

Background: TMEM16A and -16F function as a Ca2+-dependent Cl− channel and phospholipid scramblase, respectively.
Results: The N- and C-terminal domains of TMEM16A and -16F are exchangeable and are necessary for ER exit and stability.
Conclusion: TMEM16A and -16F use a similar mechanism for transporting and stabilizing protein, but their functional domains differ.
Significance: Identifying functional domains will lead to better understanding of TMEM16 family.
Keywords: Chloride Channels, Enzyme Mutation, Phosphatidylserine, Plasma Membrane, Protein Domains
Abstract
The transmembrane proteins TMEM16A and -16F each carry eight transmembrane regions with cytoplasmic N and C termini. TMEM16A carries out Ca2+-dependent Cl− ion transport, and TMEM16F is responsible for Ca2+-dependent phospholipid scrambling. Here we established assay systems for the Ca2+-dependent Cl− channel activity using 293T cells and for the phospholipid scramblase activity using TMEM16F−/− immortalized fetal thymocytes. Chemical cross-linking analysis showed that TMEM16A and -16F form homodimers in both 293T cells and immortalized fetal thymocytes. Successive deletion from the N or C terminus of both proteins and the swapping of regions between TMEM16A and -16F indicated that their cytoplasmic N-terminal (147 amino acids for TMEM16A and 95 for 16F) and C-terminal (88 amino acids for TMEM16A and 68 for 16F) regions were essential for their localization at plasma membranes and protein stability, respectively, and could be exchanged. Analyses of TMEM16A and -16F mutants with point mutations in the pore region (located between the fifth and sixth transmembrane regions) indicated that the pore region is essential for both the Cl− channel activity of TMEM16A and the phospholipid scramblase activity of TMEM16F. Some chemicals such as epigallocatechin-3-gallate and digallic acid inhibited the Cl− channel activity of TMEM16A and the scramblase activity of TMEM16F with an opposite preference. These results indicate that TMEM16A and -16F use a similar mechanism for sorting to plasma membrane and protein stabilization, but their functional domains significantly differ.
Introduction
The transmembrane protein (TMEM)2 16, also called anoctamin (Ano), family members carry eight transmembrane domains and have cytoplasmic N and C termini (1, 2). This family consists of 10 members that have 20–60% amino acid sequence identity. TMEM16A (Ano1) and -16B (Ano2) have a Ca2+-dependent Cl− channel activity (3–5) that mediates various biological processes, including transepithelial secretion, sensory transduction, and smooth muscle contraction (6). TMEM16A is expressed ubiquitously, whereas TMEM16B is expressed in a limited set of tissues such as the brain and eyes (7, 8).
The functions of other TMEM16 family members have been elusive, but we found that TMEM16F (Ano6) as well as TMEM16C (Ano3), -16D (Ano4), -16G (Ano7), and -16J (Ano9) are involved in Ca2+-dependent phospholipid scrambling (9, 10). In particular, we and others showed that TMEM16F is ubiquitously expressed in various cells, including hematopoietic cells, and is involved in the phosphatidylserine (PS) exposure on activated platelets required for blood clotting (9, 11, 12). TMEM16F is expressed in osteoblasts and was recently shown to be involved in mineralized bone matrix production during skeletal development (13). In addition, abnormalities (mutations and overexpression) in TMEM16 family members are implicated in a number of human diseases (6). For example, genetic mutations in TMEM16C, -16E, -16F, and -16K are associated with craniocervical dystonia (14), musculoskeletal disorder (15, 16), bleeding disorder (9), and cerebellar ataxia (17), respectively. TMEM16A is overexpressed in human gastrointestinal stromal tumors and in head and neck squamous carcinoma; TMEM16G is overexpressed in prostate cancer (18, 19). Thus, compounds that modulate the function of TMEM16A have been explored for the treatment of human diseases such as cystic fibrosis, hypertension, secretory diarrheas, asthma, and cancer (20). However, despite the apparently important physiological and pathological roles of TMEM16 family members, very little is known about the molecular mechanisms by which they exert their Cl− channel or phospholipid scramblase function.
In this report, we prepared a series of N- and C-terminal deletion mutants for TMEM16A and -16F and showed that these regions are essential for their localization at plasma membrane and protein stability, respectively. These essential regions, which comprise about one-third of the molecule, were exchangeable between TMEM16A and -16F. Point mutations in the “pore” domain in TMEM16F indicated that this domain is important for the scramblase activity of the protein as is true for the Cl− channel activity of TMEM16A. In addition, we found that some chemicals preferentially inhibit the Cl− channel activity of TMEM16A, whereas others preferentially inhibit the scramblase activity of TMEM16F (16F). These results indicate that TMEM16A and -16F use a similar mechanism to stabilize and localize them at plasma membranes, but their functional domains are significantly different.
EXPERIMENTAL PROCEDURES
Chemicals and Cell Lines
Tannic acid and epigallocatechin-3-gallate (EGCG) were purchased from Nacalai. Digallic acid and the aminophenylthiazole 2-(5-ethyl-4-hydroxy-6-methylpyrimidin-2-ylthio)-N-(4-(4-methoxyphenyl)thiazol-2-yl)acetamide (T16Ainh-A01) were from Santa Cruz Biotechnology and Millipore, respectively. 5-Nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), and 2-[3-(trifluoromethyl)anilino]nicotinic acid (niflumic acid) were purchased from Sigma. The cross-linker dithiobis(succinimidyl propionate) (DSP) was purchased from Thermo Scientific.
Mouse TMEM16F−/− immortalized fetal thymocytes (IFETs) were described previously (10) and maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal calf serum (FCS; Invitrogen) and 50 μm β-mercaptoethanol. Human HEK293T and Plat-E cells (21) were cultured in DMEM (Invitrogen) containing 10% FCS.
Expression Plasmids for Mouse TMEM16A and -16F cDNAs and Their Mutants
Mouse TMEM16F cDNA (GenBankTM accession number NM_175344) was described previously (9). The full-length cDNA for mouse TMEM16A (GenBank accession number BC062959.1) was described previously (10). The cDNAs for the TMEM16A and TMEM16F mutants and their chimeric molecules were prepared by recombinant PCR with the full-length TMEM16A and TMEM16F cDNAs as templates (supplemental Methods). The authenticity of the respective cDNAs was verified by sequencing.
Transformation of TMEM16F−/− IFETs
The full-length cDNA for TMEM16A, TMEM16F, or their mutants was inserted at EcoRI or between the BamHI and EcoRI sites of the mouse retroviral vector pMXs puro c-FLAG to express a protein that was FLAG-tagged at the C terminus (9). Retroviruses were produced in Plat-E cells, concentrated by centrifugation at 6,000 × g for 16 h at 4 °C, and used to infect TMEM16F−/− IFETs as described previously (10). Transformants were selected by culturing the cells in the presence of 1 μg/ml puromycin.
Analysis of Phosphatidylserine Exposure
The Ca2+-induced PS exposure was analyzed essentially as described previously (9). In brief, 5 × 105 IFETs were suspended in 1 ml of 10 mm HEPES-NaOH buffer (pH 7.5) containing 140 mm NaCl, 2.5 mm CaCl2, 5 μg/ml propidium iodide, and 1000-fold diluted Cy5-labeled Annexin V (BioVision). The cells were treated at room temperature with 3 μm A23187 (Sigma) followed by flow cytometry using a FACSCalibur (BD Biosciences). Propidium iodide-positive cells were excluded from the analysis.
Electrophysiology
The wild-type and mutant TMEM16A and -16F were tagged with FLAG at the C terminus and introduced into the BamHI or EcoRI/SalI sites of pEF-BOS-EX (22). HEK293T cells (2.5 × 105 cells) were co-transfected with 1 μg of the expression vector and 0.1 μg of pMAX-GFP (Amaxa) by lipofection using FuGENE 6 (Promega). Twenty-four hours later, the cells were reseeded on glass coverslips, and whole-cell patch clamp recording was performed within 24 h after reseeding essentially as described previously (4, 23). That is, the cells expressing GFP were voltage-clamped for 500-ms intervals between −120 and +120 mV with 10-mV increments. The holding potential was maintained at 0 mV. Data were acquired using an EPC 8 patch clamp amplifier (HEKA) and Axograph 4.8 software (Axon Instruments). All experiments were conducted at room temperature. The extracellular solution contained 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 30 mm glucose, and 10 mm HEPES-NaOH (pH 7.5). The electrodes were filled with a solution containing 140 mm NaCl, 1.12 mm EGTA, 1 mm CaCl2, 30 mm glucose, and 10 mm HEPES-NaOH (pH 7.5). The free Ca2+ concentration (500 nm) was calculated with WEBMAXC software (4).
Western Blotting
Cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl buffer (pH 8.0) containing 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, and 150 mm NaCl) supplemented with a protease inhibitor mixture (cOmplete Mini, Roche Applied Science). The lysates were centrifuged at 17,700 × g for 15 min at 4 °C, and the supernatants were mixed with 5× SDS sample buffer (200 mm Tris-HCl (pH 6.8), 10% SDS, 25% glycerol, 5% β-mercaptoethanol, and 0.05% bromphenol blue). Following incubation for 30 min at room temperature, the samples were separated by PAGE and transferred to a PVDF membrane (Millipore). The proteins were probed with mouse anti-FLAG M2-HRP mAb (Sigma) or mouse anti-α-tubulin mAb (Calbiochem) followed by HRP-conjugated goat anti-mouse Ig Ab (Dako) and visualized using the Western Lightning Plus-ECL system (PerkinElmer Life Sciences).
Chemical Cross-linking
HEK293T cells were transfected with the expression vector for FLAG-tagged TMEM16A or TMEM16F. Forty-eight hours after transfection, HEK293T cells (2 × 106 cells) or TMEM16F−/− IFET transformants (2 × 106 cells) expressing TMEM16A or TMEM16F were suspended in 1 ml of PBS (Invitrogen) and incubated with DSP for 30 min at room temperature. The reaction was stopped by adding 20 μl of 1 m glycine (pH 9.2) and incubating for 30 min on ice. After centrifugation, the cells were lysed with radioimmune precipitation assay buffer and analyzed by Western blotting using SDS sample buffer without β-mercaptoethanol.
Confocal Fluorescence Microscopy
The wild type and mutants of TMEM16A and -16F tagged with GFP at the C terminus were introduced into pMXs vector. HEK293T cells (2.5 × 105 cells) were transfected with 1 μg of the expression vector by lipofection using FuGENE 6. Twenty-four hours later, the cells were reseeded on a glass bottom dish, cultured for 24 h, and observed using a confocal microscope (FV1000-D, Olympus).
RESULTS
TMEM16A and TMEM16F
Mouse TMEM16A consists of 956 amino acids, and 16F has 911 amino acids; both carry eight transmembrane regions and have cytoplasmic N and C termini (supplemental Fig. S1). Their amino acid sequences are homologous throughout the molecule with an overall identity of 35.4% and similarity of 52.1%, and the regions around the first, fourth to fifth, and sixth to seventh transmembrane regions (amino acids 312–356, 576–641, and 698–787 on TMEM16A (16A)) are well conserved (supplemental Fig. S1) (71.1, 63.6, and 67.7% identity, respectively).
We assayed the Cl− channel activity by patch clamp analysis with human 293T cells that had been transfected with a TMEM16 expression vector (10). To assay the Ca2+-dependent phospholipid scramblase activity, we determined the A23187 Ca2+ ionophore-induced PS exposure in TMEM16F−/− IFET transformants expressing TMEM16 family members (10). As shown in Fig. 1, A and B, TMEM16A and -16F were expressed with a similar efficiency in both 293T cells and IFETs. However, 293T cells expressing TMEM16A but not -16F showed Cl− channel activity, and the A23187-induced PS-exposing activity was observed in TMEM16F-expressing but not 16A-expressing TMEM16F−/− IFET transformants. These observations confirmed that TMEM16A has Ca2+-dependent Cl− channel activity, whereas 16F functions as a Ca2+-dependent phospholipid scramblase.
FIGURE 1.
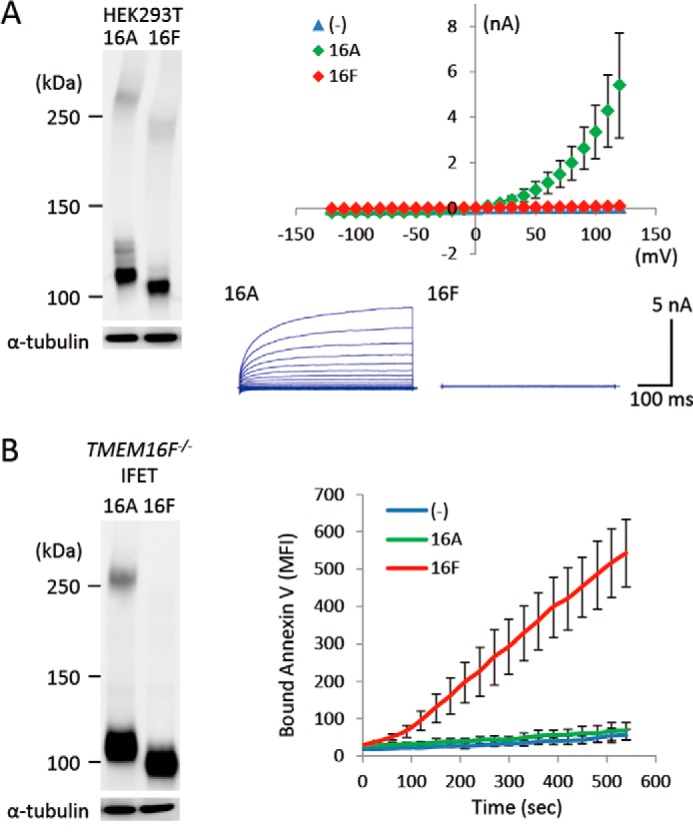
Distinct functions of TMEM16A and TMEM16F. A, Ca2+-dependent Cl− channel activity. HEK293T cells were co-transfected with the empty vector (−) or an expression vector for TMEM16A or TMEM16F together with pMAX-GFP. At left, the expression levels of TMEM16A and -16F were analyzed by Western blotting with an anti-FLAG mAb 48 h after the transfection. At right, membrane currents at the indicated voltage pulses (mV) of GFP-positive cells were measured by whole-cell patch clamp analysis. Experiments were done three to five times, and the average values are plotted with S.D. (error bars) (top). In the lower panel, the outward rectification of the Cl− current was traced at −120 to +120 mV in 10-mV steps. B, Ca2+-dependent PS-scrambling activity. TMEM16F−/− IFETs were stably transformed by the empty retroviral vector (−) or vector for the FLAG-tagged 16A or 16F. At left, the transformants were analyzed by Western blotting with an anti-FLAG mAb. At right, the cells were treated with 3 μm A23187. Their ability to bind Cy5-labeled Annexin V was followed by flow cytometry and expressed in mean fluorescence intensity (MFI).
Dimerization of TMEM16A and -16F
Sheridan et al. (24) previously reported that mouse TMEM16A expressed in 293T cells forms a dimer under non-denaturing conditions. When the cell lysates of 293T cells that had been transfected by a FLAG-tagged TMEM16A expression vector were analyzed by SDS-PAGE without a reducing agent, about half of the molecules appeared as dimers with an apparent molecular mass of about 250 kDa (Fig. 2A). Treatment of the transfected 293T cells with the cross-linker DSP dose-dependently increased the percentage of dimers without increasing higher-order complexes, confirming that most of the TMEM16A in transfected 293T cells is present as a dimer. Overexpressed TMEM16A similarly formed dimers in TMEM16F−/− IFET transformants. On the other hand, when FLAG-tagged TMEM16F expressed in 293T cells or IFETs was analyzed by SDS-PAGE, the protein was found as a monomer with molecular mass of about 100 kDa (Fig. 2B). However, like TMEM16A, when the transfected 293T cells were treated with DSP, TMEM16F appeared as a dimer, and most of the TMEM16F protein was found as a dimer after treatment with 50 μm DSP. Treatment of the IFET transformants expressing FLAG-tagged TMEM16F with DSP also revealed TMEM16F dimerization but not higher order complex formation. The TMEM16F−/− IFETs express TMEM16H and -16K (10), but the mRNA level of the endogenous TMEM16H and -16K is less than 2% of that of the exogenously introduced TMEM16A or -16F as judged by real time PCR (data not shown), indicating that TMEM16A and -16F form homodimers.
FIGURE 2.

Dimeric structure of TMEM16A and -16F. HEK293T cells that had been transfected with the expression vector for FLAG-tagged TMEM16A (A) or TMEM16F (B) and TMEM16F−/− IFET transformants expressing the FLAG-tagged TMEM16A (A) or TMEM16F (B) were treated with the indicated concentrations of DSP, washed, and lysed in radioimmune precipitation assay buffer. The lysates were separated by 5% SDS-PAGE and analyzed by Western blotting with an anti-FLAG mAb.
Successive Deletion from the N or C Terminus
Mutations or alternative splicings in the cytoplasmic N-terminal tail have a strong effect on the activity of TMEM16A and -16F (9, 25), suggesting that this region is important for the regulation of their function. To systematically analyze the contribution of the N- and C-terminal regions of TMEM16A and -16F, successive deletion mutants were prepared. Because exons are known to code for functional units (26), the mutants were prepared by successively deleting exons from the N or C terminus. As shown in Fig. 3, A and B, deletion of the first exon of TMEM16A had no effect on its Cl− channel activity. However, the mutant carrying the deletion of its second exon, which corresponds to exons 2, 3, and 4 of 16F, did not show Cl− channel activity. Similarly, deletion of exons 1 and 2 had no effect on the scramblase activity of TMEM16F, but the mutant with further deletion did not show its scramblase activity (Fig. 3, E and F). The expressed protein level of Δ1–2 of TMEM16A and Δ1–3 of TMEM16F was reduced compared with the wild type (Fig. 3, C and G), but a more dramatic change was observed for their cellular localization. That is, the wild type and the functional mutants (Δ1 mutant of TMEM16A and Δ1–2 mutant of TMEM16F) were localized at plasma membranes, whereas non-functional mutants (Δ1–2 of TMEM16A and Δ1–3 of TMEM16F) were found in the cytoplasm (Fig. 3, D and H). These results suggested that the inability of these deletion mutants to show the Cl− channel or scramblase activity was partly because they could not be localized at plasma membrane.
FIGURE 3.

N-terminal deletion of TMEM16A and -16F. A–D, effect of N-terminal deletion on the Cl− channel activity of TMEM16A. A, N-terminal deletion mutants of TMEM16A are shown schematically. Numbers in the top row and at the bottom indicate the exon number and the amino acid position where the deletion mutants start, respectively. TM1, first transmembrane region. The mutants that showed Cl− channel activity are marked by ○, and the mutants that did not are marked by ×. B, membrane currents at the indicated voltage pulses (mV) were measured by whole-cell patch clamp analysis of 293T cells transfected with the expression vector for the deletion mutant (Δexon 1, Δexon 1–2, Δexon 1–5, or Δexon 1–7 of TMEM16A). C, 293T cells that had been transfected with the expression vector for TMEM16A or its N-terminal deletion mutants were separated by 5–10% SDS-PAGE and analyzed by Western blotting with an anti-FLAG mAb. D, HEK293T cells were transfected with the expression vectors for GFP-tagged wild-type or deletion mutants (Δ1 and Δ1–2) of TMEM16A and observed using a confocal microscope. E–H, effect of N-terminal deletion on the phospholipid scramblase activity of TMEM16F. E, N-terminal deletion mutants of TMEM16F are shown. Numbers in the top row and at the bottom indicate the exon number and the amino acid position where the deletion mutants start, respectively. The mutants that showed the scramblase activity are marked by ○, and the mutants that did not are marked by ×. F, Cy5-Annexin V binding to the A23187-treated TMEM16F−/− IFET transformants expressing the full-length TMEM16F or its deletion mutant (Δexon 1, Δexon 1–2, Δexon 1–3, Δexon 1–4, Δexon 1–5, Δexon 1–6, or Δexon 1–7) was followed by FACSCalibur for 9 min and expressed as mean fluorescence intensity (MFI). G, Western blotting of TMEM16F−/− IFET transformants expressing TMEM16F or its deletion mutants. Cell lysates were separated by 5–10% SDS-PAGE and analyzed with an anti-FLAG mAb. H, HEK293T cells were transfected with the expression vectors for GFP-tagged wild-type or deletion mutants (Δ1–2 and Δ1–3) of TMEM16F and observed using a confocal microscope. Error bars represent S.D.
C-terminal deletion mutants for TMEM16A and -16F were similarly prepared, and deletion of the last exon (exon 24 of TMEM16A and exon 20 of TMEM16F) completely abrogated protein expression in both cases (Fig. 4). The mRNA for the exon 24-deleted TMEM16F was present at a level similar to that for the wild-type TMEM16F (data not shown), suggesting that the drastic loss of C-terminal-deleted TMEM16A and -16F protein expression was because of the extreme instability of the mutant proteins.
FIGURE 4.
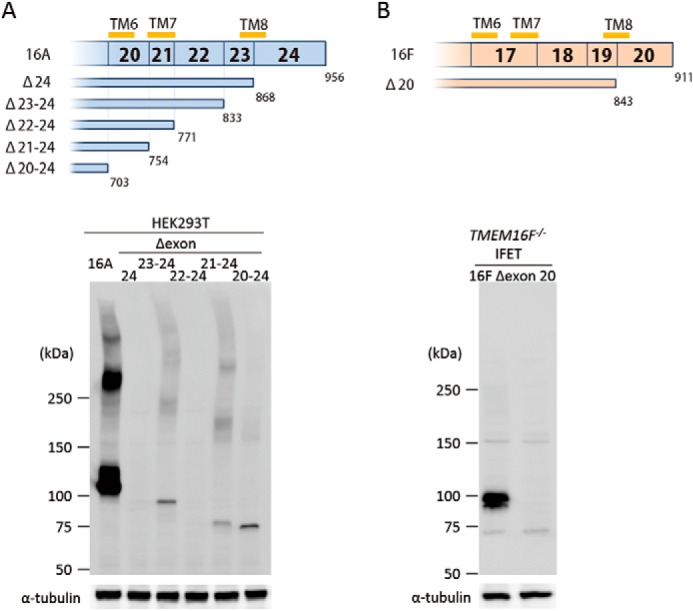
C-terminal deletion of TMEM16A and -16F. A, effect of C-terminal deletion on the expression of TMEM16A. In the upper panel, structures of the C-terminal deletion mutants of TMEM16A are schematically shown. Numbers in the top row indicate the exon number. TM6, TM7, and TM8 represent the sixth, seventh, and eighth transmembrane regions. Numbers at the bottom indicate the amino acid position where the deletion mutants end. In the lower panel, the cell lysates from 293T cells that had been transfected with the vector for full-length 16A or its C-terminal deletion mutants (Δexon 24, Δexon 23–24, Δexon 22–24, Δexon 21–24, and Δexon 20–24) were separated by 5–10% SDS-PAGE and analyzed by Western blotting with an anti-FLAG mAb. As a control, Western blotting was performed with an anti-α-tubulin mAb, and the results are shown at the bottom. B, effect of C-terminal deletion on the expression of TMEM16F. The structure of the C-terminal deletion mutant of TMEM16F is shown. Numbers in the top row indicate the exon number. TM6, TM7, and TM8 represent the sixth, seventh, and eighth transmembrane regions. Numbers at the bottom indicate the amino acid position where the deletion mutant ends. In the lower panel, the lysates from TMEM16F−/− IFET transformants expressing the full-length 16F or its C-terminal deletion mutant (Δexon 20) were separated by 5–10% SDS-PAGE and analyzed by Western blotting with an anti-FLAG mAb. As a control, Western blotting was performed with an anti-α-tubulin mAb, and the results are shown at the bottom.
Functional Swapping between TMEM16A and -16F at the N and C Termini
TMEM16A and -16F perform different functions, Cl− channel activity and phospholipid-scrambling activity; however, the deletion of N-terminal and C-terminal regions had similar effects on them. To analyze the exchangeability of these regions between TMEM16A and -16F, their exons were swapped at the corresponding regions (Figs. 5A and 6A). For protein expression, the cytoplasmic N-terminal and C-terminal tails were fully exchangeable. That is, the protein level of the chimeric molecules carrying the N- or C-terminal sequence of 16F in the nFA or AFc series chimera or the N- or C-terminal sequence of 16A in the nAF or FAc series chimera was similar to or better than that of authentic TMEM16A or -16F (Figs. 5B and 6B). Most of the N-terminal cytoplasmic tail (amino acids 1–285 of 16A and 1–250 of 16F) was also functionally exchangeable. That is, the nFA3 chimera carrying the N-terminal tail of 16F was fully active for the Cl− channel activity (Fig. 5C), and the PS-exposing activity of the nAF3 chimera carrying the N-terminal tail of 16A was about 70% of that of the authentic TMEM16F (Fig. 5D). Notably, the nAF1 and nAF2 chimeras in which the 16A-derived region was shorter than that of nAF3 did not show the PS-scrambling activity. Analyses of cellular localization with GFP-tagged proteins indicated that nAF3 but not nAF1 and nAF2 chimeras was localized at plasma membrane (Fig. 5E).
FIGURE 5.

N-terminal chimeric mutants between TMEM16A and -16F. A, structures of the two sets of N-terminal chimeric molecules (nFA and nAF series) are schematically shown. In each chimera, the regions in blue are from TMEM16A, and those in brown are from TMEM16F. Numbers in the rows indicate the exon number, and the position at which the chimeric molecule was prepared is indicated by a red bar with the construct number. The constructs with which Cl− channel or phospholipid scramblase activity was detected are marked by ○, whereas those that did not show the channel or scramblase activity are marked by ×. B, the expression levels of the chimeric proteins in 293T cells for the nFA series and in TMEM16F−/− IFETs for the nAF series were analyzed by Western blotting with an anti-FLAG mAb. C, the Cl− channel activity was measured by whole-cell patch clamp analysis of 293T cells transfected with the nFA series of chimeric constructs. D, Cy5-Annexin V binding to A23187-treated TMEM16F−/− IFET transformants expressing empty vector (−), 16F, or the nAF series of chimeric constructs was followed for 9 min using FACSCalibur and expressed as mean fluorescence intensity (MFI). E, HEK293T cells were transfected with the expression vectors for GFP-tagged nAF1, -2, or -3 and observed using a confocal microscope. Error bars represent S.D. TM, transmembrane region.
FIGURE 6.

C-terminal chimeric mutants between TMEM16A and -16F. A, structures of the two sets of C-terminal chimeric molecules (AFc and FAc series) are shown schematically. In each chimera, the regions in blue are from TMEM16A, and those in brown are from TMEM16F. Numbers in the row indicate the exon number, and the position at which the chimeric molecule was prepared is indicated by a red bar with the construct number. The constructs with which Cl− channel or phospholipid scramblase activity was detected are marked by ○, whereas those that did not show the channel or scramblase activity are marked by ×. B, the expression level of the chimeric proteins in 293T cells for the AFc series and in TMEM16F−/− IFETs for the FAc series was analyzed by Western blotting with an anti-FLAG mAb. C, membrane currents at the indicated voltage pulses (mV) were measured by whole-cell patch clamp analysis of 293T cells transfected with the AFc series of chimeric constructs. D, Cy5-Annexin V binding to the A23187-treated TMEM16F−/− IFET transformants expressing empty vector (−), full-length 16F, or the FAc series of chimeric constructs was followed for 9 min using FACSCalibur and expressed as mean fluorescence intensity (MFI). Error bars represent S.D. TM, transmembrane region.
The functional exchangeability was observed at the C terminus as well (amino acids 869–956 of TMEM16A and 844–911 of 16F). The AFc1 chimera carrying the C-terminal cytoplasmic tail of 16F and the FAc1 chimera carrying the C-terminal cytoplasmic tail of 16A worked as a Cl− channel and phospholipid scramblase, respectively (Fig. 6, C and D). It is notable that the AFc2 chimera carrying a longer 16F-derived C-terminal region (809–911 of 16F) had Cl− channel activity, whereas the FAc 2 chimera carrying the corresponding 16A-derived C-terminal region (834–956 of 16A) failed to show the phospholipid scramblase activity (Fig. 6, C and D).
Point Mutations in the Pore Region
A region called the pore is present between the fifth and sixth transmembrane regions of TMEM16A (1), and point mutations in this region affect the ion permeability of TMEM16A (5). This pore region (the entire region between the fifth and sixth transmembrane regions) is well conserved between TMEM16A and 16F with 44.3% identity in the amino acid sequence (Fig. 7A). To examine whether similar amino acids are involved in the Cl− channel activity of TMEM16A and the phospholipid scramblase activity of TMEM16F, point mutations were introduced into TMEM16A and TMEM16F at three positions (amino acid positions 617, 641, and 664 in TMEM16A and 592, 616, and 638 in TMEM16F). As shown in Fig. 7C, the replacement of arginine 617 and lysine 641 by glutamic acid in TMEM16A severely inactivated its Cl− channel activity, whereas the mutation of lysine 664 by glutamic acid had no effect. Similarly, replacing arginine 592 and lysine 616 but not lysine 638 with glutamic acid significantly inactivated the scramblase activity of 16F (Fig. 7D), and replacing both arginine 592 and lysine 616 almost completely destroyed its activity. These results suggest that, analogous to its function in TMEM16A, the pore region of TMEM16F plays an important role in its phospholipid-scrambling activity.
FIGURE 7.
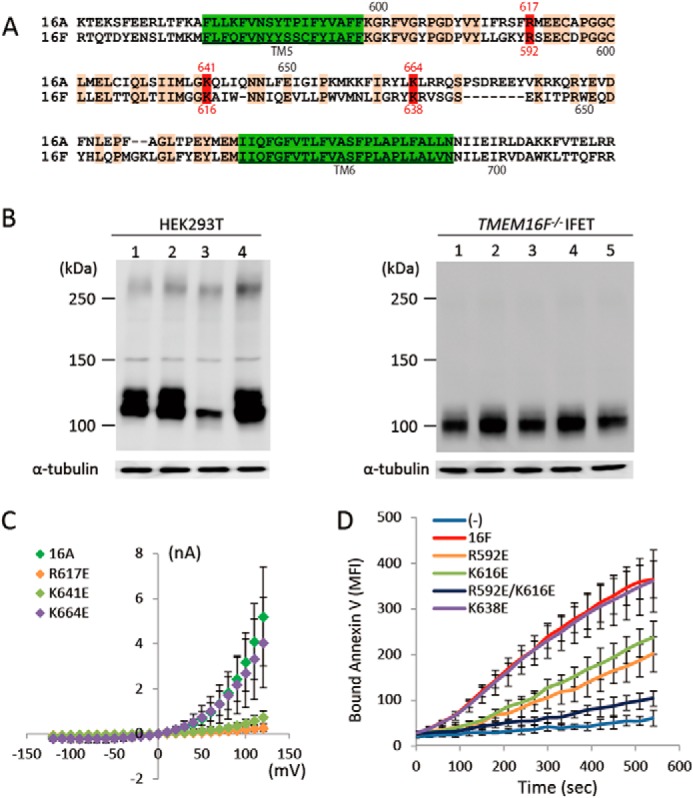
Point mutations in TMEM16A and -16F. A, the amino acid sequences of mouse TMEM16A (positions from 599 to 702) and -16F (positions from 574 to 671) were aligned to obtain maximum homology. Amino acids identical between TMEM16A and -16F are orange, and the transmembrane regions are green. The numbers above and below the sequence indicate the amino acid positions in TMEM16A and -16F, respectively. Amino acids mutated to glutamic acid are in red. B, cell lysates of HEK293T cells that had been transfected with the expression vector for TMEM16A or its indicated mutants (left panel) or the cell lysates of TMEM16F−/− IFET transformants expressing TMEM16F or its indicated mutants (right panel) were analyzed by Western blot with an anti-FLAG antibody. Left panel, lane 1, TMEM16A; lane 2, R617E mutant; lane 3, K641E mutant; lane 4, K664E mutant. Right panel, lane 1, TMEM16F; lane 2, R592E mutant; lane 3, K616E mutant; lane 4, R592E/K616E mutant; lane 5, K638E mutant. C, membrane currents at the indicated voltage pulses (mV) were measured by the whole-cell patch clamp analysis of 293T cells transfected with the expression vector for TMEM16A or its indicated point mutants. D, Cy5-Annexin V binding to the A23187-treated TMEM16F−/− IFET transformants expressing empty vector (−), 16F, or its indicated point mutants was followed for 9 min using FACSCalibur and expressed as mean fluorescence intensity (MFI). Error bars represent S.D.
Effect of Chemical Inhibitors
NPPB, DIDS, and niflumic acid are classical inhibitors of Ca2+-dependent Cl− channel activity (20); they inhibit the channel activity of TMEM16A with IC50 values of 10 μm for NPPB, 20 μm for DIDS, and 30 μm for niflumic acid (3, 4). Recently, several other compounds were found to inhibit the Cl− channel activity of 16A (27, 28). Tannic acid, EGCG, digallic acid, and T16Ainh-A01 inhibit the Cl− channel activity of 16A with IC50 values of 6.4, 100, 3.6, and 1.1 μm, respectively. To examine the effect of these inhibitors on the PS scramblase activity of TMEM16F, TMEM16F−/− IFETs expressing TMEM16F were pretreated with various concentrations of each compound for 5 min and treated with A23187 in the presence of the compound, and the PS exposure was monitored by FACS analysis. As shown in Fig. 8A, tannic acid and EGCG efficiently inhibited the scramblase activity of 16F with IC50 values of less than 1.0 μm, whereas NPPB, DIDS, digallic acid, and T16Ainh-A01 inhibited the activity of 16F with IC50 values of 80–300 μm. In particular, niflumic acid inhibited the scramblase only at high concentrations with an IC50 of more than 1000 μm. We then examined the effect of tannic acid, EGCG, digallic acid, and T16Ainh-A01 on the Cl− channel activity of TMEM16A. That is, 293T cells were transfected with the expression vector for TMEM16A, pretreated for 5 min with each compound, and subjected to the patch clamp analysis. In agreement with previous reports (27, 28), the IC50 values of tannic acid, digallic acid, and T16Ainh-A01 for the Cl− channel activity of TMEM16A were less than 10 μm, whereas that of EGCG was higher than 100 μm (Fig. 8B). A plot of IC50 of TMEM16F versus TMEM16A (Fig. 8C) indicated that some chemicals (T16Ainh-A01 and digallic acid) efficiently inhibit the channel activity of TMEM16A but not the scramblase activity of 16F, whereas another compound (EGCG) inhibits the activity of TMEM16F more efficiently than the activity of 16A.
FIGURE 8.

Effect of various compounds on the PS scramblase activity of TMEM16F and on the Cl− channel activity of TMEM16A. A, TMEM16F−/− IFET transformants expressing TMEM16F were pretreated for 5 min with the indicated concentrations of various Cl− channel inhibitors (tannic acid, EGCG, digallic acid, T16Ainh-A01, NPPB, DIDS, and niflumic acid) followed by stimulation with A23187 and flow cytometry analysis with Cy5-labeled Annexin V. The mean fluorescence intensity observed at 5 min was determined, and the data are expressed as the percentage of fluorescence intensity detected without the compound. Experiments were carried out at least three times, and the average values are plotted with S.D. (error bars). B, 293T cells transfected with the expression vector for TMEM16A were incubated for 5 min with the indicated concentrations of tannic acid, EGCG, digallic acid, or T16Ainh-A01, and membrane currents at 120 mV were measured by whole-cell patch clamp analysis. The data are expressed as the percentage of Cl− current detected without compound. Experiments were carried out at least three times, and the average values are plotted with S.D. (error bars). C, IC50 values of tannic acid, EGCG, digallic acid, and T16Ainh-A01 for TMEM16A (x axis) and -16F (y axis) were determined and plotted.
DISCUSSION
The function of TMEM16F has so far been very controversial. Several groups, including ours, reported that TMEM16F has little or no Ca2+-dependent Cl− channel activity (7, 10, 29), whereas Yang et al. (11) showed that it functions as a cation channel. On the other hand, other reports have claimed that it works as a Cl− channel in dendritic cells (30), contributes to the outward rectifying Cl− current for cell shrinkage during apoptosis (31), and functions as a volume-sensitive outwardly rectifying anion channel (32). Finally, Shimizu et al. (33) recently reported that TMEM16F functions as a Ca2+-dependent Cl− channel at high intracellular Ca2+ concentrations but that it does not support volume-sensitive outwardly rectifying currents activated by osmotic swelling or apoptotic stimulation. We found that a TMEM16F deficiency in human and mouse cells impairs Ca2+-dependent phospholipid scrambling and that its point mutation causes the constitutive scrambling of phospholipids on plasma membranes, leading us to conclude that TMEM16F is a phospholipid scramblase or at least a subunit of such a scramblase (9, 34). This PS-scrambling activity of TMEM16F was recently confirmed by Yang et al. (11) with mouse platelets and by Ehlen et al. (13) with developing bone cells.
Here, we confirmed that TMEM16F was required for Ca2+ ionophore-induced PS exposure in a mouse fetal thymocyte line. The defective PS exposure in TMEM16F−/− cells could not be rescued by TMEM16A. In contrast, TMEM16A but not -16F carries a strong Ca2+-dependent Cl− channel activity in human 293T cells. Although we cannot formally rule out the possibility that TMEM16F has a Cl− channel activity in IFETs, these results suggest that TMEM16F does not require Cl− channel activity for its phospholipid scramblase activity and that TMEM16A and -16F carry out distinct functions, Cl− channel activity and phospholipid scramblase activity, respectively.
The N-terminal deletion and swapping in TMEM16A and -16F indicated that a 200–250-amino acid N-terminal domain that is necessary for the Cl− channel activity of TMEM16A and the lipid scramblase activity of 16F is exchangeable between TMEM16A and -16F. Both TMEM16A and -16F are localized at plasma membranes. The N-terminal deletion of TMEM16A and -16F impaired their localization to the plasma membranes, and the deleted mutants stayed in the cytoplasm, likely in endoplasmic reticulum. In fact, a well conserved signal for the protein export from endoplasmic reticulum (dibasic motif (R/K)X(R/K)) (35) was found in the N-terminal domain of TMEM16A and -16F (supplemental Fig. S1). An unexpected inability of nAF1 and nAF2 chimeras to be localized at plasma membranes suggested that the dibasic motif for the endoplasmic reticulum export alone is not sufficient for transporting proteins to plasma membranes. Indeed, some proteins require multiple endoplasmic reticulum exit signals for efficient transport (36).
One of the common features of TMEM16A and -16F is the Ca2+ requirement for their activities, and an Arg-to-Leu point mutation at amino acid position 211 in mouse TMEM16F sensitizes it for Ca2+.3 It is therefore possible that the N-terminal domain of each protein is responsible for the Ca2+ responsiveness as well. Although we cannot find an EF hand motif for Ca2+ binding, a conserved putative calmodulin-binding site ((F/I/L/V/W)XXX(F/A/I/L/V/W)XX(F/A/I/L/V/W)XXXXX(F/I/L/V/W)) (37) exists in the N-terminal domain of TMEM16A and -16F (supplemental Fig. S1). Mutational analysis of this calmodulin-binding site as well as other well conserved motifs (GLYFXDGXRKVDYILVY and SLFFXDGXRRIDFILVY) with unknown function (supplemental Fig. S1) will be necessary to understand the function of the N-terminal region. A domain in the C-terminal region of TMEM16A and -16F was required for their stable protein expression and was also exchangeable between the two proteins. Protein sumoylation is known to stabilize proteins (38, 39). A putative sumoylation site ((V/I/L/M/F/P/C)KXE or (E/D)XK(L/V/I/F/P)) can be found in this region of TMEM16A and -16F (supplemental Fig. S1). Whether these motifs contribute to the stability of TMEM16A and -16F remains to be studied.
P4-type ATPase catalyzes phospholipid transport in an ATP-dependent manner and is a member of the P-type ATPases, which pump small cations and metal ions (40). In P4-type ATPase, the anionic residues that typically inhabit the ligand-binding pocket in cation pumps are replaced by a mixture of hydrophobic and polar uncharged residues, indicating that the same region may be involved in lipid transport (41). Similarly, because positively charged amino acids in the reentrance loop located between the fifth and sixth transmembrane regions in TMEM16A are essential for its ion permeability, Yang et al. (5) proposed that this domain works as a pore and filter for Cl− ions. We showed here that two positively charged residues conserved between TMEM16A and -16F in the pore region are required for the Cl− channel activity of 16A and the phospholipid-scrambling activity of 16F. However, swapping this region between TMEM16A and -16F inactivated channel and scramblase activities.4 How the similar “pocket” in TMEM16A and -16F can act on both small ions and bulky phospholipids is not clear and will be a challenging topic for future research.
The excessive extracellular transport of Cl− ions appears to cause hypertension, diarrhea, cancer, and asthma, whereas its deficiency causes salivary gland dysfunction, cystic fibrosis, dry eye syndrome, intestinal hypomotility, and other diseases (20). TMEM16A, therefore, has been focused on as a drug target, and small molecules that function as its inhibitors or activators have been developed (27, 28, 42). However, because TMEM16F is involved in the PS exposure of activated platelets and its defect causes bleeding disorders, inhibitors or activators of TMEM16A should be specific for TMEM16A. Here we showed that among seven different compounds (NPPB, DIDS, niflumic acid, tannic acid, EGCG, digallic acid, and T16Ainh-A01) that inhibit the Cl− channel activity of TMEM16A (3, 4, 27, 28), tannic acid and EGCG efficiently inhibited the scramblase activity of TMEM16F, whereas the inhibitory activity of other compounds against TMEM16F was weak. These results suggest that the active sites of TMEM16A and -16F significantly differ and are in agreement with previous observations that tannic acid and EGCG have antithrombotic activities (43). The assignment of the binding sites for these compounds in TMEM16A and -16F will be useful for understanding how TMEM16A and -16F work as a Cl− channel and phospholipid scramblase. Our results also indicate that more careful characterization of TMEM16 family members is necessary to develop better, more specific drugs.
Acknowledgments
We are grateful to H. Ohmori (Department of Physiology, Graduate School of Medicine, Kyoto University) for valuable advice on electrophysiology. We thank M. Fujii for secretarial assistance.
This work was supported in part by grants-in-aid from the Ministry of Education, Science, Sports, and Culture in Japan.

This article contains supplemental methods and Fig. S1.
J. Suzuki and S. Nagata, unpublished result.
T. Suzuki, J. Suzuki, and S. Nagata, unpublished results.
- TMEM
- transmembrane protein
- IFET
- immortalized fetal thymocyte
- PS
- phosphatidylserine
- EGCG
- epigallocatechin-3-gallate
- T16Ainh-A01
- 2-(5-ethyl-4-hydroxy-6-methylpyrimidin-2-ylthio)-N-(4-(4-methoxyphenyl)thiazol-2-yl)acetamide
- NPPB
- 5-nitro-2-(3-phenylpropylamino)benzoic acid
- DIDS
- 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid
- niflumic acid
- 2-[3-(trifluoromethyl)anilino]nicotinic acid
- DSP
- dithiobis(succinimidyl propionate)
- Ano
- anoctamin
- 16A
- TMEM16A
- 16F
- TMEM16F.
REFERENCES
- 1. Hartzell H. C., Yu K., Xiao Q., Chien L. T., Qu Z. (2009) Anoctamin/TMEM16 family members are Ca2+-activated Cl− channels. J. Physiol. 587, 2127–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flores C. A., Cid L. P., Sepúlveda F. V., Niemeyer M. I. (2009) TMEM16 proteins: the long awaited calcium-activated chloride channels? Braz. J. Med. Biol. Res. 42, 993–1001 [DOI] [PubMed] [Google Scholar]
- 3. Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L. (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594 [DOI] [PubMed] [Google Scholar]
- 4. Schroeder B. C., Cheng T., Jan Y. N., Jan L. Y. (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y. D., Cho H., Koo J. Y., Tak M. H., Cho Y., Shim W. S., Park S. P., Lee J., Lee B., Kim B. M., Raouf R., Shin Y. K., Oh U. (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215 [DOI] [PubMed] [Google Scholar]
- 6. Duran C., Hartzell H. C. (2011) Physiological roles and diseases of tmem16/anoctamin proteins: are they all chloride channels? Acta Pharmacol. Sin. 32, 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schreiber R., Uliyakina I., Kongsuphol P., Warth R., Mirza M., Martins J. R., Kunzelmann K. (2010) Expression and function of epithelial anoctamins. J. Biol. Chem. 285, 7838–7845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang F., Rock J. R., Harfe B. D., Cheng T., Huang X., Jan Y. N., Jan L. Y. (2009) Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl. Acad. Sci. U.S.A. 106, 21413–21418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki J., Umeda M., Sims P. J., Nagata S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 [DOI] [PubMed] [Google Scholar]
- 10. Suzuki J., Fujii T., Imao T., Ishihara K., Kuba H., Nagata S. (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang H., Kim A., David T., Palmer D., Jin T., Tien J., Huang F., Cheng T., Coughlin S. R., Jan Y. N., Jan L. Y. (2012) TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lhermusier T., Chap H., Payrastre B. (2011) Platelet membrane phospholipid asymmetry: from the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J. Thromb. Haemost. 9, 1883–1891 [DOI] [PubMed] [Google Scholar]
- 13. Ehlen H. W., Chinenkova M., Moser M., Munter H. M., Krause Y., Gross S., Brachvogel B., Wuelling M., Kornak U., Vortkamp A. (2013) Inactivation of Anoctamin-6/Tmem16f, a regulator of phosphatidylserine scrambling in osteoblasts, leads to decreased mineral deposition in skeletal tissues. J. Bone Miner. Res. 28, 246–259 [DOI] [PubMed] [Google Scholar]
- 14. Charlesworth G., Plagnol V., Holmström K. M., Bras J., Sheerin U.-M., Preza E., Rubio-Agusti I., Ryten M., Schneider S. A., Stamelou M., Trabzuni D., Abramov A. Y., Bhatia K. P., Wood N. W. (2012) Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am. J. Hum. Genet. 91, 1041–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mizuta K., Tsutsumi S., Inoue H., Sakamoto Y., Miyatake K., Miyawaki K., Noji S., Kamata N., Itakura M. (2007) Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 357, 126–132 [DOI] [PubMed] [Google Scholar]
- 16. Tsutsumi S., Kamata N., Vokes T. J., Maruoka Y., Nakakuki K., Enomoto S., Omura K., Amagasa T., Nagayama M., Saito-Ohara F., Inazawa J., Moritani M., Yamaoka T., Inoue H., Itakura M. (2004) The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). Am. J. Hum. Genet. 74, 1255–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vermeer S., Hoischen A., Meijer R. P., Gilissen C., Neveling K., Wieskamp N., de Brouwer A., Koenig M., Anheim M., Assoum M., Drouot N., Todorovic S., Milic-Rasic V., Lochmüller H., Stevanin G., Goizet C., David A., Durr A., Brice A., Kremer B., van de Warrenburg B. P., Schijvenaars M. M., Heister A., Kwint M., Arts P., van der Wijst J., Veltman J., Kamsteeg E.-J., Scheffer H., Knoers N. (2010) Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am. J. Hum. Genet. 87, 813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kashyap M. K., Marimuthu A., Kishore C. J., Peri S., Keerthikumar S., Prasad T. S., Mahmood R., Rao S., Ranganathan P., Sanjeeviah R. C., Vijayakumar M., Kumar K. V., Montgomery E. A., Kumar R. V., Pandey A. (2009) Genomewide mRNA profiling of esophageal squamous cell carcinoma for identification of cancer biomarkers. Cancer Biol. Ther. 8, 36–46 [DOI] [PubMed] [Google Scholar]
- 19. Das S., Hahn Y., Nagata S., Willingham M. C., Bera T. K., Lee B., Pastan I. (2007) NGEP, a prostate-specific plasma membrane protein that promotes the association of LNCaP cells. Cancer Res. 67, 1594–1601 [DOI] [PubMed] [Google Scholar]
- 20. Verkman A. S., Galietta L. J. (2009) Chloride channels as drug targets. Nat. Rev. Drug Discov. 8, 153–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morita S., Kojima T., Kitamura T. (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 22. Murai K., Murakami H., Nagata S. (1998) Myeloid-specific transcriptional activation by murine myeloid zinc finger protein-2. Proc. Natl. Acad. Sci. U.S.A. 95, 3461–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuba H., Yamada R., Ohmori H. (2003) Evaluation of the limiting acuity of coincidence detection in nucleus laminaris of the chicken. J. Physiol. 552, 611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sheridan J. T., Worthington E. N., Yu K., Gabriel S. E., Hartzell H. C., Tarran R. (2011) Characterization of the oligomeric structure of the Ca2+-activated Cl− channel Ano1/TMEM16A. J. Biol. Chem. 286, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferrera L., Caputo A., Ubby I., Bussani E., Zegarra-Moran O., Ravazzolo R., Pagani F., Galietta L. (2009) Regulation of TMEM16A chloride channel properties by alternative splicing. J. Biol. Chem. 284, 33360–33368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Traut T. W. (1988) Do exons code for structural or functional units in proteins? Proc. Natl. Acad. Sci. U.S.A. 85, 2944–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Namkung W., Phuan P.-W., Verkman A. S. (2011) TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J. Biol. Chem. 286, 2365–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Namkung W., Thiagarajah J. R., Phuan P. W., Verkman A. S. (2010) Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 24, 4178–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duran C., Qu Z., Osunkoya A. O., Cui Y., Hartzell H. C. (2012) ANOs 3–7 in the anoctamin/Tmem16 Cl− channel family are intracellular proteins. Am. J. Physiol. Cell Physiol. 302, C482–C493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szteyn K., Schmid E., Nurbaeva M. K., Yang W., Münzer P., Kunzelmann K., Lang F., Shumilina E. (2012) Expression and functional significance of the Ca2+-activated Cl− channel ANO6 in dendritic cells. Cell. Physiol. Biochem. 30, 1319–1332 [DOI] [PubMed] [Google Scholar]
- 31. Martins J. R., Faria D., Kongsuphol P., Reisch B., Schreiber R., Kunzelmann K. (2011) Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc. Natl. Acad. Sci. U.S.A. 108, 18168–18172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Almaça J., Tian Y., Aldehni F., Ousingsawat J., Kongsuphol P., Rock J. R., Harfe B. D., Schreiber R., Kunzelmann K. (2009) TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J. Biol. Chem. 284, 28571–28578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimizu T., Iehara T., Sato K., Fujii T., Sakai H., Okada Y. (2013) TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am. J. Physiol. Cell Physiol. 304, C748–C759 [DOI] [PubMed] [Google Scholar]
- 34. Segawa K., Suzuki J., Nagata S. (2011) Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. U.S.A. 108, 19246–19251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giraudo C. G., Maccioni H. J. (2003) Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol. Biol. Cell 14, 3753–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barlowe C. (2003) Signals for COPII-dependent export from the ER: what's the ticket out? Trends Cell Biol. 13, 295–300 [DOI] [PubMed] [Google Scholar]
- 37. Rhoads A. R., Friedberg F. (1997) Sequence motifs for calmodulin recognition. FASEB J. 11, 331–340 [DOI] [PubMed] [Google Scholar]
- 38. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 39. Klenk C., Humrich J., Quitterer U., Lohse M. J. (2006) SUMO-1 controls the protein stability and the biological function of phosducin. J. Biol. Chem. 281, 8357–8364 [DOI] [PubMed] [Google Scholar]
- 40. Folmer D. E., Elferink R. P., Paulusma C. C. (2009) P4 ATPases—lipid flippases and their role in disease. Biochim. Biophys. Acta 1791, 628–635 [DOI] [PubMed] [Google Scholar]
- 41. Muthusamy B.-P., Natarajan P., Zhou X., Graham T. R. (2009) Linking phospholipid flippases to vesicle-mediated protein transport. Biochim. Biophys. Acta 1791, 612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Namkung W., Yao Z., Finkbeiner W. E., Verkman A. S. (2011) Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 25, 4048–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang W.-S., Lim I.-H., Yuk D.-Y., Chung K.-H., Park J.-B., Yoo H.-S., Yun Y.-P. (1999) Antithrombotic activities of green tea catechins and (−)-epigallocatechin gallate. Thromb. Res. 96, 229–237 [DOI] [PubMed] [Google Scholar]