeptides in temporomandibular joints with rheumatoid arthritis: a clinical study. Scand J Dent Res. 1991;99:519–521. doi: 10.1111/j.1600-0722.1991.tb01063.x. [DOI] [PubMed] [Google Scholar]
- Attal N, Jazat F, Kayser V, Guilbaud G. Further evidence for “pain-related” behaviours in a model of unilateral peripheral mononeuropathy. Pain. 1990;41:235–251. doi: 10.1016/0304-3959(90)90022-6. [DOI] [PubMed] [Google Scholar]
- Ault B, Hildebrand LM. Effects of excitatory amino acid receptor antagonists on a capsaicin-evoked nociceptive reflex: a comparison with morphine, clonidine, and baclofen. Pain. 1993;52:341–349. doi: 10.1016/0304-3959(93)90168-O. [DOI] [PubMed] [Google Scholar]
- Bileviciute I, Lundeberg T, Edblom A, Theodorssan E. Bilateral changes of substance P-, neurokinin A-, calcitonin gene-related peptide- and neuropeptide Y-like immunoreactivity in rat knee joint synovial fluid during acute monoarthritis. Neurosci Lett. 1993;153:37–40. doi: 10.1016/0304-3940(93)90071-r. [DOI] [PubMed] [Google Scholar]
- Brain SD, Williams TJ. Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br J Pharm. 1985;86:855–860. doi: 10.1111/j.1476-5381.1985.tb11107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter D, Lundberg A, Norrsell U. Primary afferent depolarization evoked from the sensorimotor cortex. Acta Physiol Scand. 1963;59:126–142. doi: 10.1111/j.1748-1716.1963.tb02729.x. [DOI] [PubMed] [Google Scholar]
- Chung K, Lee WT, Carlton SM. The effects of dorsal rhizotomy and spinal cord isolation on calcitonin gene-related peptide-labeled terminals in the rat lumbar dorsal horn. Neurosci Lett. 1988;90:27–32. doi: 10.1016/0304-3940(88)90781-1. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Hong KAP, Langford LA, Schaible H-G, Schmidt RF. Discharge characteristics of fine medial articular afferents at rest and during passive movements of inflamed knee joints. Brain Res. 1983;272:185–188. doi: 10.1016/0006-8993(83)90379-7. [DOI] [PubMed] [Google Scholar]
- Craig AD, Heppelmann B, Schaible H-G. The projection of the medial and posterior articular nerves of the cat’s knee to the spinal cord. J Comp Neurol. 1988;276:279–288. doi: 10.1002/cne.902760210. [DOI] [PubMed] [Google Scholar]
- Davies SN, Lodge D. Evidence for involvement of N-methylaspartate receptors in “wind-up” of class 2 neurons in the dorsal horn of the rat. Brain Res. 1987;424:402–406. doi: 10.1016/0006-8993(87)91487-9. [DOI] [PubMed] [Google Scholar]
- Donaldson LF, McQueen DS, Seckl JR. Abstract, XXXII Congress of the International Union of Physiological Sciences. 1993. Inhibition of spread of inflammation by perineural capsaicin: involvement of the nervous system in experimental arthritis. [Google Scholar]
- Dougherty PM, Palecek J, Palecekova V, Sorkin LS, Willis WD. The role of NMDA and non-NMDA excitatory amino acid receptors in the excitation of primate spinothalamic tract neurons by mechanical, chemical, thermal, and electrical stimuli. J Neurosci. 1992a;12:3025–3041. doi: 10.1523/JNEUROSCI.12-08-03025.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Sluka KA, Sorkin LS, Westlund KN, Willis WD. Neural changes in acute arthritis in monkeys I. Parallel enhancement of responses of spinothalamic tract neurons to mechanical stimuli and excitatory amino acids. Brain Res Rev. 1992b;17:1–13. doi: 10.1016/0165-0173(92)90002-4. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Schmidt RF, Willis WD. Pharmacological studies on presynaptic inhibition. J Physiol. 1963;168:500–530. doi: 10.1113/jphysiol.1963.sp007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Schaible H-G, Schmidt RF. Changes in extracellular postassium concentration in cat spinal cord in response to innocuous and noxious stimulation of legs with healthy and inflamed knee joints. Exp Brain Res. 1990;79:283–292. doi: 10.1007/BF00608237. [DOI] [PubMed] [Google Scholar]
- Hurley JV. Acute Inflammation. Churchill Livingstone; New York: 1983. [Google Scholar]
- Jimenez I, Rudomin P, Solodkin M. Mechanisms involved in the depolarization of cutaneous afferents produced by segmental and descending inputs in the cat spinal cord. Exp Brain Res. 1987;69:195–207. doi: 10.1007/BF00247042. [DOI] [PubMed] [Google Scholar]
- Lam FY, Ferrell WR. Inhibition of carrageenan induced inflammation in the rat knee by substance P antagonist. Ann Rheum Dis. 1989a;48:928–932. doi: 10.1136/ard.48.11.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FY, Ferrell WR. Capsaicin suppresses substance P-induced joint inflammation in the rat. Neurosci Lett. 1989b;105:155–158. doi: 10.1016/0304-3940(89)90028-1. [DOI] [PubMed] [Google Scholar]
- Larsson J, Ekblom A, Henriksson K, Lundeberg T, Theodorsson E. Concentration of substance P, neurokinin A, calcitonin gene-related peptide, neuropeptide Y and vasoactive intestinal polypeptide in synovial fluid from knee joints in patients suffering from rheumatoid arthritis. Scand J Rheumatol. 1991;20:326–335. doi: 10.3109/03009749109096808. [DOI] [PubMed] [Google Scholar]
- Levine JD, Clark R, Devor M, Helms C, Moskowitz MA, Basbaum AI. Intraneuronal substance P contributes to the severity of experimental arthritis. Science. 1984;226:547–549. doi: 10.1126/science.6208609. [DOI] [PubMed] [Google Scholar]
- Levine JD, Dardick SJ, Roizen MF, Helms C, Basbaum AI. Contribution of sensory afferents and sympathetic afferents to joint injury in experimental arthritis. J Neurosci. 1986;6:3423–3429. doi: 10.1523/JNEUROSCI.06-12-03423.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Coderre TJ, Basbaum AI. The peripheral nervous system and the inflammatory process. In: Dubner R, Gebhart GF, Bond MR, editors. Proceedings of the Vth World Congress on Pain. Pain Research and Clinical Management; Amsterdam: Elsevier; 1988. pp. 33–43. [Google Scholar]
- Mapp PI, Kidd BL, Gibson SJ, Terry JM, Revell PA, Ibrahim NBN, Blake DR, Polak JM. Substance P-, calcitonin gene-related peptide- and C-flanking peptide of neuropeptide Y-immunoreactive fibres are present in normal synovium but depleted in patients with rheumatoid arthritis. Neuroscience. 1990;37:143–153. doi: 10.1016/0306-4522(90)90199-e. [DOI] [PubMed] [Google Scholar]
- Marshall KW, Chiu B, Inman RD. Substance P and arthritis: analysis of plasma and synovial fluid levels. Arthritis Rheum. 1990;33:87–90. doi: 10.1002/art.1780330111. [DOI] [PubMed] [Google Scholar]
- Martin RF, Haber LH, Willis WD. Primary afferent depolarization of identified cutaneous fibers following stimulation in medial brain stem. J Neurophys. 1979;42:779–790. doi: 10.1152/jn.1979.42.3.779. [DOI] [PubMed] [Google Scholar]
- Näsström J, Karlsson U, Post C. Antinociceptive actions of different classes of excitatory amino-acid receptor antagonists in mice. Europ J Pharmacol. 1992;212:21–29. doi: 10.1016/0014-2999(92)90067-e. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Schaible H-G. Evidence for a central component in the sensitization of spinal neurons with joint input during development of acute arthritis in cat’s knee. J Neurophys. 1990;64:303–310. doi: 10.1152/jn.1990.64.1.299. [DOI] [PubMed] [Google Scholar]
- Pereira da Silva JA, Carmo-Fonseca M. Peptide containing nerves in human synovium: immunohistochemical evidence for decreased innervation in rheumatoid arthritis. J Rheumatol. 1990;17:1592–1599. [PubMed] [Google Scholar]
- Rees H, Sluka KA, Westlund KN, Willis WD. Do dorsal root reflexes augment peripheral inflammation? Neuroreport. 1994;5:821–824. doi: 10.1097/00001756-199403000-00021. [DOI] [PubMed] [Google Scholar]
- Ren K, Williams GM, Hylden JLK, Ruda MA, Dubner R. The intrathecal administration of excitatory amino-acid receptor antagonists selectively attenuated carrageenan-induced behavioral hyperalgesia in rats. Europ J Pharmacol. 1992;219:235–243. doi: 10.1016/0014-2999(92)90301-j. [DOI] [PubMed] [Google Scholar]
- Schaible H-G, Schmidt RF, Willis WD. Enhancement of the responses of ascending tract cells in the cat spinal cord by acute inflammation of the knee joint. Exp Brain Res. 1987;66:466–489. doi: 10.1007/BF00270681. [DOI] [PubMed] [Google Scholar]
- Schaible H-G, Jarrot B, Hope PJ, Duggan AW. Release of immunoreactive substance P in the spinal cord during development of acute arthritis in the knee joint of the cat: a study with antibody microprobes. Brain Res. 1990;529:214–223. doi: 10.1016/0006-8993(90)90830-5. [DOI] [PubMed] [Google Scholar]
- Schaible H-G, Grubb BD, Neugebauer V, Oppman M. The effects of NMDA antagonists on neuronal activity in cat spinal cord evoked by acute inflammation in the knee joint. Europ J Neurosci. 1991;3:981–991. doi: 10.1111/j.1460-9568.1991.tb00034.x. [DOI] [PubMed] [Google Scholar]
- Skilling SR, Smullin HD, Beitz AJ, Larsson AA. Extracellular amino acid concentrations in the dorsal spinal cord of freely moving rats following veratridine and nociceptive stimulation. J Neurochem. 1988;51:127–132. doi: 10.1111/j.1471-4159.1988.tb04845.x. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Westlund KN. An experimental arthritis in rat: dorsal horn aspartate and glutamate increases. Neurosci Lett. 1992;145:141–144. doi: 10.1016/0304-3940(92)90006-s. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Westlund KN. An experimental arthritis in rat: the effects on non-NMDA and NMDA receptor antagonists. Neurosci Lett. 1993a;149:99–102. doi: 10.1016/0304-3940(93)90357-q. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Westlund KN. Centrally administered non-NMDA but not NMDA receptor antagonists block peripheral knee joint inflammation. Pain. 1993b;55:217–225. doi: 10.1016/0304-3959(93)90150-N. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Westlund KN. Spinal cord amino acid release and content in an arthritis model: the effects of pretreatment with non-NMDA, NMDA, and NK1 receptor antagonists. Brain Res. 1993c;627:89–103. doi: 10.1016/0006-8993(93)90752-9. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Westlund KN. Behavioral and immunohistochemical changes in an experimental arthritis model in rats. Pain. 1993d;55:367–377. doi: 10.1016/0304-3959(93)90013-F. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Dougherty PM, Sorkin LS, Willis WD, Westlund KN. Neural changes in acute arthritis in monkeys III. Changes in substance P, calcitonin gene-related peptide and glutamate in the dorsal horn of the spinal cord. Brain Res Rev. 1992;17:29–38. doi: 10.1016/0165-0173(92)90004-6. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Willis WD, Westlund KN. Joint inflammation and hyperalgesia are reduced by spinal bicuculline. Neuroreport. 1993;5:109–112. doi: 10.1097/00001756-199311180-00003. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Steinman JL, Hughes MG, Willis WD, McAdoo DJ. Microdialysis recovery of serotonin released in spinal cord dorsal horn. J Neurosci Methods. 1988;23:131–138. doi: 10.1016/0165-0270(88)90185-9. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Sluka KA, Dougherty PM, Westlund KN, Willis WD. Neural changes in acute arthritis in monkeys IV. Time-course of amino acid release into the lumbar dorsal horn. Brain Res Rev. 1992;17:39–50. doi: 10.1016/0165-0173(92)90005-7. [DOI] [PubMed] [Google Scholar]
- Svoboda J, Motin V, Hajek I, Sykova E. Increase in extracellular potassium level in rat spinal dorsal horn induced by noxious stimulation and peripheral injury. Brain Res. 1988;458:97–105. doi: 10.1016/0006-8993(88)90500-8. [DOI] [PubMed] [Google Scholar]
- Weihe E, Nohr D, Millan MJ, Stein C, Müller S, Gramsch C, Herz A. Peptide neuroanatomy of adjuvant-induced arthritic inflammation in rat. Agents Actions. 1988;25:255–259. doi: 10.1007/BF01965027. [DOI] [PubMed] [Google Scholar]
- Westlund KN, Sun YC, Sluka KA, Dougherty PM, Sorkin LS, Willis WD. Neural changes in acute arthritis in monkeys II. Increased glutamate immunoreactivity in the medial articular nerve. Brain Res Rev. 1992;17:15–27. doi: 10.1016/0165-0173(92)90003-5. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Thompson SWN. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–299. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of diffferent origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci. 1986;6:1433–1442. doi: 10.1523/JNEUROSCI.06-05-01433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Bailey J, Roddy DR, Harty GJ. Peripheral release of substance P from primary afferents. In: Dubner R, Gebhart GF, Bond MR, editors. Proceedings of the Vth World Congress on Pain. Pain Research and Clinical Management; Amsterdam: Elsevier; 1988. pp. 51–54. [Google Scholar]