

Background: Ubiquitination and degradation promote signal termination of the Ret receptor.
Results: Cbl-3/c and CD2AP are both required for the ubiquitination of Ret51 on Lys1060 and Lys1107.
Conclusion: The Ret51 isoform-specific ubiquitination on Lys1060 and Lys1107 may underlie its rapid degradation.
Significance: Regulation of CD2AP·Cbl-3/c activity may underlie the diversity of functional outcomes of Ret signal transduction.
Keywords: Neurodevelopment, Neurotrophic Factor, Protein Degradation, Receptor Tyrosine Kinase, Ubiquitination
Abstract
Ret is the receptor tyrosine kinase for the glial cell line-derived neurotrophic factor (GDNF) family of neuronal growth factors. Upon activation by GDNF, Ret is rapidly polyubiquitinated and degraded. This degradation process is isoform-selective, with the longer Ret51 isoform exhibiting different degradation kinetics than the shorter isoform, Ret9. In sympathetic neurons, Ret degradation is induced, at least in part, by a complex consisting of the adaptor protein CD2AP and the E3-ligase Cbl-3/c. Knockdown of Cbl-3/c using siRNA reduced the GDNF-induced ubiquitination and degradation of Ret51 in neurons and podocytes, suggesting that Cbl-3/c was a predominant E3 ligase for Ret. Coexpression of CD2AP with Cbl-3/c augmented the ubiquitination of Ret51 as compared with the expression of Cbl-3/c alone. Ret51 ubiquitination by the CD2AP·Cbl-3/c complex required a functional ring finger and TKB domain in Cbl-3/c. The SH3 domains of CD2AP were sufficient to drive the Cbl-3/c-dependent ubiquitination of Ret51, whereas the carboxyl-terminal coiled-coil domain of CD2AP was dispensable. Interestingly, activated Ret induced the degradation of CD2AP, but not Cbl-3/c, suggesting a potential inhibitory feedback mechanism. There were only two major ubiquitination sites in Ret51, Lys1060 and Lys1107, and the combined mutation of these lysines almost completely eliminated both the ubiquitination and degradation of Ret51. Ret9 was not ubiquitinated by the CD2AP·Cbl-3/c complex, suggesting that Ret9 was down-regulated by a fundamentally different mechanism. Taken together, these results suggest that only the SH3 domains of CD2AP were necessary to enhance the E3 ligase activity of Cbl-3/c toward Ret51.
Introduction
Ret is a receptor tyrosine kinase that is critical for the development of the nervous system and kidneys (1, 2). Ret is activated by a family of four growth factors known as the glial cell line-derived neurotrophic factor (GDNF)3 family ligands (GFLs). Each GFL binds to a preferred glycerophosphatidylinositol-anchored coreceptor, which are collectively known as the GFRαs. The GFRα·GFL complex binds and activates Ret (3). Ret autophosphorylation on multiple tyrosine residues initiates the association of adaptor proteins and enzymes that trigger second messenger cascades (4). Receptor tyrosine kinase activity in general is tightly regulated, and the two predominant mechanisms of down-regulation of receptor tyrosine kinase signaling are dephosphoryation of the receptor by tyrosine phosphatases and ubiquitination and subsequent degradation of the receptor. Often both of these processes occur simultaneously to the same receptor, and mutations in either of these down-regulation mechanisms can lead to several types of cancers. In the case of Ret, degradation appears to be the predominant mechanism of down-regulation for the longer and more potently transforming isoform of Ret, Ret51 (5–7). The shorter isoform of Ret, Ret9, has 9 carboxyl-terminal amino acids that are different from the 51 carboxyl-terminal amino acids of Ret51. Interestingly, Ret9 is not as rapidly degraded as Ret51 is in sympathetic neurons, which is one of the most well studied cell types for Ret signal transduction. The mechanism of Ret9 down-regulation is poorly understood (5–7).
The rapid degradation of Ret51 is initiated by ubiquitination, which triggers its degradation predominantly by proteasomes (5, 6). The Cbl family of E3 ligases appears to be critical for the ubiquitination of Ret51, with both c-Cbl and Cbl-3/c capable of binding and ubiquitinating Ret51, thereby promoting its degradation (6, 8). In primary sympathetic neurons, Ret51 degradation occurs primarily via Cbl-3/c, although Cbl-b also can associate with Ret51 (5, 8). Cbl-3/c differs significantly from the other two mammalian Cbls in that it lacks a large portion of the carboxyl-terminal region present in c-Cbl and Cbl-b. This region contains an ubiquitin-associated domain that is involved in ubiquitin binding, several regulatory phosphorylation sites, and the majority of a proline-rich domain involved in the binding of adapter proteins (9–12). Nevertheless, Cbl-3/c has an active catalytic ring finger domain and can ubiquitinate proteins, thus targeting them for internalization and degradation (10, 11). In sympathetic neurons, Cbl-3/c associates with the adaptor protein CD2AP, which appears to promote Ret51 degradation via Cbl-3/c (8). CD2AP, along with its paralog CIN85, regulate the association of Cbl proteins with receptors, thereby linking these receptor complexes to the internalization machinery (13–17). Interestingly, in glomerular podocytes, CD2AP deficiency induces the up-regulation of CIN85, leading to an excessive inhibition of receptor tyrosine kinase signaling (18). In the case of Ret51, it is not known how CD2AP augments the degradative function of Cbl-3/c or whether CD2AP has any effect on the E3 ligase activity of Cbl-3/c.
The turnover of activated Ret complexes is of paramount importance to the development and maintenance of peripheral neurons. In sensory neurons of the dorsal root ganglia, Ret51 is not degraded nearly as rapidly as in sympathetic neurons (19). In dorsal root ganglia sensory neurons, GFLs can promote survival over long axonal distances from axon terminals to cell bodies, which requires the internalization and retrograde transport of activated Ret complexes (19). Early postnatal sympathetic neurons, in contrast, cannot be supported over long axonal distances by GFLs because activated Ret complexes are degraded rapidly prior to any significant amount of Ret51 being retrogradely transported (19). Thus, mechanisms of Ret turnover, at a subcellular level, dictate what type of functional outcome that GFL will have. Mechanisms of Ret turnover are also likely to be a point of regulation for coincidence detection, and recent evidence suggests that reverse signaling by ephrin receptors promotes motor neuron axon guidance via the augmentation of local Ret signaling events (20).
In this study, we sought to illuminate further the mechanism by which the CD2AP·Cbl-3/c complex promotes the degradation of Ret. The ubiquitination activity of the CD2AP·Cbl-3/c complex was analyzed in detail, and it was discovered that CD2AP enhances Cbl-3/c-dependent ubiquitination of Ret51 via the SH3 domains of CD2AP, possibly by associating with Cbl-3/c and relieving intramolecular autoinhibition of Cbl-3 (21). Ret9 was not ubiquitinated by the CD2AP·Cbl-3/c complex, suggesting a mechanism to explain the more rapid degradation of Ret51, as compared with Ret9, in several populations of neurons. Interestingly, there are only two major ubiquitination sites in Ret51, Lys1060 and Lys1107, which upon mutation led to the markedly increased stability of Ret51. These data suggest that CD2AP and Cbl-3/c coordinately act to promote Ret51 ubiquitination and degradation upon its activation and are likely to be a regulatory point by which other signaling pathways modulate Ret activity for the purposes of promoting axon growth and guidance. The recent discovery that CD2AP is a susceptibility locus for Alzheimer disease underscores the potential relevance of this ubiquitination mechanism in neurodegeneration (39, 40).
EXPERIMENTAL PROCEDURES
Mammalian Cell Cultures
NIH3T3 cells were maintained in DMEM supplemented with FBS (10% by volume) and a penicillin/streptomycin/glutamine mixture. These cells were maintained at 37 °C with 5% CO2. NIH3T3 cells were grown on 4-well chambered coverglass slides (Lab-Tek, ThermoFisher Scientific, Inc.) for live cell imaging or in 6-well tissue culture plates (Falcon) for biochemical studies. The cells were allowed to proliferate until an approximate density of 70% confluence was obtained prior to transfection. Primary sympathetic neurons were produced and maintained as described previously (22). Cultures of differentiated murine podocytes were also produced and maintained in vitro as described previously (23, 24).
Transfections and Plasmids
Transfections of NIH3T3 cells were performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). For experiments that monitored ubiquitination, 48 h after transfection the cells were treated with both concanamycin (200 nm) and epoxomicin (5 μm) for 6–12 h to inhibit lysosomal and proteasomal degradation, respectively. Even in the presence of these inhibitors, we still occasionally observed a loss of Ret51 protein in conditions of high ubiquitination, such as in Fig. 1G. For inhibition of Ret kinase activity, two structurally distinct inhibitors (RPI-1 and vatalanib; Santa Cruz) were added to the culture medium at final concentrations of 60 and 30 μm, respectively, at the same time that protease inhibitors were added (6–12 h). For biochemical experiments on Ret51 turnover, the NIH3T3 cells were treated with cycloheximide (1 μm) to inhibit protein translation, thereby revealing the half-life of Ret51. The plasmids encoding CD2AP, CD2AP 1–328, and CD2AP 561–681 were generously provided by Mireille Cormont (13). The plasmids encoding FLAG-tagged Cbl-3, Cbl-3 C351A, Cbl-3 G276E, and Cbl-3 TKB were a kind gift from Tadashi Yamamoto (25). Plasmids encoding point mutations in Cbl-3 (R390Q, W378A, and Y341F) were produced by site-directed mutagenesis using the QuikChange II kit (Agilent Technologies).
FIGURE 1.

Cbl-3 and CD2AP together promote the ubiquitination of Ret51. A, differentiated mouse podocytes were transfected with siRNA to either Cbl-3/c, or a scrambled, nonsilencing control siRNA (indicated above blots). After 48 h the cells were then stimulated with GDNF, or medium alone, for 24 h. Ret51 was immunoprecipitated from detergent extracts of cells subjected to these treatments, and the level of Ret degradation was determined by immunoblotting these immunoprecipitates with Ret51 antibodies (top panel). Cbl-3 immunoblotting (middle panel) of the supernatants displayed a reduction in Cbl-3 protein upon transfection with Cbl-3 siRNA. Actin immunoblotting of the supernatants confirmed that similar amounts of protein extracts were analyzed (bottom panel). B, primary sympathetic neurons were subjected to siRNA silencing of Cbl-3/c, as was done in A, and were stimulated with either GDNF or medium alone for 10 min, prior to any significant reduction in Ret51 levels. The level of Ret51 ubiquitination was determined by immunoblotting Ret51 immunoprecipitates derived from these neurons with antibodies to multiubiquitin (top panel). The amount of Ret51 in each condition was determined by reprobing the Ret51 immunoprecipitates with Ret 51 antibodies (upper middle panel). Cbl-3 immunoblotting of the supernatants from the immunoprecipitations confirmed that the transfection of siRNA targeted to Cbl-3/c reduced Cbl-3/c levels (lower middle panel), and actin immunoblotting of the supernatants again confirmed the analysis of equal amounts of protein (bottom panel). C, duplicate experiments were quantified and graphed as the means ± range. D, sympathetic neurons were subjected to Cbl-3 silencing, as in A, and ubiquitinated proteins were immunoprecipitated using polyubiquitin antibodies and probed with Ret51 antibodies. Actin confirmed the analysis of equal amounts of protein. E, NIH3T3 cells were transfected with CD2AP, Cbl-3/c, or CD2AP + Cbl-3/c along with Ret51 and HA-tagged ubiquitin. After 48 h, the cells were treated with proteasome and lysosome inhibitors and then detergent-extracted after an additional 6–12 h. The level of Ret51 ubiquitination in each condition was ascertained by immunoblotting Ret51 immunoprecipitates derived from these transfected cells with HA epitope tag antibodies (top panel). The level of Ret51, along with the levels of Cbl-3/c and CD2AP, were determined by immunoblotting, as was done in the middle panels of B. Actin immunoblotting of the supernatants served as a confirmation that similar amounts of protein were analyzed (bottom panel). F, quantifications of this experiment are shown as the means ± S.E. (three experiments were quantified). The asterisk indicates a statistically significant difference between the two conditions (p < 0.05), and N.S. indicates no significant difference. G, NIH3T3 cells were transfected with CD2AP and Cbl-3/c, or neither, along with Ret51 and HA-tagged ubiquitin. After 48 h, the cells were treated with proteasome and lysosome inhibitors along with the Ret kinase inhibitor RPI-1 (60 μm) and then detergent-extracted after an additional 6–12 h. H, Ret ubiquitination was monitored as in E, and the results were quantified. The data are presented as the means ± range. The experiments in this figure were repeated two or three times with similar results. IP, immunoprecipitation; W, Western blot.
In transfection experiments, the relative molecular mass of Cbl-3/c varied somewhat depending on whether it contained a FLAG tag. Lysine mutations in Ret51 (K1060R, K1107R, and dual mutant K1060R/K1107R) were also produced using site-directed mutagenesis. For the production of Ret51-kikume fusion proteins, human Ret51 was amplified by PCR and inserted into the BamHI and NotI restriction sites of a mammalian expression vector encoding humanized kikume, thereby placing kikume in frame on the carboxyl terminus of Ret51 (CoralHue Kikume, pKikGR1, MBL International Corp.). Both strands of the inserted cDNA were sequenced to confirm the absence of any mutations introduced during the cloning process.
Gene silencing in sympathetic neurons and podocytes was accomplished using siRNA, as we have done previously (8, 23). Briefly, siRNA against Cbl-3/c, or a scrambled control, were transfected into the cells using i-Fect according to the manufacturer's instructions (Neuromics). Biochemical experiments were performed 48–72 h after the transfection. Immunoblotting of Cbl-3/c confirmed that the siRNA transfection reduced the levels of Cbl-3/c by more than 60%. Cotransfection of a fluorescently labeled nontargeting siRNA confirmed that the transfection resulted in greater than 90% of the cells taking up the siRNA (siGLO RISC-free siRNA; Dharmacon RNA Technologies).
Immunoprecipitations
After the described transfections, cells were washed twice with ice-cold PBS (pH 7.4) and extracted with immunoprecipitation buffer (10 mm Tris-buffered saline, pH 7.4, 1% Nonidet P-40, 10% glycerol, 500 μm sodium vanadate, and protease inhibitors) by gentle agitation for 25 min at 4 °C. The cells were transferred into microcentrifuge tubes using cell scrapers. The lysates were cleared of nuclei and collagen debris by centrifuging at 13,000 × g for 5 min, and the supernatants were then transferred to new microcentrifuge tubes. To these extracts anti-Ret51 (C20, Santa Cruz Biotechnology), anti-multiubiquitin (FK2, Enzo Life Sciences), anti-Myc tag (Upstate Cell Signaling Solutions), or anti-FLAG (Sigma) was added along with 25 μl of protein A-agarose and 50 μl of protein G-agarose (Roche Applied Sciences). After incubation for 3 h at 4 °C, the immunoprecipitates were washed three times with immunoprecipitation buffer. The complexes were prepared for SDS-PAGE by adding 25 μl of 2× sample buffer (125 mm Tris, pH 6.8, 20% glycerol, 10% β-mercaptoethanol, 4% SDS, and 0.016% bromphenol blue) and heating the immunoprecipitates for 6 min in a boiling water bath.
Immunoblotting
Protein samples were loaded and separated on 4–12% gradient gels, or on 8 and 10% gels. After SDS-PAGE the proteins were transferred to PVDF membranes (Millipore). The blots were then blocked for 1 h with either 5% milk or 3% BSA (both in Tris-buffered saline containing 0.1% Tween 20 (TBST)). The blots were next incubated in the primary antibody in 3% BSA/TBST for 2 h at room temperature or overnight at 4 °C. The blots were washed three times and incubated in the appropriate secondary antibody for 1 h at room temperature. Lastly, the blots were washed and developed using an enhanced chemiluminescent substrate (Pierce-ThermoFisher). The antibodies for immunoblotting were as follows: anti-HA (1:1000 dilution; HA.11 Clone 16B12, Covance), anti-actin (1:1000 dilution; I-19, Santa Cruz Biotechnology), anti-CD2AP (1:500 dilution; H-290, Santa Cruz), anti-Cbl-C (1:500 dilution; Orbigen), anti-ubiquitin (1:200 dilution, U5379, Sigma), anti-multiubiquitin (1:2000 dilution, Clone FK2, Enzo Life Sciences), anti-Myc tag (1:500 dilution, Clone 9E10, Millipore), and anti-DYKDDDDK (FLAG, 1:1000 dilution, Clone 9A3, Cell Signaling). The migration characteristics of Ret may vary depending upon the percentage of gel it is run on and the extent of ubiquitination. In addition, Ret can appear as a doublet if it is not fully processed to the mature, glycosylated form, as occurs in some cell types (see Fig. 1). The immunoblots were stripped and reprobed by incubating them in glycine (100 mm, pH 2.75) twice for 15 min and neutralizing them with four washes of TBST. The immunoprecipitation immunoblots were reprobed with anti-Ret51 (rabbit, 1:1000 dilution), or anti-ectodomain Ret (1:250 dilution), both of which have been reported previously (7). Immunoblots were quantified using ImageJ software (National Institutes of Health) and graphed as the means ± range or S.E.
Live Cell Imaging
Transfected cells were imaged using a TCS SP5 II confocal microscope in resonance scan mode (Leica Microsystems, Inc.). Fluorescent cells were exposed to UV light (380 nm) for 12 s to permanently photoconvert the kikume fusion proteins from the green to the red photoisomer. The rate of degradation of the Ret51-kikume fusion proteins within individual cells was quantified by measuring the integrated intensity of the red fluorescence over the course of 40 min at intervals of 15 s. Images of the green and red photoisomers were observed using 488- and 546-nm lasers, respectively, and were viewed using the Leica Application Suite software. A resonance scanner was used to reduce the effects of photobleaching because of the rapid scan time (8000 Hz). The cells were maintained in a ZILCS environmental chamber (5% CO2 at 37 °C) during the entire period of data acquisition (Tokai Hit). Integrated intensity values from the Ret51-kikume degradation data from multiple trials were analyzed using SigmaPlot software.
Immunofluorescent Labeling
Transfected NIH3T3 cells were fixed with 1% paraformaldehyde for 30 min at room temperature, permeabilized with 0.2% Triton X-100, and blocked for 1 h using the mouse on mouse blocking kit according to the manufacturer's instructions (Vector Labs). The cells were then incubated with anti-FLAG (1:200 dilution) or anti-Myc (1:200 dilution) overnight at 4 °C. The cells were washed, incubated with a fluorescent secondary antibody (anti-mouse CF633, Biotium), washed again, and mounted with a fluorescence mounting medium (Vectashield, Vector Labs). The cells were imaged with a SP5 II confocal microscope in resonance scanning mode (Leica Microsystems).
RESULTS
CD2AP Promotes the Cbl-3/c-dependent Ubiquitination of Ret51
Previous experiments examining Ret degradation have focused on neurons. To determine whether non-neuronal cell types utilize Cbl-3/c for the ubiquitination and degradation of Ret, differentiated podocytes from the glomeruli of kidneys were subjected to siRNA silencing of Cbl-3/c. Podocytes under normal, uninjured conditions express Ret at low levels, along with the coreceptor for GDNF, GFRα1 (23). Podocytes transfected with siRNA were stimulated with GDNF, or medium alone, for 24 h to induce Ret activation and degradation. The cells were then detergent-extracted, and Ret51 was immunoprecipitated. Similar to sympathetic neurons (8), knockdown of Cbl-3/c inhibited the degradation of Ret triggered by GDNF stimulation (Fig. 1A). These data suggest that Cbl-3/c, most likely as part of a protein complex, regulates Ret51 turnover in multiple cell types and not just in sympathetic neurons (8). Silencing of CD2AP in podocytes did not alter Ret51 degradation (data not shown), which is consistent with the observation that CD2AP deficiency in podocytes leads to a compensatory up-regulation of its paralog, CIN85 (18).
Cbl-3/c differs significantly from the two other mammalian Cbl family members, c-Cbl and Cbl-b, allowing for the possibility that Cbl-3/c may function in Ret51 degradation as an adaptor protein, rather than a bona fide E3-ligase. To determine whether Cbl-3/c endogenously promotes the ubiquitination of Ret51, siRNA silencing was performed on primary sympathetic neurons derived from the superior cervical ganglion. Sympathetic neurons were stimulated with GDNF, or medium alone, to activate Ret51 and initiate its ubiquitination, as has been shown previously (5). After 15 min of stimulation, Ret51 was immunoprecipitated, and its extent of ubiquitination was determined by immunoblotting the immunoprecipitates with polyubiquitin antibodies. Knockdown of Cbl-3/c blocked GDNF-initiated ubiquitination of Ret51 but did not appear to alter the basal level of Ret51 ubiquitination in the absence of GDNF (Fig. 1B). Quantification of the immunoblots confirmed that the increase in the ubiquinated Ret51:total Ret51 ratio upon GDNF stimulation was inhibited when Cbl-3 expression was reduced by siRNA (Fig. 1C). To confirm these results, this experiment was also performed by immunoprecipitating ubiquitinated proteins using polyubiquitin antibodies followed by Ret51 immunoblotting. Similar to what was observed in Fig. 1B, silencing of Cbl-3 reduced the amount of ubiquitinated Ret51 that was induced by GDNF stimulation (Fig. 1D). Because the siRNA transfection did not completely silence Cbl-3/c (Fig. 1B), it is possible that the basal level of Ret ubiquitination was due to the remaining Cbl-3/c that was not silenced. It is also possible that other E3 ligases such as c-Cbl and Cbl-b, both of which are expressed in superior cervical ganglion neurons, were also able to ubiquitinate Ret51. Taken together, Cbl-3/c was responsible for a significant proportion of GDNF-initiated Ret51 ubiquitination in sympathetic neurons.
As a means of performing mechanistic analyses of the Ret51 ubiquitination complex, NIH3T3 cells were used as a “blank slate” for the expression of individual pathway constituents. NIH3T3 cells were chosen, rather than HEK293 cells, HELA cells, or CHO cells, because NIH3T3 cells did not express Ret9 or CD2AP and only expressed low levels of Ret51 and Cbl-3/c (data not shown). Primary sympathetic neurons and differentiated podocytes could not be used for these experiments because they cannot be transfected at levels sufficient for biochemical studies. When Ret51 was transiently transfected into cell lines, it became highly autophosphorylated, even in the absence of GFLs, which obviated the need to cotransfect GFRαs and perform ligand stimulations (7). It was difficult, in fact, to transiently transfect Ret51 or Ret9 without inducing a high basal level of autophosphorylation. Upon expression of Ret51 alone, a detectible level of ubiquitination occurred (Fig. 1E). In all of the experiments examining Ret51 ubiquitination in transfected NIH3T3 cells, the cells were treated with proteasome and lysosome inhibitors to block Ret51 degradation, which made the levels of Ret51 more consistent between conditions, thereby allowing for a more accurate assessment of the extent of Ret51 ubiquitination. Cotransfection of Ret51 with either CD2AP or with Cbl-3/c had no consistent effect on Ret ubiquitination (Fig. 1E). CIN85 was expressed in these NIH3T3 cells (data not shown), which may partially explain why overexpression of CD2AP alone had no apparent effect. Interestingly, cotransfection of both CD2AP and Cbl-3/c with Ret51 resulted in an increase in Ret51 ubiquitination (Fig. 1E), which was confirmed further by quantification of the immunoblots (Fig. 1F). To confirm that Ret51 ubiquitination observed in NIH3T3 cells was dependent upon Ret51 activation, cells transfected with Ret51 alone or Ret51 along with CD2AP and Cbl-3/c were exposed to the Ret kinase inhibitors RPI-1 (Fig. 1G) and vatalanib (data not shown) for 6–12 h. Ret51 kinase inhibition dramatically reduced ubiquitination in both conditions, although some basal level of ubiquitination persisted (Fig. 1, G and H). These data confirmed that, in transfected NIH3T3 cells, Ret51 ubiquitination is largely dependent upon its activation and autophosphorylation. Taken together, these data suggested that both CD2AP and Cbl-3/c together regulated Ret51 ubiquitination in both neuronal and non-neuronal cells.
CD2AP Promotes the Degradation of Ret51 in a Cbl-3/c-dependent Manner in Living Cells
To confirm that the ubiquitination of Ret51 observed in multiple cell types correlated with the degradation of Ret51 in NIH3T3 cells, Ret51 turnover was monitored using fluorescent pulse-chase labeling of Ret51. To this end, a cDNA encoding a Ret51-kikume fusion protein was transfected into NIH3T3 cells. Because kikume can be permanently photoconverted from green to red upon exposure to 350–400-nm light, the half-life of Ret51-kikume fusion proteins could be monitored in living cells by imaging the loss of red fluorescence as a function of time after exposure of the cells to UV light (Fig. 2A). Kikume was fused to the carboxyl terminus of human Ret51 such that intracellular trafficking and degradation kinetics could be monitored (“Experimental Procedures”). Ret51-kikume, as well as wild type Ret51, when transfected into NIH3T3 cells, was properly routed to the plasma membrane (data not shown). Using this method, it was determined that transfected Ret51-kikume had an average half-life of 5.6 min (Fig. 2B). Cotransfection of Ret51-kikume with CD2AP did not significantly accelerate the degradation of Ret51-kikume (Fig. 2B). Coexpression of both CD2AP with Cbl-3/c with Ret51-kikume reduced significantly the half-life of Ret51-kikume (t½ = 3.0 min; Fig. 2, A and B), consistent with the increased ubiquitination of Ret51 upon expression of both CD2AP and Cbl-3/c (Fig. 1, C and D). Interestingly, expression of Cbl-3/c alone reduced the degradation of Ret51-kikume (Fig. 2B). This was similar to what was observed previously upon the coexpression of Ret51 with Cbl-3/c in HEK293 cells (8). This suggests that Cbl-3/c alone is unable to promote Ret51 degradation in the absence of CD2AP and may actually inhibit degradation upon overexpression by sequestering away necessary components of the degradation process from Ret51, or by binding to Ret51 in an inactive state and reducing the association of other proteins necessary for the degradation process. The coexpression of CD2AP (Myc-tagged) and Cbl-3/c (FLAG-tagged) in the cells that expressed Ret51-kikume was confirmed by immunolabeling these cells with Myc and FLAG antibodies, respectively (Fig. 2C). To confirm that the loss of fluorescence over time was not due solely to photobleaching, this experiment was repeated with fewer time points. Rather than imaging the cells frequently, the selected cells were only imaged twice, once immediately after photoconversion and once more after 30 min. As before, Ret51-kikume fluorescence declined after 30 min (Fig. 2, D and E). In the presence of CD2AP and Cbl-3, however, Ret51-kikume fluorescence was lost significantly faster (Fig. 2, D and E), confirming that CD2AP and Cbl-3 promote Ret51 degradation rapidly in living cells.
FIGURE 2.

CD2AP and Cbl-3/c alter Ret51 degradation in living cells. NIH3T3 cells were transfected with a Ret51-kikume fusion protein, and the presence of this protein was monitored by live cell, resonance scanning confocal microscopy. At t = 0, the kikume protein was photoconverted from green to red by exposure to 380-nm light, and z-stack images were taken every 15 s. In A, a typical time course of the loss of Ret51-kikume fluorescence from the cell is shown in cells transfected with Ret51-kikume alone and Ret51-kikume along with CD2AP and Cbl-3/c. This imaging method was applied to cells expressing Ret51-kikume alone, Ret51-kikume along with CD2AP, Cbl-3/c, or both CD2AP and Cbl-3/c. B, relative fluorescence intensity was measured from 12–15 cells from three to five independent transfections and graphed as the means ± S.E. (data points were only shown for each minute). Cells expressing CD2AP and Cbl-3/c (red triangles) displayed a significant increase in the rate of Ret51 degradation. Asterisks below the trace indicate a statistically significant difference (p < 0.05) as compared with Ret alone (black circles). Cells expressing Cbl-3/c (blue triangles) had a significantly slower rate of Ret51-kikume degradation, and expression of CD2AP was not significantly different from Ret51-kikume alone (green and black circles, respectively). C, to confirm the coexpresssion of all three proteins in these experiments, NIH3T3 cells were transfected with Ret51-kikume or Ret51-kikume along with CD2AP (Myc-tagged) and Cbl-3 (FLAG-tagged) and then fixed and immunolabeled with antibodies to Myc or FLAG (red). Because anti-Myc and anti-FLAG were both mouse monoclonal antibodies, triple labeling could not be performed. D, the cells were transfected with either Ret51-kikume alone or Ret51-kikume with CD2AP and Cbl-3. At t = 0, the cells were photoconverted but only imaged twice, once immediately following photoconversion and once after 30 min. E, the data from multiple cells of each condition were quantified. The relative fluorescence of Ret51-kikume was significantly lower (p < 0.05) in the presence of CD2AP and Cbl-3.
Ret9 Is Not Ubiquitinated by the CD2AP·Cbl-3/c Complex
Although Ret9 is not degraded as rapidly as Ret51, it is still degraded in sympathetic neurons upon its activation (5, 6). To determine whether the CD2AP·Cbl-3/c complex could ubiquitinate Ret9, cell transfection studies were carried out. Coexpression of CD2AP and Cbl-3/c with Ret9 did not induce the ubiquitination of Ret9, even though Ret51 ubiquitination was dramatically increased by CD2AP·Cbl-3/c (Fig. 3). In fact, there was little, if any, ubiquitination of Ret9 that could be detected in NIH3T3 cells (Fig. 3). These data suggested that Ret9 may not be degraded as rapidly as Ret51 because it was not a substrate for ubiquitination by the CD2AP·Cbl-3/c complex.
FIGURE 3.
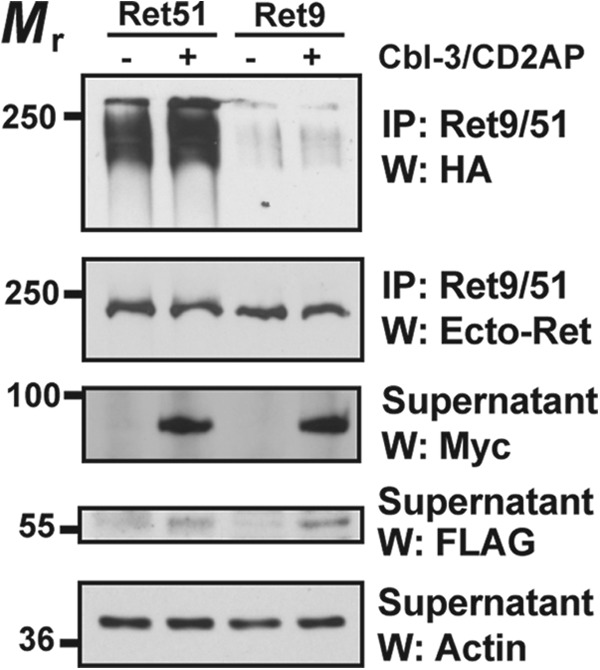
CD2AP and Cbl-3 are not regulators of Ret9 ubiquitination. NIH3T3 cells were transfected with either Ret51 or Ret9, along CD2AP and Cbl-3/c (indicated as +) or with neither (indicated as −). All conditions were also transfected with HA-ubiquitin. The extent of Ret9 and Ret51 ubiquitination was ascertained as in Fig. 1E (top panel). Immunoblotting with antibodies to the shared extracellular domain of Ret was performed on the immunoprecipitates to confirm that similar amounts of Ret51 and Ret9 were expressed (middle panel). Proteasome and lysosome inhibitors were applied to the cells 6–12 h before detergent extraction to stabilize the amount of Ret in each condition, as was done in Fig. 1E. Actin Westerns of the supernatants indicated that there were equal amounts of protein in each sample (bottom panel). The expression of CD2AP and Cbl-3/c were confirmed by Myc and FLAG immunoblotting, respectively, of the supernatants. This experiment was repeated four times with similar results. IP, immunoprecipitation; W, Western blot.
Ubiquitination of Ret51 on Lys1060 and Lys1107 Is Necessary for Degradation
Receptor tyrosine kinases are typically monoubiquitinated on multiple lysine residues, which targets them for internalization and intracellular trafficking to the lysosome for degradation (26–28). Ret51, in contrast, is polyubiquitinated and degraded predominantly by proteasomes (5). To determine which lysine residues in Ret were ubiquitinated, site-directed mutagenesis was performed on Ret51. Two lysines were initially targeted for mutation to arginine: Lys1060 and Lys1107. Lys1107 was chosen because it is in the unique carboxyl terminus of Ret51 and may be responsible, at least in part, for the more rapid degradation of Ret51 as compared with Ret9. Lys1060 was similarly chosen because it is only 4 amino acids away from the carboxyl-terminal splice site and may be recognized differently between Ret9 and Ret51. Ret51, K1060R-Ret51, K1107R-Ret51, and a K1060R/K1107R-Ret51 double mutant were transfected into NIH3T3 cells along with CD2AP and Cbl-3/c. As before, Ret51 was heavily ubiquitinated under these conditions (Fig. 4A). There was only a modest decrease in the ubiquitination of K1060R-Ret51 and K1107R-Ret51 (Fig. 4A). In stark contrast, there was a great reduction in the ubiquitination of the K1060R/1107R-Ret51 double mutant in the presence of CD2AP and Cbl-3/c (Fig. 4A). In fact, K1060R/K1107R-Ret51 migrated at a smaller molecular mass as compared with the other forms of Ret, consistent with the lack of ubiquitin incorporation (Fig. 4A). Quantification of these experiments indicated a 70% decrease in the relative ubiquitination of K1060R/1107R-Ret51 as compared with wild type Ret51 (Fig. 4B). These results indicate that both Lys1060 and Lys1107 are ubiquitinated in Ret51 and that these two lysines account for the majority of Ret51 ubiquitination by CD2AP and Cbl-3/c.
FIGURE 4.
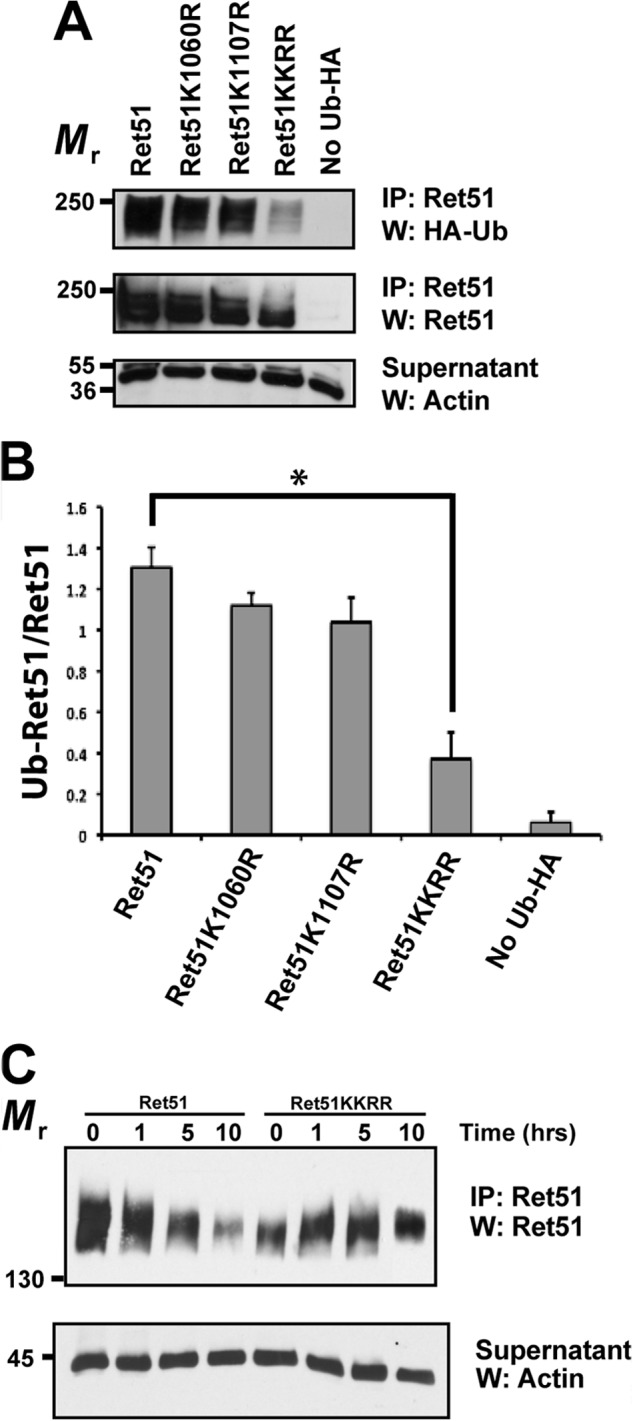
The major ubiquitination sites in Ret51 are lysines 1060 and 1107. A, NIH3T3 cells were transfected with CD2AP, Cbl-3, HA-ubiquitin, and either Ret51, Ret51-K1060R, Ret51-K1107R, or Ret51-K1060R/K1107R (indicated as KKRR). The amount of Ret51 ubiquitination was then ascertained as in Fig. 1E, including the use of proteasome and lysosome inhibitors to stop Ret51 degradation. Ret51 reprobing of the immunoprecipitates (middle panel), along with actin immunoblotting of the supernatants (bottom panel), were used to confirm that a similar amount of Ret51 and total protein were analyzed in each condition. B, the results in A were quantified as in Fig. 1F. The asterisk indicates a significant difference, p < 0.05. C, NIH3T3 cells were transfected with either Ret51-KKRR or Ret51, along with CD2AP and Cbl-3. After 48 h, the cells were given cycloheximide (1 μm) for 1, 5, or 10 h. The level of Ret51 in each condition was ascertained by immunoprecipitation of Ret51 followed by immunoblotting with Ret51 antibodies. Actin Westerns of the supernatants again confirmed the analysis of similar amounts of protein. Cycloheximide treatment inhibits protein synthesis and any further Ret51 production, thereby allowing the rate at which Ret51 is degraded to be determined. These experiments were repeated three times with similar results. IP, immunoprecipitation; W, Western blot.
To determine whether Lys1060 and Lys1107 together were required for the degradation of Ret51 triggered by CD2AP and Cbl-3/c, the degradation of the K1060R/1107R-Ret51 mutant was compared with Ret51. NIH3T3 cells were transfected with either Ret51, or K1060R/K1107R-Ret51, in the presence of CD2AP and Cbl-3/c. After 48 h, the cells were treated with cycloheximide to stop any further protein synthesis and were not treated with the proteasome and lysosome inhibitors that were used in the ubiquitination experiments. The degradation of Ret51 was then monitored by Ret51 immunoprecipitation followed by Ret immunoblotting. As seen before, Ret51 was degraded rapidly, with most Ret51 being lost after 10 h of cycloheximide exposure (Fig. 4C). It was noted that K1060R/1107R-Ret51 was not initially expressed as highly as was Ret51 was in NIH3T3 cells, even though equivalent amounts of plasmid were used in each transfection (Fig. 4C). Nevertheless, K1060R/1107R-Ret51 was not significantly degraded over this same 10-h period of cycloheximide exposure (Fig. 4C), indicating that ubiquitination on these two lysines was responsible for targeting Ret51 for degradation. There was still some degradation of K1060R/1107R-Ret51 that occurred over longer time periods (data not shown), suggesting that either additional lysines can be ubiquitinated in Ret51 or that other mechanisms exist for Ret51 degradation.
The Ring Finger and TKB Domains of Cbl-3/c Are Required for Ret51 Ubiquitination
To determine which portions of Cbl-3/c were required for Ret51 ubiquitination, a series of point mutants of Cbl-3/c were expressed in NIH3T3 cells along with CD2AP and Ret51. Interestingly, a point mutation in the TKB domain that significantly impairs its ability to bind to tyrosine-phosphorylated proteins (G276E-Cbl-3/c) almost completely eliminated the ubiquitination of Ret51, as compared with wild type Cbl-3/c (Fig. 5, A and B). Three different ring finger mutations in Cbl-3/c were examined that reduce E3 ligase activity, C351A-Cbl-3/c, R390Q-Cbl-3/c, and W378A-Cbl-3/c. Two of these mutants, C351A-Cbl-3/c and R390Q-Cbl-3/c, were significantly impaired in their ability to ubiquitinate Ret51 (Fig. 5, A and B). W378A-Cbl-3/c was not significantly impaired, which may be related to the previous observation that although this mutation does significantly reduce E3-ligase activity in c-Cbl, it is considered a structurally less disruptive mutation than other ring finger mutations (29, 30). Consistent with these ring finger point mutants, a truncated version of Cbl-3/c that removes the ring finger entirely, leaving only the TKB domain, had no ubiquitination activity toward Ret51 (Fig. 5, A and B). As expected, these results suggested that Cbl-3 directly ubiquitinated Ret51 and that it did not act as an adaptor to recruit another E3 ligase to ubiquitinate Ret51. Lastly, a highly conserved tyrosine in Cbl-3/c, Tyr341, was mutated and examined for its requirement for Cbl-3/c to ubiquitinate Ret51. Surprisingly, the Y341F-Cbl-3/c mutant was fully capable of ubiquitinating Ret51 (Fig. 5, A and B), even though this mutation reduces E3-ligase activity in Cbl-3/c in cell free systems (21). These results were confirmed by quantification of the immunoblots (Fig. 5B). In NIH3T3 cells, we observed a basal level of E3 ligase activity toward Ret without transfected Cbl-3/c (Fig. 1, E and F). This basal level of ubiquitination appeared to be diminished by the G276E and TKB only Cbl-3/c mutants (Fig. 5, A and B), raising the interesting possibility that these mutants may function in a dominant inhibitory manner.
FIGURE 5.

Cbl-3/c-mediated ubiquitination of Ret51 requires both its ring finger and TKB domains. Cbl-3/c mutants were transfected into NIH3T3 cells along with CD2AP, Ret51, and HA-ubiquitin. These inactivating mutations included three point mutations in the ring finger domain of the E3 ligase (C351A, R390Q, and W378A) as well as a deletion of the entire ring finger (TKB only), a mutation of the phosphotyrosine-binding domain of the TKB region (G276E) and a Y341F mutant that eliminates a phosphorylation site that is conserved among Cbl family members. Wild type Cbl-3/c was also included as a positive control (each variant is indicated above the top panel). A, the extent of Ret51 ubiquitination was evaluated as was done in Fig. 1E and displayed. B, three representative experiments were quantified and graphed as the means ± S.E. Asterisks indicated a statistically significant difference (p < 0.05) between the indicated condition and wild type Cbl-3 (WT). C, identical transfections and inhibitor treatments were performed as in A. The cell extracts were subjected to FLAG immunoprecipitation to isolate the transfected Cbl-3 proteins that were all FLAG-tagged. These immunoprecipitations were then immunoblotted with Ret51 antibodies followed by Cbl-3 antibodies. Although the TKB domain of Cbl-3 has a smaller relative molecular mass, it is displayed in the panel along with the other Cbl-3/c mutants (box). Actin was used as a loading control. D, as a control for the specificity of the immunoprecipitations, cells were transfected with Ret and CD2AP in the presence or absence of FLAG-Cbl-3/c. FLAG immunoprecipitations and subsequent immunoblot analysis were performed as in C. E, primary sympathetic neurons were exposed to PP2, PP3, or vehicle alone (DMSO) for 1 h prior to GDNF stimulation for 10 min (indicated above blots). Ret51 was immunoprecipitated and then analyzed with polyubiquitin immunoblotting, as was done in Fig. 1B. Phospho-Src immunoblotting of the supernatants (third panel) confirmed inhibition of Src with PP2. The experiments shown in this figure were performed two or three times with similar results. IP, immunoprecipitation; W, Western blot.
To determine whether any of these Cbl-3/c mutations that reduced Ret51 ubiquitination were due to the loss of association between them and Ret51, coimmunoprecipitation studies were performed. Interestingly, all of the Cbl-3/c point mutants associated with Ret51 at levels similar to native Cbl-3/c (Fig. 5C). The specificity of the coimmunoprecipitations was confirmed by performing FLAG-Cbl-3/c immunoprecipitations in the absence of transfected Cbl-3/c (Fig. 5D). In this case, Ret51 was not coimmunoprecipitated in the absence of Cbl-3/c. Taken together, these results suggested that the G276E mutation in the TKB domain, and the ring finger mutations, did not impair the ability of Cbl-3/c to ubiquitinate Ret51 because of the loss of physical association with Ret51. Furthermore, these results indicated that both the ring finger and TKB domains of Cbl-3/c were critically important for the CD2AP-enhanced ubiquitination of Ret51.
Because Tyr341 in Cbl-3/c is phosphorylated by Src, we tested whether inhibition of Src altered the ubiquitination of Ret51. Primary sympathetic neurons were subjected to Src inhibition using the selective inhibitor PP2, or the inactive, structurally related compound PP3 as a negative control, followed by stimulation with GDNF or medium alone. Ret51 was then immunoprecipitated, and the level of ubiquitination was analyzed by polyubiquitin immunoblotting. Inhibition of Src with PP2 did not alter Ret51 ubiquitination, and the level of Ret51 ubiquitination was similar to that observed in the presence of PP3 or the vehicle alone (Fig. 5E). Thus, Src activation was not necessary for GDNF-mediated Ret51 ubiquitination in sympathetic neurons.
The SH3 Domains of CD2AP Are Sufficient for Mediating Cbl-3/c-dependent Ubiquitination of Ret51
To identify the regions of CD2AP that augment the ubiquitination of Ret51 by Cbl-3/c, we transfected two truncated versions of CD2AP into the Ret51 ubiquitination assay system: CD2AP encoding amino acids 1–328 and the carboxyl-terminal 561–681 amino acids of CD2AP. The amino-terminal 328 residues of CD2AP encompass the three SH3 domains of CD2AP (SH3-CD2AP), whereas the carboxyl-terminal 561–681 residues of CD2AP contain only the coiled-coil domain of CD2AP (CC-CD2AP). Interestingly, the SH3 domains of CD2AP were sufficient to enhance Ret51 ubiquitination, but the coiled-coil domain of CD2AP on its own had very little activity in this assay (Fig. 6A). Quantification of the immunoblots determined that the CC-CD2AP construct was significantly impaired in its ability to trigger Ret51 ubiquitination, in contrast to SH3-CD2AP that was not significantly altered (Fig. 6B). The apparently high basal level of Ret ubiquitination in the “no CD2AP” condition reflected a high level of total Ret in this particular condition as compared with the others (Fig. 6A). These observations suggested that CD2AP associated with the Ret51 complex via the amino-terminal SH3 domains, thereby activating Cbl-3/c that also associates with Ret51. To test this possibility, the CD2AP truncations, or full-length CD2AP, were cotransfected into NIH3T3 cells along with Ret51 and Cbl-3/c. CD2AP was then immunoprecipitated and analyzed for its association with Ret51 by immunoblotting these immunoprecipitates with Ret51 antibodies. Consistent with the Ret51 ubiquitination results, SH3-CD2AP and full-length CD2AP associated with Ret51, but CC-CD2AP did not (Fig. 6C). These results indicated that the SH3 domains of CD2AP associated with the Ret51 complex, which is similar to how CD2AP interacts with many signaling proteins, such as c-Cbl (14). Taken together, these results demonstrated that the SH3 domains of CD2AP were sufficient for its association with the Ret51 complex and were sufficient to enhance the ubiquitination of Ret51 by Cbl-3. In contrast, the carboxyl-terminal coiled-coil domain of CD2AP was neither necessary nor sufficient for Ret51 ubiquitination.
FIGURE 6.

The SH3 domains of CD2AP are sufficient for Ret association and Cbl-3-mediated ubiquitination. A, cells were transfected with full-length CD2AP, with the amino-terminal three SH3 domains of CD2AP (1–328 amino acids, SH3-CD2AP) or the carboxyl-terminal 120 amino acids that encodes the coiled-coil domain of CD2AP (561–681, CC-CD2AP). All of the cells were also cotransfected with Cbl-3, Ret51, and HA-ubiquitin. Ret ubiquitination was ascertained as in Fig. 1E. Supernatants from the first Ret immunoprecipitation were subsequently immunoprecipitated using anti-Myc and probed for Myc to confirm similar expression of the CD2AP variants. Asterisks on the right side of the Myc-CD2AP blot indicate the position of full-length CD2AP and the two truncations of CD2AP. B, the experiments in A were quantified and graphed as the means ± S.E. (n = 4). The asterisk indicates a significant difference between the WT CD2AP and CC-CD2AP conditions, p < 0.05. C, NIH3T3 cells were transfected with the same plasmids as in A. The cells were then subjected to Myc (CD2AP) immunoprecipitation followed by immunoblotting for Ret51. Myc immunoblotting of the immunoprecipitation supernatants confirmed the expression of the CD2AP variants (middle panels). The relative molecular masses of CD2AP, SH3-CD2AP, and CC-CD2AP are indicated with asterisks as in A (79–90, 45–55, and 10–15 kDa, respectively). Actin served as a loading control. D, NIH3T3 cells were transfected with Ret51 (WT) or kinase inactive Ret51 (KD) along with CD2AP, with or without Cbl-3. After 24 h, detergent extracts were produced from the transfected cells, and these extracts were immunoblotted with antibodies to CD2AP (Myc), Cbl-3, or actin as a loading control. The cells were not treated with protease inhibitors such that differences in protein degradation could be observed. The experiments in this figure were performed two or three times with similar results. IP, immunoprecipitation; W, Western blot.
To determine whether CD2AP or Cbl-3/c are targeted for degradation along with Ret51, CD2AP and Cbl-3 were transfected into NIH3T3 cells along with Ret51 or a kinase-dead mutant of Ret51. After 24 h, the cells were then detergent-extracted, and the levels of CD2AP and Cbl-3/c were determined. Interestingly, CD2AP levels were reduced when coexpressed with Ret51 but not when coexpressed with kinase-dead Ret51 (Fig. 6D). Cbl-3 levels were unchanged, and the expression of Cbl-3 did not alter CD2AP levels (Fig. 6D). These data suggest that Ret kinase activity promotes the degradation of CD2AP, but not Cbl-3, along with itself.
DISCUSSION
In the results presented here, we have demonstrated that Cbl-3/c was required for the ubiquitination and degradation of Ret51 in both sympathetic neurons and differentiated podocytes. Cbl-3/c alone did not efficiently ubiquitinate Ret51, and it required the adaptor protein CD2AP for maximal ubiquitination and degradation. One of the most significant observations is that there are only two predominant ubiquitination sites in Ret51, Lys1060 and Lys1107. The ubiquitination of a Ret51 K1060R/K1107R double mutant by Cbl-3/c is markedly reduced, and this mutant has a much greater half-life. Importantly, CD2AP is also degraded during Ret degradation, revealing a potential negative feedback loop that could serve to limit the extent to which GFL activation of Ret induces its degradation.
The ring finger domain and the phosphotyrosine binding capacity of the TKB domain of Cbl-3/c were required for Ret51 ubiquitination, as well as the SH3 domains of CD2AP. The association of Cbl-3/c to Ret51 is not phosphorylation-dependent (8), and yet the phosphotyrosine binding function of the TKB domain was required. These data suggest that another tyrosine-phosphorylated protein must associate with Cbl-3 via the TKB domain, possibly CD2AP itself. The requirement of the SH3 domains of CD2AP for Ret51 ubiquitination, however, raises the possibility that CD2AP may associate with the proline rich region of Cbl-3/c. This is unlikely, however, given that CD2AP and CIN85 interact with the extreme carboxyl termini of c-Cbl and Cbl-b, which does not exist in Cbl-3/c (14, 31). In fact, CIN85 does not interact with Cbl-3/c, although CD2AP does (8, 32), suggesting that perhaps other proteins may be involved in this interaction. It is not certain, however, why CD2AP is necessary for the efficient Cbl-3/c ubiquitination of Ret51. In cell-free systems, the amino-terminal TKB domain of Cbl-3/c negatively regulates its E3 ligase activity (21), providing the possibility that CD2AP may act to relieve this intramolecular inhibition. Alternatively, CD2AP may enhance the association of the loaded E2 to Cbl-3/c or enhance the release of the unloaded E2 (21).
In Cbl-3/c, Tyr341 phosphorylation enhances its E3 ligase activity, and mutation of this residue reduces Cbl-mediated ubiquitination of substrates (33–35). Interestingly, Src phosphorylates Tyr341 of Cbl-3/c, thereby fully activating it (21, 25). Surprisingly, in the results reported here, the Y341F mutation of Cbl-3/c did not reduce its ability to ubiquitinate Ret51. In our NIH3T3 expression, an E2 ligase was not cotransfected with Ret51, Cbl-3, and CD2AP, which could potentially make the E2 levels limiting. Src-mediated phosphorylation of Cbl-3 on Tyr341 only occurs under conditions in which E2 levels are greater than E3 and are not limiting to the reaction (21). It is therefore possible that in the experiments presented here, the E2 ligase levels were limiting, thereby reducing the need for Src phosphorylation of Cbl-3/c. Alternatively, CD2AP may act to enhance Cbl-3/c-mediated ubiquitination of Ret51 by inducing Cbl-3/c activation in the absence of Src phosphorylation. Although Src associates with autophosphorylated Tyr981 of Ret and is activated upon this association (36), Src activation was not necessary for the GDNF-induced ubiquitination of Ret51 in sympathetic neurons (Fig. 5). GFL-mediated Src activation, however, is necessary for the survival-promoting effects of Ret activation (36, 37). It is possible that Src-mediated activation of Cbl-3 is substrate-dependent or context-dependent, providing an additional mechanism for the regulation of receptor degradation.
There were only two predominant ubiquitination lysines in Ret51, Lys1060 and Lys1107, and mutation of both Lys1060 and Lys1107 in Ret51 almost completely eliminated its autophosphorylation-dependent degradation. The observation that two lysines account for the majority of Ret51 ubiquitination is consistent with the fact that Ret51 is polyubiquitinated, rather than being heavily monoubiquitinated on multiple lysines, as is the case for many receptor tyrosine kinases. In neurons, for example, Ret51 activation induces a shift in its relative molecular mass by as much as 100 kDa, suggesting that Lys1060 and Lys1107 must be polyubiquitinated (5). Surprisingly, the other isoform of Ret, Ret9, was not ubiquitinated by the CD2AP·Cbl-3/c complex, even though Cbl-3/c and CD2AP can associate with Ret9 in transfected HEK293 cells (8). These data suggest that perhaps Cbl-3/c and CD2AP do not normally associate with Ret9 under physiologic, nonoverexpressed conditions or that the association of this complex with Ret9 is not sufficient to trigger its ubiquitination. Interestingly, the observation that the CD2AP·Cbl-3/c complex predominantly ubiquitinates Lys1060 and Lys1107 suggests an explanation for the lack of Ret9 ubiquitination by the CD2AP·Cbl-3/c complex: Lys1107 does not exist in Ret9, and the surrounding amino acid context of Lys1060 is different in Ret51 than in Ret9. Identification of the degradation pathway of Ret9 awaits further analysis but is likely to aid in our understanding of the molecular basis for the dramatic differences between the turnover and signaling capacities of these two Ret isoforms, which often vary among different types of cells. It was recently reported that Ret9 and Ret51 undergo differential intracellular trafficking and that Ret51 can undergo recycling back to the plasma membrane after its activation with GDNF (38). This alternative trafficking pathway for Ret51 contributes to a slower rate of Ret51 degradation in primary enteric neurons and cultured SH-SY5Y cells (38). These data suggest that the remarkable differences between the mechanisms of Ret trafficking and degradation in different types of neurons likely accounts for the cell type-specific functions of the GFLs. One possible mechanism to explain this diversity is if the degradation of CD2AP that was induced by activated Ret51 (Fig. 6D) is dependent upon cell type or subcellular localization. Thus, Ret activation and ubiquitination that triggers subsequent CD2AP degradation would likely alter the intracellular trafficking of ubiquitinated Ret51 or the overall kinetics of Ret51 degradation. The recent observation that variants of CD2AP are associated with late onset Alzheimer disease suggests that CD2AP-regulated protein turnover may be central to neuronal homeostasis (39, 40), and further characterization of the molecular mechanisms of CD2AP-mediated protein ubiquitination may underscore the importance of these pathways in neuronal maintenance and neurodegeneration.
Acknowledgments
We thank Sheena Koushik and Monzurul Chowdhury for technical assistance. We also thank Hideki Enomoto for helpful scientific discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01, NS058510 (B. A. P.) and National Institutes of Health Career Development Award K08, DK084210 (to C. C. T.).
- GDNF
- glial cell line-derived neurotrophic factor
- GFL
- GDNF family ligand
- SH
- Src homology.
REFERENCES
- 1. Airaksinen M. S., Saarma M. (2002) The GDNF family. Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 3, 383–394 [DOI] [PubMed] [Google Scholar]
- 2. Baloh R. H., Enomoto H., Johnson E. M., Jr., Milbrandt J. (2000) The GDNF family ligands and receptors-implications for neural development. Curr. Opin. Neurobiol. 10, 103–110 [DOI] [PubMed] [Google Scholar]
- 3. Bespalov M. M., Saarma M. (2007) GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. Sci. 28, 68–74 [DOI] [PubMed] [Google Scholar]
- 4. Wells S. A., Jr., Santoro M. (2009) Targeting the RET pathway in thyroid cancer. Clin. Cancer Res. 15, 7119–7123 [DOI] [PubMed] [Google Scholar]
- 5. Pierchala B. A., Milbrandt J., Johnson E. M., Jr. (2006) Glial cell line-derived neurotrophic factor-dependent recruitment of Ret into lipid rafts enhances signaling by partitioning Ret from proteasome-dependent degradation. J. Neurosci. 26, 2777–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scott R. P., Eketjäll S., Aineskog H., Ibáñez C. F. (2005) Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J. Biol. Chem. 280, 13442–13449 [DOI] [PubMed] [Google Scholar]
- 7. Tsui-Pierchala B. A., Ahrens R. C., Crowder R. J., Milbrandt J., Johnson E. M., Jr. (2002) The long and short isoforms of Ret function as independent signaling complexes. J. Biol. Chem. 277, 34618–34625 [DOI] [PubMed] [Google Scholar]
- 8. Tsui C. C., Pierchala B. A. (2008) CD2AP and Cbl-3/Cbl-c constitute a critical checkpoint in the regulation of Ret signal transduction. J. Neurosci. 28, 8789–8800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duan L., Reddi A. L., Ghosh A., Dimri M., Band H. (2004) The Cbl family and other ubiquitin ligases. Destructive forces in control of antigen receptor signaling. Immunity 21, 7–17 [DOI] [PubMed] [Google Scholar]
- 10. Keane M. M., Ettenberg S. A., Nau M. M., Banerjee P., Cuello M., Penninger J., Lipkowitz S. (1999) cbl-3. A new mammalian cbl family protein. Oncogene 18, 3365–3375 [DOI] [PubMed] [Google Scholar]
- 11. Kim M., Tezuka T., Suziki Y., Sugano S., Hirai M., Yamamoto T. (1999) Molecular cloning and characterization of a novel cbl-family gene, cbl-c. Gene 239, 145–154 [DOI] [PubMed] [Google Scholar]
- 12. Swaminathan G., Tsygankov A. Y. (2006) The Cbl family proteins. Ring leaders in regulation of cell signaling. J. Cell Physiol. 209, 21–43 [DOI] [PubMed] [Google Scholar]
- 13. Cormont M., Metón I., Mari M., Monzo P., Keslair F., Gaskin C., McGraw T. E., Le Marchand-Brustel Y. (2003) CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 4, 97–112 [DOI] [PubMed] [Google Scholar]
- 14. Kirsch K. H., Georgescu M. M., Shishido T., Langdon W. Y., Birge R. B., Hanafusa H. (2001) The adapter type protein CMS/CD2AP binds to the proto-oncogenic protein c-Cbl through a tyrosine phosphorylation-regulated Src Homology 3 domain interaction. J. Biol. Chem. 276, 4957–4963 [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi S., Sawano A., Nojima Y., Shibuya M., Maru Y. (2004) The c-Cbl/CD2AP complex regulates VEGF-induced endocytosis and degradation of Flt-1 (VEGFR-1). FASEB J. 18, 929–931 [DOI] [PubMed] [Google Scholar]
- 16. Petrelli A., Gilestro G. F., Lanzardo S., Comoglio P. M., Migone N., Giordano S. (2002) The endophilin-CIN85-Cbl complex mediates ligand-dependent down regulation of c-Met. Nature 416, 187–190 [DOI] [PubMed] [Google Scholar]
- 17. Soubeyran P., Kowanetz K., Szymkiewicz I., Langdon W. Y., Dikic I. (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416, 183–187 [DOI] [PubMed] [Google Scholar]
- 18. Tossidou I., Kardinal C., Peters I., Kriz W., Shaw A., Dikic I., Tkachuk S., Dumler I., Haller H., Schiffer M. (2007) CD2AP/CIN85 balance determines receptor tyrosine kinase signaling response in podocytes. J. Biol. Chem. 282, 7457–7464 [DOI] [PubMed] [Google Scholar]
- 19. Tsui C. C., Pierchala B. A. (2010) The differential axonal degradation of Ret accounts for cell-type-specific function of glial cell line-derived neurotrophic factor as a retrograde survival factor. J. Neurosci. 30, 5149–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonanomi D., Chivatakarn O., Bai G., Abdesselem H., Lettieri K., Marquardt T., Pierchala B. A., Pfaff S. L. (2012) Ret is a multifunctional coreceptor that integrates diffusable- and contact-axon guidance signals. Cell 148, 568–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ryan P. E., Sivadasan-Nair N., Nau M. M., Nicholas S., Lipkowitz S. (2010) The N terminus of Cbl-c regulates ubiquitin ligase activity by modifying affinity for the ubiquitin-conjugating enzyme. J. Biol. Chem. 285, 23687–23698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsui-Pierchala B. A., Ginty D. D. (1999) Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J. Neurosci. 19, 8207–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsui C. C., Shankland S. J., Pierchala B. A. (2006) Glial cell line-derived neurotrophic factor and its receptor Ret is a novel ligand-receptor complex critical for survival response during podocyte injury. J. Am. Soc. Nephrol. 17, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 24. Pierchala B. A., Muñoz M. R., Tsui C. C. (2010) Proteomic analysis of the slit diaphragm complex. CLIC5 is a protein critical for podocyte morphology and function. Kidney Int. 78, 868–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim M., Tezuka T., Tanaka K., Yamamoto T. (2004) Cbl-c suppresses v-Src-induced transformation through ubiquitin-dependent protein degradation. Oncogene 23, 1645–1655 [DOI] [PubMed] [Google Scholar]
- 26. Aguilar R. C., Wendland B. (2003) Ubiquitin. Not just for proteasomes anymore. Curr. Opin. Cell Biol. 15, 184–190 [DOI] [PubMed] [Google Scholar]
- 27. Bonifacino J. S., Weissman A. M. (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 14, 19–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hicke L. (2001) Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell. Biol. 2, 195–201 [DOI] [PubMed] [Google Scholar]
- 29. Freemont P. S. (2000) Ubiquitination. RING for destruction? Curr. Biol. 10, R84–R87 [DOI] [PubMed] [Google Scholar]
- 30. Joazeiro C. A., Wing S. S., Huang H., Leverson J. D., Hunter T., Liu Y. C. (1999) The tyrosine kinase negative regulator c-Cbl is a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312 [DOI] [PubMed] [Google Scholar]
- 31. Kowanetz K., Szymkiewicz I., Haglund K., Kowanetz M., Husnjak K., Taylor J. D., Soubeyran P., Engstrom U., Ladbury J. E., Dikic I. (2003) Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and down-regulation of epidermal growth factor receptors. J. Biol. Chem. 278, 39735–39746 [DOI] [PubMed] [Google Scholar]
- 32. Szymkiewicz I., Kowanetz K., Soubeyran P., Dinarina A., Lipkowitz S., Dikic I. (2002) CIN85 participates in Cbl-b-mediated down-regulation of receptor tyrosine kinases. J. Biol. Chem. 277, 39666–39672 [DOI] [PubMed] [Google Scholar]
- 33. Sanada M., Suzuki T., Shih L. Y., Otsu M., Kato M., Yamazaki S., Tamura A., Honda H., Sakata-Yanagimoto M., Kumano K., Oda H., Yamagata T., Takita J., Gotoh N., Nakazaki K., Kawamata N., Onodera M., Nobuyoshi M., Hayashi Y., Harada H., Kurokawa M., Chiba S., Mori H., Ozawa K., Omine M., Hirai H., Nakauchi H., Koeffler H. P., Ogawa S. (2009) Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460, 904–908 [DOI] [PubMed] [Google Scholar]
- 34. Thien C. B., Walker F., Langdon W. Y. (2001) RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol. Cell 7, 355–365 [DOI] [PubMed] [Google Scholar]
- 35. Levkowitz G., Waterman H., Ettenberg S. A., Katz M., Tsygankov A. Y., Alroy I., Lavi S., Iwai K., Reiss Y., Ciechanover A., Lipkowitz S., Yarden Y. (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 36. Encinas M., Crowder R. J., Milbrandt J., Johnson E. M., Jr. (2004) Tyrosine 981, a novel Ret autophosphorylation site, binds c-Src to mediate neuronal survival. J. Biol. Chem. 279, 18262–18269 [DOI] [PubMed] [Google Scholar]
- 37. Encinas M., Tansey M. G., Tsui-Pierchala B. A., Comella J. X., Milbrandt J., Johnson E. M., Jr. (2001) c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J. Neurosci. 21, 1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Richardson D. S., Rodrigues D. M., Hyndman B. D., Crupi M. J., Nicolescu A. C., Mulligan L. M. (2012) Alternative splicing results in RET isoforms with distinct trafficking properties. Mol. Biol. Cell 23, 3838–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., Gallins P. J., Buxbaum J. D., Jarvik G. P., Crane P. K., Larson E. B., Bird T. D., Boeve B. F., Graff-Radford N. R., De Jager P. L., Evans D., Schneider J. A., Carrasquillo M. M., Ertekin-Taner N., Younkin S. G., Cruchaga C., Kauwe J. S., Nowotny P., Kramer P., Hardy J., Huentelman M. J., Myers A. J., Barmada M. M., Demirci F. Y., Baldwin C. T., Green R. C., Rogaeva E., St George-Hyslop P., Arnold S. E., Barber R., Beach T., Bigio E. H., Bowen J. D., Boxer A., Burke J. R., Cairns N. J., Carlson C. S., Carney R. M., Carroll S. L., Chui H. C., Clark D. G., Corneveaux J., Cotman C. W., Cummings J. L., DeCarli C., DeKosky S. T., Diaz-Arrastia R., Dick M., Dickson D. W., Ellis W. G., Faber K. M., Fallon K. B., Farlow M. R., Ferris S., Frosch M. P., Galasko D. R., Ganguli M., Gearing M., Geschwind D. H., Ghetti B., Gilbert J. R., Gilman S., Giordani B., Glass J. D., Growdon J. H., Hamilton R. L., Harrell L. E., Head E., Honig L. S., Hulette C. M., Hyman B. T., Jicha G. A., Jin L. W., Johnson N., Karlawish J., Karydas A., Kaye J. A., Kim R., Koo E. H., Kowall N. W., Lah J. J., Levey A. I., Lieberman A. P., Lopez O. L., Mack W. J., Marson D. C., Martiniuk F., Mash D. C., Masliah E., McCormick W. C., McCurry S. M., McDavid A. N., McKee A. C., Mesulam M., Miller B. L., Miller C. A., Miller J. W., Parisi J. E., Perl D. P., Peskind E., Petersen R. C., Poon W. W., Quinn J. F., Rajbhandary R. A., Raskind M., Reisberg B., Ringman J. M., Roberson E. D., Rosenberg R. N., Sano M., Schneider L. S., Seeley W., Shelanski M. L., Slifer M. A., Smith C. D., Sonnen J. A., Spina S., Stern R. A., Tanzi R. E., Trojanowski J. Q., Troncoso J. C., Van Deerlin V. M., Vinters H. V., Vonsattel J. P., Weintraub S., Welsh-Bohmer K. A., Williamson J., Woltjer R. L., Cantwell L. B., Dombroski B. A., Beekly D., Lunetta K. L., Martin E. R., Kamboh M. I., Saykin A. J., Reiman E. M., Bennett D. A., Morris J. C., Montine T. J., Goate A. M., Blacker D., Tsuang D. W., Hakonarson H., Kukull W. A., Foroud T. M., Haines J. L., Mayeux R., Pericak-Vance M. A., Farrer L. A., Schellenberg G. D. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., Abraham R., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Jones N., Stretton A., Thomas C., Richards A., Ivanov D., Widdowson C., Chapman J., Lovestone S., Powell J., Proitsi P., Lupton M. K., Brayne C., Rubinsztein D. C., Gill M., Lawlor B., Lynch A., Brown K. S., Passmore P. A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A. D., Beaumont H., Warden D., Wilcock G., Love S., Kehoe P. G., Hooper N. M., Vardy E. R., Hardy J., Mead S., Fox N. C., Rossor M., Collinge J., Maier W., Jessen F., Rüther E., Schürmann B., Heun R., Kolsch H., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frolich L., Hampel H., Gallacher J., Hull M., Rujescu D., Giegling I., Goate A. M., Kauwe J. S., Cruchaga C., Nowotny P., Morris J. C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P. P., Van Broeckhoven C., Livingston G., Bass N. J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C. E., Tsolaki M., Singleton A. B., Guerreiro R., Muhleisen T. W., Nothen M. M., Moebus S., Jockel K. H., Klopp N., Wichmann H. E., Pankratz V. S., Sando S. B., Aasly J. O., Barcikowska M., Wszolek Z. K., Dickson D. W., Graff-Radford N. R., Petersen R. C., van Duijn C. M., Breteler M. M., Ikram M. A., DeStefano A. L., Fitzpatrick A. L., Lopez O., Launer L. J., Seshadri S., Berr C., Campion D., Epelbaum J., Dartigues J. F., Tzourio C., Alperovitch A., Lathrop M., Feulner T. M., Friedrich P., Riehle C., Krawczak M., Schreiber S., Mayhaus M., Nicolhaus S., Wagenpfeil S., Steinberg S., Stefansson H., Stefansson K., Snaedal J., Bjornsson S., Jonsson P. V., Chouraki V., Genier-Boley B., Hiltunen M., Soininen H., Combarros O., Zelenika D., Delepine M., Bullido M. J., Pasquier F., Mateo I., Frank-Garcia A., Porcellini E., Hanon O., Coto E., Alvarez V., Bosco P., Siciliano G., Mancuso M., Panza F., Solfrizzi V., Nacmias B., Sorbi S., Bossu P., Piccardi P., Arosio B., Annoni G., Seripa D., Pilotto A., Scarpini E., Galimberti D., Brice A., Hannequin D., Licastro F., Jones L., Holmans P. A., Jonsson T., Riemenschneider M., Morgan K., Younkin S. G., Owen M. J., O'Donovan M., Amouyel P., Williams J. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]