

Background: Noscapine is a safe orally available anticough medicine also known to bind microtubules and induce cancer cell death.
Results: Noscapine inhibits myofibroblast differentiation and pulmonary fibrosis through prostaglandin receptors and activation of PKA.
Conclusion: Noscapine is an antifibrotic drug acting through PKA activation via EP2 prostaglandin receptors.
Significance: This study describes a novel antifibrotic function and novel mechanism of action of noscapine.
Keywords: Fibrosis, Microtubules, Myofibroblast, Prostaglandins, Protein Kinase A (PKA), Noscapine, SRF, Stress Fibers
Abstract
Myofibroblast differentiation is a key process in the pathogenesis of fibrotic disease. We have shown previously that differentiation of myofibroblasts is regulated by microtubule polymerization state. In this work, we examined the potential antifibrotic effects of the antitussive drug, noscapine, recently found to bind microtubules and affect microtubule dynamics. Noscapine inhibited TGF-β-induced differentiation of cultured human lung fibroblasts (HLFs). Therapeutic noscapine treatment resulted in a significant attenuation of pulmonary fibrosis in the bleomycin model of the disease. Noscapine did not affect gross microtubule content in HLFs, but inhibited TGF-β-induced stress fiber formation and activation of serum response factor without affecting Smad signaling. Furthermore, noscapine stimulated a rapid and profound activation of protein kinase A (PKA), which mediated the antifibrotic effect of noscapine in HLFs, as assessed with the PKA inhibitor, PKI. In contrast, noscapine did not activate PKA in human bronchial or alveolar epithelial cells. Finally, activation of PKA and the antifibrotic effect of noscapine in HLFs were blocked by the EP2 prostaglandin E2 receptor antagonist, PF-04418948, but not by the antagonists of EP4, prostaglandin D2, or prostacyclin receptors. Together, we demonstrate for the first time the antifibrotic effect of noscapine in vitro and in vivo, and we describe a novel mechanism of noscapine action through EP2 prostaglandin E2 receptor-mediated activation of PKA in pulmonary fibroblasts.
Introduction
Idiopathic pulmonary fibrosis is a progressive, fatal disease characterized by parenchymal fibrosis and structural distortion of the lungs. Age-adjusted mortality due to pulmonary fibrosis is increasing (1), and it poses a vexing clinical challenge given the lack of proven efficacious therapy. Idiopathic pulmonary fibrosis is thought to be a disorder of abnormal wound healing (2, 3), in which the initial trigger to the fibrotic response is injury to the alveolar epithelial cells, followed by an exuberant, nonresolving wound healing response (4–6) driven by myofibroblasts, specialized fibroblasts with enhanced contractile and matrix gene expression (7–9). Disrupting cellular mechanisms responsible for the acquisition of the myofibroblast phenotype may be a potential strategy to attenuate the ongoing fibrotic response in pulmonary fibrosis.
Transforming Growth Factor-β1 (TGF-β1) is a key mediator of myofibroblast differentiation and tissue fibrosis (10–12). We have demonstrated previously that TGF-β utilizes stress fiber-dependent signaling driving nuclear accumulation of megakaryoblastic leukemia-1 (MKL1)2 and activation of serum response factor (SRF) to fully induce the myofibroblast phenotype, and that activators of protein kinase A (PKA) attenuate myofibroblast differentiation through inhibition of SRF, but not Smad signaling (13, 14). These data suggest that targeting the MKL1/SRF pathway could be an attractive approach for preventing myofibroblast differentiation and pulmonary fibrosis without affecting the initial Smad signaling of TGF-β.
Previous studies using the bleomycin model of pulmonary fibrosis have shown the protective effects of the prostacyclin analog iloprost or of prostaglandin E2 (15, 16), both of which are known to act through cAMP/PKA signaling. We have shown a similar protective effect of adrenomedullin in a mouse model with genetic sensitization of adrenomedullin signaling in myofibroblasts (17). We also have shown recently that myofibroblast differentiation is regulated by microtubule dynamics through a control of actin stress fiber/SRF signaling, independent of Smads (18). Unfortunately, the prototypical microtubule stabilizer, paclitaxel (Taxol), has significant toxicity which limits its potential as a therapeutic in fibrotic disease. A search for modulators of microtubule dynamics with low toxicity in vivo identified noscapine as a potential candidate compound, and we sought to investigate its antifibrotic potential.
Noscapine is a nonsedative, nonaddictive alkaloid found in opium latex, which has been used with low toxicity as an antitussive medication in humans (19–21). The mechanism of the antitussive action of noscapine remains unclear but may involve the central nervous system (22, 23) and more specifically, sigma receptors, as this effect of noscapine was inhibited by the sigma receptor antagonist, rimcazole (24). More recently, studies by Joshi's group have shown that noscapine binds to tubulin, changes its conformation, and arrests dividing mammalian cells in mitosis through its effects on microtubule assembly (25). Therefore, noscapine has been studied extensively in cancer research, and its antitumor activity has been demonstrated in multiple in vivo studies (25–28).
In this study, we show for the first time that noscapine is an effective antifibrotic agent both in vitro and in vivo and describe a novel mechanism of noscapine action through a rapid activation of cAMP/PKA signaling mediated by EP2 prostaglandin E2 receptors in pulmonary fibroblasts.
EXPERIMENTAL PROCEDURES
Primary Culture of Human and Mouse Lung Fibroblasts
Human lung fibroblasts were cultured as described previously (13). Briefly, tissue samples from explanted lungs from patients undergoing lung transplantation were placed in DMEM with antibiotics. Lung tissue was minced to ∼1-mm3 pieces, washed, and plated on 10-cm plates in growth medium containing DMEM supplemented with 10% FBS and antibiotics. The medium was changed twice a week. After ∼2 weeks, the explanted and amplified fibroblasts were cleared from the tissue pieces, trypsinized, and further amplified as passage 1. Mouse lung fibroblasts were cultured in a similar manner. For experiments, cells were grown in 12-well plates at a density of 1 × 105 cells/well in a growth medium for 24 h, starved in DMEM containing 0.1% bovine serum albumin (BSA) for 48 h, and treated with desired drugs for various times as indicated in the figure legends. All primary cultures were used from passage 3 to 10.
Cytotoxicity Assay
Cytotoxicity of drugs was examined by measuring the release of lactate dehydrogenase in the media, using the CytoTox 96 assay (Promega) following the manufacturer's protocol.
Cell Lysis and Western Blotting
Cells were lysed in radioimmunoprecipitation assay buffer containing 25 mm HEPES (pH 7.5), 150 mm NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 0.5% sodium deoxycholate, 2 mm EDTA, 2 mm EGTA, 10% glycerol, 1 mm NaF, 200 μm sodium orthovanadate, and protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF), scraped, sonicated for 5 s, mixed with Laemmli buffer, and boiled for 5 min. Samples were then subjected to polyacrylamide gel electrophoresis and Western blotting with desired primary antibodies and corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies, and developed by chemiluminescence reaction (Pierce). Digital chemiluminescent images below the saturation level were obtained with a LAS-4000 analyzer, and the light intensity was quantified using Multi Gauge software (Fujifilm).
Bleomycin-induced Pulmonary Fibrosis
8–10 week old C57BL/6 were intratracheally instilled with 1 unit/kg bleomycin (Bleocip, Cipla Ltd.). Noscapine was administered intraperitoneally daily at a dose of 100 mg/kg beginning on day 7 after bleomycin treatment. Lungs were removed 21 days after bleomycin administration. Left lungs were formalin-fixed and paraffin embedded, and immunohistochemistry was performed on lung sections. Images of the stained sections were obtained on a CRi Pannoramic whole slide scanner. Right lungs were used for hydroxyproline content measurements.
Hydroxyproline Assay
The hydroxyproline assay was performed as described previously (17). Briefly, right lungs were homogenized in 6 n hydrochloric acid and hydrolyzed for 12 h at 110 °C. An aliquot was evaporated, resuspended in citrate-acetate buffer with chloramine T, and left at room temperature for 20 min. Ehrlich's solution was then added, and samples were heated at 65 °C for 15 min. After cooling to room temperature, absorbance was measured at 550 nm. Hydroxyproline content was determined against a standard curve generated from pure hydroxyproline.
Transfection and Luciferase Assay
Subconfluent cells were co-transfected with desired firefly luciferase reporter plasmid, and thymidine kinase (TK) promoter-driven Renilla luciferase plasmid (TK-Rl). Cells were serum-starved followed by stimulation with the desired agonists. Cells were washed and then lysed in protein extraction reagent. Lysates were assayed for firefly and Renilla luciferase activity using the dual luciferase assay kit (Promega). To account for differences in transfection efficiency, firefly luciferase activity of each sample was normalized to Renilla luciferase activity.
In Vitro Isolation of Stress Fibers
Stress fibers were isolated as described previously (14, 18). All of the procedures were performed on ice using buffers containing protease inhibitors (Sigma). After stimulation with desired agonists, cells were washed and then extracted with buffer containing 2.5 mm triethanolamine (pH 8.2), followed by extraction with 0.05% Nonidet P-40 (pH 7.2) and subsequent extraction with 0.5% Triton X-100 (pH 7.2). Cells were washed, PBS was added, and the cells were scraped and centrifuged at 100,000 × g for 1 h. Supernatant was removed, and the pellet was sonicated in PBS.
Isolation of nuclear and cytoplasmic fractions was performed using NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific) following the manufacturer's protocol. The purity of isolated fractions was confirmed by Western blotting with the nuclear lamin A/C and cytoplasmic 14-3-3β.
In Vitro Isolation of Soluble and Insoluble Microtubule Fractions
Soluble and insoluble fractions of microtubules were isolated using a method described previously (29). Cells were rinsed twice with warm Dulbecco's phosphate-buffered saline and lysed at room temperature in microtubule-stabilizing buffer containing 2 m glycerol, 20 mm Tris-HCl (pH 6.8), 140 mm NaC1, 0.5% Nonidet P-40, 1 mm MgC12, 2 mm EGTA, 10 μm Taxol, and protease inhibitors mixture. The plates were then put on ice; cells were scraped and transferred to a microcentrifuge tube. Each well was rinsed with a second aliquot of stabilization buffer, combined with the first aliquot, and briefly vortexed. Lysates were centrifuged at 20,000 × g for 10 min at 4 °C. Supernatants containing soluble tubulin were removed, and the pellets with polymerized tubulin were lysed in radioimmunoprecipitation assay buffer and sonicated.
Indirect Immunofluorescence Microscopy
Cells were fixed in 4% paraformaldehyde in PBS followed by permeabilization with 0.2% Triton X-100 in PBS. Cells were then incubated in 2% BSA in PBS, followed by incubation with mouse α-tubulin antibodies at 4 °C overnight, washed, and incubated with anti-mouse FITC-conjugated secondary antibodies, washed, and coverslips were mounted using Vectashield mounting medium containing DAPI nuclear stain (Vector Laboratories, Burlingame, CA). Immunofluorescence images were obtained using an Olympus 1X71 fluorescent microscope.
Adenovirus-mediated DNA Transduction
Adenovirus (0.2 × 109 viral particles/ml) was incubated with cells for 24 h with Lipofectamine LTX reagent (Stratagene, La Jolla, CA) to facilitate the efficiency of transduction. Cells were then serum-starved and stimulated with the desired agonists for the indicated times.
Reagents
Noscapine hydrochloride hydrate was from Sigma-Aldrich. TGF-β was from Calbiochem. Pharmaceutical grade bleomycin (Bleocip) was from Cipla LTD. SC-560 and NS-298 were from Sigma-Aldrich. AH-6809, RO-1138452, PF-04418948, L-161,982 and BW A868C were from Cayman Chemicals. Rimcazole was from Santa Cruz Biotechnology. Antibodies against SM α-actin, β-actin, and α-tubulin were from Sigma-Aldrich; collagen-1 antibodies used for Western blotting were from Cedarlane; vasodilator-stimulated phosphoprotein (VASP) antibodies were from Calbiochem; MKL1 antibodies were from Bethyl Laboratories; PKA-substrate antibodies were from Cell Signaling Technologies. Antibodies against collagen I (ab21286) and collagen III (ab7778) were obtained from Abcam and were used for immunohistochemistry at the dilution of 1:300 and 1:600, respectively. The firefly luciferase reporter for SRF activity (SRF-Luc) was a gift from Tatyana Voyno-Yasenetskaya (University of Illinois at Chicago). The firefly luciferase reporter for the −125 base pair SM α-actin promoter was a kind gift of Dr. Joseph Miano (University of Rochester). The plasmid for luciferase reporter driven by Smad-binding elements (SBE-Luc) was provided by Dr. Bert Vogelstein (The Johns Hopkins University). Ad-PKI and Ad-LacZ were described previously (30).
Statistical Analysis
Quantitative data from three independent experiments were analyzed by Student's t test. Values of p < 0.05 (*) were considered statistically significant.
RESULTS
Antifibrotic Effects of Noscapine in Vitro and in Vivo
We first examined the effect of noscapine on TGF-β-induced differentiation of cultured human lung fibroblasts (HLFs). As shown in Fig. 1A, noscapine significantly attenuated TGF-β-induced expression of SM-α-actin and collagen-1 in cultured HLFs. Given the reported effect of noscapine on apoptosis of cancer cells (25), we examined the cytotoxic effect of noscapine on HLF by measuring the release of lactate dehydrogenease. As shown in Fig. 1B, noscapine treatment promoted a small although statistically significant release of lactate dehydrogenase compared with a profound effect of hydrogen peroxide.
FIGURE 1.

Antifibrotic effect of noscapine in vitro and in vivo. A, inhibition of myofibroblast differentiation by noscapine in vitro. Quiescent HLF were treated with 1 ng/ml TGF-β1 in the presence of 100 μm noscapine for 48 h. Cell lysates were analyzed by Western blotting with the indicated antibodies. Shown are the representative images and the quantitative analysis of ECL from at least three independent experiments. B, effect of 100 μm noscapine or 1 mm H2O2 (48 h) on lactate dehydrogenase (LDH) release by HLF. C and D, attenuation of bleomycin-induced pulmonary fibrosis by noscapine. Mice were treated with 1 unit/kg bleomycin intratracheally. 7 days after bleomycin administration, 100 mg/kg noscapine or vehicle was delivered intraperitoneally daily for 14 more days. Animals were sacrificed, and lungs were removed. C, right lungs were processed for hydroxyproline assay. DMSO, dimethyl sulfoxide. D, left lungs were formalin-fixed, paraffin-embedded, sectioned, and processed for H&E or trichrome staining or for immunohistochemistry with collagen I, collagen III, or SM-α-actin (SMA) antibodies. The specificity of primary antibodies is demonstrated by the lack of staining in airway epithelial cells. No staining was observed in the absence of primary antibodies or with the use of normal IgG (data not shown). Representative images are shown. Error bars, S.E.; *, p < 0.05.
To examine the effect of noscapine on fibrinogenesis in vivo, we used the bleomycin model of pulmonary fibrosis in mice. In this model, intratracheal administration of bleomycin results in the initial injury of alveolar epithelial cells followed by an inflammatory response during the first week and development of pulmonary fibrosis at 2–3 weeks after bleomycin administration. As shown in Fig. 1, intratracheal administration of bleomycin resulted in a significant pulmonary fibrosis accompanied by accumulation myofibroblasts, as determined by hydroxyproline assay for collagen content (Fig. 1C) and by histological assessment of lung sections stained with H&E, trichrome, or with antibodies against collagen I, collagen III, or SM-α-actin (Fig. 1D). To avoid a potential effect of noscapine on the initial injury and inflammatory response, we used a “therapeutic” protocol, wherein noscapine was administered daily beginning on day 7 after bleomycin administration, and the lungs were analyzed on day 21. As shown in Fig. 1, administration of noscapine resulted in a significant decrease of pulmonary fibrosis as assessed by the hydroxyproline assay of mouse lungs and by immunohistochemical staining of mouse lung sections with antibodies against collagen I, collagen III, or SM-α-actin. Importantly, noscapine-treated lungs had a clearly decreased number of SM-α-actin-positive cells (Fig. 1D), which is consistent with the regulation of myofibroblast differentiation by noscapine in vitro (Fig. 1A).
Noscapine Inhibits TGF-β1-induced SRF Activity without Affecting Smad-dependent Gene Transcription
We next examined the mechanism by which noscapine inhibits myofibroblast differentiation in response to TGF-β. We and others have established previously that this process is initiated by Smad-dependent expression of intermediate signaling molecules driving the activation of SRF that is required for the expression of SM-α-actin, collagen 1, and other myofibroblast differentiation markers (13, 14, 31). We also showed that activation of SRF by TGF-β in HLF is mediated by increased expression and nuclear accumulation of SRF co-activator MKL1 and that this process is blocked by microtubule stabilizer, Taxol (14, 18). As shown in Fig. 2A, noscapine attenuated TGF-β-induced expression and accumulation of MKL1 in the nuclear fraction. Furthermore, treatment of HLFs with noscapine resulted in a significantly decreased activation of SRF by TGF-β, as assessed by a SRF-driven luciferase reporter (Fig. 2B). Finally, noscapine blocked TGF-β-induced activation of −125-base pair fragment of SM-α-actin promoter (Fig. 2C) known to contain two SRF binding sites (32). In contrast, noscapine did not affect TGF-β-induced Smad2 phosphorylation (Fig. 2D), nuclear accumulation (Fig. 2E), or Smad-dependent gene transcription, as determined by a luciferase reporter driven by Smad-binding elements (Fig. 2F).
FIGURE 2.

Noscapine inhibits SRF activation without affecting Smad signaling in HLF. Effect of 100 μm noscapine (Nosc) on TGF-β1-induced MKL1 expression and accumulation in the nuclear fraction (A), on SRF-luciferase activity (B), on −125-bp SMA promoter activity (C), on Smad2 phosphorylation (D), on Smad2 nuclear accumulation (E), and on SBE-luciferase activity (F) is shown. Error bars, S.E.; *, p < 0.05.
Noscapine Does Not Affect the Gross Microtubule Content but Inhibits Actin Polymerization in Response to TGF-β
In vitro studies by others suggest that noscapine does not promote or inhibit the gross microtubule polymerization, but rather affects the steady-state dynamics of microtubule assembly, by increasing the ”pause” state of microtubules in cancer cells (33). Nevertheless, given our previously published data showing that microtubule stabilization by Taxol inhibits myofibroblast differentiation (18), we examined whether noscapine affected the gross microtubule content of HLFs. Immunocytochemical analysis showed that noscapine clearly altered the architecture of microtubules, causing their concentration at the perinuclear space (Fig. 3A). However, it was not evident from this analysis whether noscapine promoted gross microtubule polymerization, whereas Taxol treatment showed a clear increase in immunohistochemical staining of microtubules (Fig. 3A). Therefore, we performed in vitro microtubule fractionation from cultured HLFs as a quantitative assay for the gross microtubule content. As shown in Fig. 3B, the microtubule fraction was not significantly enriched with α-tubulin by noscapine following 24-h treatment, whereas Taxol treatment clearly resulted in microtubule polymerization. We also failed to detect microtubule stabilization by noscapine at earlier time points (1–3 h) using this assay (data not shown), whereas Taxol was effective in microtubule polymerization at these time points (14). These data suggests that noscapine and Taxol inhibit myofibroblast differentiation by distinct mechanisms.
FIGURE 3.
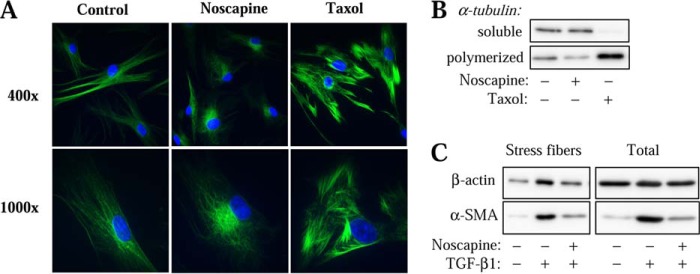
Noscapine has no effect on gross microtubule polymerization state but inhibits stress fiber formation in HLF. A, HLFs were treated with 100 μm noscapine or 10 μm Taxol for 24 h, followed by immunostaining with β-tubulin antibodies (green) and counterstaining for nuclei with DAPI (blue). Shown are the representative images at ×400 and ×1000 magnification. B, HLFs were treated with 100 μm noscapine or 10 μm Taxol for 24 h. Soluble and polymerized β-tubulin were in vitro fractionated and detected by Western blotting. C, HLFs were treated with 1 ng/ml TGF-β1 with or without 100 μm noscapine for 24 h. In vitro stress fiber preparations or total cell lysates were analyzed by Western blotting for β-actin or SMA as indicated.
Therefore, we then further examined the possible mechanism of regulation of myofibroblast differentiation by noscapine. Given that noscapine inhibited SRF activation but not Smad-dependent gene transcription in response to TGF-β (Fig. 2), and given our previous studies pointing to the role of actin stress fiber formation in TGF-β-induced myofibroblast differentiation downstream of Smad signaling (14), we examined the effect of noscapine on this process by in vitro isolation of stress fibers. As shown in Fig. 3C, noscapine appreciably attenuated TGF-β-induced accumulation of the housekeeping β-actin as well as of the newly expressed SM-α-actin in the stress fiber fraction.
Activation of PKA Mediates the Regulation of Myofibroblast Differentiation by Noscapine
We and others have previously established that myofibroblast differentiation can be regulated by activators of cAMP/PKA signaling through inhibition of SRF but not of Smad-dependent gene transcription (13, 34). Therefore, we examined whether the effect of noscapine is dependent on PKA activation. Strikingly, we found that noscapine stimulated a rapid and prolonged stimulation of PKA activity in HLFs, as assessed by the electrophoretic mobility shift of PKA substrate, VASP (Fig. 4A). We have previously established that such a shift of VASP is mediated by a PKA-dependent phosphorylation and serves as a sensitive reporter for PKA activity (35). Supporting this, noscapine induced phosphorylation of multiple proteins phosphorylated by PKA, as assessed by Western blotting with PKA-substrate antibodies (Fig. 4A), the specificity of which was demonstrated by us previously (36). Importantly, a 1-h stimulation with Taxol, which resulted in a complete microtubule polymerization (18), had no effect on PKA activation (Fig. 4A), suggesting that the effect of noscapine on myofibroblast differentiation may be not related to microtubule stabilization.
FIGURE 4.

Activation of PKA mediates the regulation of myofibroblast differentiation by noscapine. A, quiescent HLFs were treated with 10 μm Taxol or 100 μm noscapine for the indicated times. Lysates were analyzed by Western blotting with antibodies against VASP or against PKA-substrate (PKAS) antibodies. B, HLFs, mouse lung fibroblasts (MLF), human bronchial epithelial cells (16HBE), or human alveolar epithelial cells (A549) were treated with 100 μm noscapine or 10 μm forskolin for the indicated times. Lysates were analyzed by Western blotting for VASP shift. C and D, HLF were transduced with Ad-PKI or control Ad-LacZ adenoviruses. C, cells were then treated with 100 μm noscapine for 30 min, and equal amounts of protein were assessed by Western blotting for VASP shift. D, cells were treated with 100 μm noscapine followed by 1 ng/ml TGF-β1 for 48 h. Equal amounts of cell lysates were analyzed by Western blotting for the desired proteins. Cell lysates were analyzed by Western blotting with the indicated antibodies. Shown are the representative images and the quantitative analysis of enhanced chemiluminescence from three independent experiments. Error bars, S.E.; *, p < 0.05.
Noscapine also induced a rapid VASP shift in primary cultured mouse lung fibroblasts, albeit to a lesser extent (Fig. 4B). Interestingly, noscapine failed to induce VASP shift in a human bronchial epithelial cell line 16-HBE or in the alveolar epithelial cancer cell line A549 (Fig. 4B), suggesting a cell-specific effect of noscapine on PKA activation.
To examine the role of PKA in the regulation of myofibroblast differentiation by noscapine, we used adenovirus-mediated expression of the PKA inhibitor protein, PKI (Ad-PKI), which we and others have shown to be an effective and specific tool for the assessment of the role of PKA (30, 37). As shown in Fig. 4C, transduction of HLFs with Ad-PKI abolished the noscapine-induced VASP shift, whereas the control Ad-LacZ did not. Importantly, Ad-PKI but not Ad-LacZ rescued HLF cells from the inhibition of TGF-β-induced expression of SM-α-actin and collagen-1 by noscapine (Fig. 4D).
EP2 Prostaglandin Receptors Mediate Regulation of Myofibroblast Differentiation by Noscapine in HLF
Previous studies have demonstrated that human lung fibroblasts are highly responsive to prostaglandin E2 (PGE2), prostaglandin D2 (PGD2), and prostacyclin (PGI2) in terms of cAMP activation and inhibition of myofibroblast activation (13, 34, 38). We also showed that PKA can be rapidly activated by endothelin-1 or by ATP through cyclooxygenase (COX)-mediated prostanoid synthesis in vascular smooth muscle cells (30, 35, 37). In addition, the effects of noscapine on the cough reflex may involve opioid sigma receptors (24), although there is no evidence for activation of PKA through these receptors. To test the potential involvement of these mechanisms in the action of noscapine, we used pharmacological inhibitors of the corresponding receptors and enzymes. As shown in Fig. 5A, noscapine-induced VASP phosphorylation was not affected by the sigma receptor antagonist, rimcazole, by the specific antagonist of prostacyclin receptors, RO-1138452, or by the combined inhibition of COX-I and COX-II with SC-560 and NS-398, respectively. In contrast the PGE2/PGD2 receptor antagonist, AH6809, completely blocked the noscapine-induced VASP shift (Fig. 5A).
FIGURE 5.

Activation of PKA and regulation of myofibroblast differentiation by noscapine are mediated by EP2 receptors. A, HLFs were pretreated for 1 h with the EP/DP receptor antagonist, AH-6809 (10 μm), with the IP receptor antagonist, RO-1138452 (20 μm), with the COX-I and COX-II inhibitors, SC-560 (3 μm) and NS-398 (3 μm), or with the sigma opioid receptor antagonist, rimcazole (10 μm), followed by 100 μm noscapine for 10 min. Equal amounts of lysates were analyzed by Western blotting for VASP shift. B, HLFs were treated with EP2 receptor antagonist, PF-04418948 (1 μm), with EP4 receptor antagonist, L-161,982 (10 μm), or with DP receptor antagonist, BW A868C (10 μm) for 1 h, followed by stimulation with 100 μm noscapine for 10 min. Equal amounts of lysates were analyzed by Western blotting for VASP shift. C, HLFs were treated with EP2 receptor antagonist PF-04418948 (1 μm) for 1 h, followed by stimulation with 1 ng/ml TGF-β with or without 100 μm noscapine for 48 h. Cell lysates were analyzed by Western blotting with the indicated antibodies. Shown are the representative images and the quantitative analysis of ECL from three independent experiments. Error bars, S.E.; *, p < 0.05.
We then examined the effect of more selective antagonists of prostanoid receptors known to mediate cAMP production on the regulation of VASP shift and on myofibroblast differentiation by noscapine. Fig. 5B shows that noscapine-induced VASP shift was blocked by the selective antagonist of EP2 receptors for PGE2, PF-04418948. In contrast, noscapine-induced VASP shift was unaffected by the antagonist of EP4 receptors for PGE2 (L-161,982), or of DP receptors for PGD2 (BW A868C) (Fig. 5B). Furthermore, PF-04418948 rescued cells from the inhibition of TGF-β-induced expression of SM-α-actin and collagen-1 by noscapine (Fig. 5C). Together, these data suggest that regulation of myofibroblast differentiation by noscapine is, at least in part, mediated by activation of PKA through EP2 receptors.
DISCUSSION
Noscapine has been studied extensively in cancer research due to its effects on microtubule dynamics, and its antitumor activity has been demonstrated in multiple in vivo studies (25–28). Our study demonstrates for the first time that (i) noscapine attenuates fibrotic responses both in cultured human pulmonary fibroblasts and in an experimental model of pulmonary fibrosis; (ii) noscapine activates PKA specifically in pulmonary fibroblasts but not in bronchial or alveolar epithelial cells; (iii) PKA activation mediates the antifibrotic effects of noscapine; and (iv) the mechanism of PKA stimulation by noscapine involves activation of EP2 prostanoid receptors, which mediate regulation of myofibroblast differentiation by noscapine.
The antifibrotic effect of noscapine found in our study is similar to the stable prostacyclin analog, iloprost, in the bleomycin model (15). However, the therapeutic potential of iloprost maybe limited due to the side effects associated with vasodilation of vascular pulmonary beds. Of note, noscapine had no effect on PKA activation in cultured human pulmonary artery smooth muscle cells (data not shown). Thus, noscapine may have a greater therapeutic potential for treatment of pulmonary fibrosis, as it has been already used for decades as an antitussive drug with low toxicity in several countries throughout Europe, Asia, and South America, and there are no reports on pulmonary vasodilation in response to noscapine treatment.
Our studies show for the first time that noscapine induces a rapid and profound activation of PKA in human lung fibroblasts, which we reproduced using noscapine from two independent sources (Sigma-Aldrich and R&D Systems). We also show that PKA activation by noscapine is causatively associated with inhibition of myofibroblast differentiation. Given numerous studies on the mechanism of noscapine action, one would be surprised that this effect of noscapine has not been described before. In fact, one earlier study suggested that noscapine can promote cAMP accumulation in response to the activator of adenylyl cyclase, forskolin, in brain slices, while having no effect on cAMP accumulation in the absence of forskolin (23). Our data suggest a cell-specific effect of noscapine on PKA activation, wherein it had no effect on PKA activity in epithelial (bronchial or alveolar) cell lines (Fig. 4), as well as in pulmonary artery smooth muscle cells (data not shown), likely due to a lack of EP2 receptor expression in these cells. Given that most studies on noscapine as related to its effects on microtubule assembly and regulation of cell growth were focused on cancer cells from epithelial and lymphoid origins (25, 26, 39–41), it is possible that PKA activation by noscapine was overlooked, at least in fibroblasts. A broader investigation of cell-specific effects of noscapine will be performed in the future.
Our data also suggest that the antifibrotic effect of noscapine on lung fibroblast activation is mediated by EP2 receptors for PGE2, as the selective EP2 receptor antagonist, PF-04418948, blocked the activation of PKA and rescued the inhibition of myofibroblast differentiation by noscapine (Fig. 5). Furthermore, the antifibrotic effect of noscapine in our experiments (Fig. 1) was similar to that shown for PGE2, also in the bleomycin model (16). Of note, multiple groups have demonstrated dysregulation of PGE2 synthesis (as a result of down-regulation of COX-II expression) in the lung of human patients with pulmonary fibrosis as well as in animal models, which was associated with severity of the disease (42–47), suggesting that activation of PGE2 receptors could be an attractive approach for treatment of pulmonary fibrosis. Thus, noscapine could be a novel, nontoxic and orally available agonist of EP2 receptor signaling with antifibrotic action.
The precise mechanism of EP2 receptor activation by noscapine requires further investigation. It remains to be determined whether (and how) noscapine binds EP2 receptors and whether it acts as an EP2 agonist compared with PGE2 and with the other EP2 agonists. Given that noscapine has no structural homology to PGE2, it would be important to identify a structural moiety of noscapine responsible for EP2 activation and antifibrotic action compared with its effect on microtubule dynamics. Finally, a number of noscapine analogs are being developed for anticancer therapy based on its microtubule binding properties (48–50); and it would be important to compare them with noscapine in terms of PKA activation, regulation of microtubule dynamics, and antifibrotic activity, both in vitro and in vivo.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM85058 (to N. O. D), K08 HL093367-01A1 (to N. S.), and T32HL007237 (to J. K.). This work was also supported by American Heart Association Fellowship 10PRE4190120 (to J. K.).
- MKL1
- megakaryoblastic leukemia-1
- DP
- D-prostanoid receptor
- EP
- E-prostanoid receptor
- HLF
- human lung fibroblast
- SM
- smooth muscle
- SMA
- SM-α-actin
- SRF
- serum response factor
- VASP
- vasodilator-stimulated phosphoprotein.
REFERENCES
- 1. Olson A. L., Swigris J. J., Lezotte D. C., Norris J. M., Wilson C. G., Brown K. K. (2007) Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am. J. Respir. Crit. Care Med. 176, 277–284 [DOI] [PubMed] [Google Scholar]
- 2. White E. S., Lazar M. H., Thannickal V. J. (2003) Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol. 201, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardie W. D., Glasser S. W., Hagood J. S. (2009) Emerging concepts in the pathogenesis of lung fibrosis. Am. J. Pathol. 175, 3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noble P. W., Homer R. J. (2005) Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 33, 113–120 [DOI] [PubMed] [Google Scholar]
- 5. Selman M., Pardo A. (2002) Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir. Res. 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thannickal V. J., Toews G. B., White E. S., Lynch J. P., 3rd, Martinez F. J. (2004) Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 55, 395–417 [DOI] [PubMed] [Google Scholar]
- 7. Desmoulière A., Chaponnier C., Gabbiani G. (2005) Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 13, 7–12 [DOI] [PubMed] [Google Scholar]
- 8. Hinz B., Phan S. H., Thannickal V. J., Galli A., Bochaton-Piallat M. L., Gabbiani G. (2007) The myofibroblast: one function, multiple origins. Am. J. Pathol. 170, 1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Powell D. W., Mifflin R. C., Valentich J. D., Crowe S. E., Saada J. I., West A. B. (1999) Myofibroblasts. I. Paracrine cells important in health and disease. Am. J. Physiol. 277, C1–9 [DOI] [PubMed] [Google Scholar]
- 10. Broekelmann T. J., Limper A. H., Colby T. V., McDonald J. A. (1991) Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 88, 6642–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaminski N., Allard J. D., Pittet J. F., Zuo F., Griffiths M. J., Morris D., Huang X., Sheppard D., Heller R. A. (2000) Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc. Natl. Acad. Sci. U.S.A. 97, 1778–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sime P. J., Xing Z., Graham F. L., Csaky K. G., Gauldie J. (1997) Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sandbo N., Kregel S., Taurin S., Bhorade S., Dulin N. O. (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-β. Am. J. Respir. Cell Mol. Biol. 41, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sandbo N., Lau A., Kach J., Ngam C., Yau D., Dulin N. O. (2011) Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L656–L666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu Y., Liu Y., Zhou W., Xiang R., Jiang L., Huang K., Xiao Y., Guo Z., Gao J. (2010) A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 11, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dackor R. T., Cheng J., Voltz J. W., Card J. W., Ferguson C. D., Garrett R. C., Bradbury J. A., DeGraff L. M., Lih F. B., Tomer K. B., Flake G. P., Travlos G. S., Ramsey R. W., Jr., Edin M. L., Morgan D. L., Zeldin D. C. (2011) Prostaglandin E2 protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L645–L655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kach J., Sandbo N., Sethakorn N., Williams J., Reed E. B., La J., Tian X., Brain S. D., Rajendran K., Krishnan R., Sperling A. I., Birukov K., Dulin N. O. (2013) Regulation of myofibroblast differentiation and bleomycin-induced pulmonary fibrosis by adrenomedullin. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L757–L764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandbo N., Ngam C., Torr E., Kregel S., Kach J., Dulin N. (2013) Control of Myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J. Biol. Chem. 288, 15466–15473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Empey D. W., Laitinen L. A., Young G. A., Bye C. E., Hughes D. T. (1979) Comparison of the antitussive effects of codeine phosphate 20 mg, dextromethorphan 30 mg and noscapine 30 mg using citric acid-induced cough in normal subjects. Eur. J. Clin. Pharmacol. 16, 393–397 [DOI] [PubMed] [Google Scholar]
- 20. Dahlström B., Mellstrand T., Löfdahl C. G., Johansson M. (1982) Pharmacokinetic properties of noscapine. Eur. J. Clin. Pharmacol. 22, 535–539 [DOI] [PubMed] [Google Scholar]
- 21. Karlsson M. O., Dahlström B., Eckernäs S. A., Johansson M., Alm A. T. (1990) Pharmacokinetics of oral noscapine. Eur. J. Clin. Pharmacol. 39, 275–279 [DOI] [PubMed] [Google Scholar]
- 22. Karlsson M. O., Dahlström B., Neil A. (1988) Characterization of high-affinity binding sites for the antitussive [3H]noscapine in guinea pig brain tissue. Eur. J. Pharmacol. 145, 195–203 [DOI] [PubMed] [Google Scholar]
- 23. Mourey R. J., Dawson T. M., Barrow R. K., Enna A. E., Snyder S. H. (1992) [3H]noscapine binding sites in brain: relationship to indoleamines and the phosphoinositide and adenylyl cyclase messenger systems. Mol. Pharmacol. 42, 619–626 [PubMed] [Google Scholar]
- 24. Kamei J., Iwamoto Y., Misawa M., Kasuya Y. (1993) Effects of rimcazole, a specific antagonist of sigma sites, on the antitussive effects of nonnarcotic antitussive drugs. Eur. J. Pharmacol. 242, 209–211 [DOI] [PubMed] [Google Scholar]
- 25. Ye K., Ke Y., Keshava N., Shanks J., Kapp J. A., Tekmal R. R., Petros J., Joshi H. C. (1998) Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc. Natl. Acad. Sci. U.S.A. 95, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landen J. W., Lang R., McMahon S. J., Rusan N. M., Yvon A. M., Adams A. W., Sorcinelli M. D., Campbell R., Bonaccorsi P., Ansel J. C., Archer D. R., Wadsworth P., Armstrong C. A., Joshi H. C. (2002) Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 62, 4109–4114 [PubMed] [Google Scholar]
- 27. Barken I., Geller J., Rogosnitzky M. (2008) Noscapine inhibits human prostate cancer progression and metastasis in a mouse model. Anticancer Res. 28, 3701–3704 [PubMed] [Google Scholar]
- 28. Jackson T., Chougule M. B., Ichite N., Patlolla R. R., Singh M. (2008) Antitumor activity of noscapine in human non-small cell lung cancer xenograft model. Cancer Chemother. Pharmacol. 63, 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minotti A. M., Barlow S. B., Cabral F. (1991) Resistance to antimitotic drugs in Chinese hamster ovary cells correlates with changes in the level of polymerized tubulin. J. Biol. Chem. 266, 3987–3994 [PubMed] [Google Scholar]
- 30. Hogarth D. K., Sandbo N., Taurin S., Kolenko V., Miano J. M., Dulin N. O. (2004) Dual role of PKA in phenotypic modulation of vascular smooth muscle cells by extracellular ATP. Am. J. Physiol. Cell Physiol. 287, C449–456 [DOI] [PubMed] [Google Scholar]
- 31. Small E. M., Thatcher J. E., Sutherland L. B., Kinoshita H., Gerard R. D., Richardson J. A., Dimaio J. M., Sadek H., Kuwahara K., Olson E. N. (2010) Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 107, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mack C. P., Thompson M. M., Lawrenz-Smith S., Owens G. K. (2000) Smooth muscle α-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ. Res. 86, 221–232 [DOI] [PubMed] [Google Scholar]
- 33. Zhou J., Panda D., Landen J. W., Wilson L., Joshi H. C. (2002) Minor alteration of microtubule dynamics causes loss of tension across kinetochore pairs and activates the spindle checkpoint. J. Biol. Chem. 277, 17200–17208 [DOI] [PubMed] [Google Scholar]
- 34. Huang S., Wettlaufer S. H., Hogaboam C., Aronoff D. M., Peters-Golden M. (2007) Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L405–L413 [DOI] [PubMed] [Google Scholar]
- 35. Davis A., Hogarth K., Fernandes D., Solway J., Niu J., Kolenko V., Browning D., Miano J. M., Orlov S. N., Dulin N. O. (2003) Functional significance of protein kinase A activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell. Signal. 15, 597–604 [DOI] [PubMed] [Google Scholar]
- 36. Taurin S., Sandbo N., Qin Y., Browning D., Dulin N. O. (2006) Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 281, 9971–9976 [DOI] [PubMed] [Google Scholar]
- 37. Taurin S., Hogarth K., Sandbo N., Yau D. M., Dulin N. O. (2007) Gβγ-mediated prostacyclin production and cAMP-dependent protein kinase activation by endothelin-1 promotes vascular smooth muscle cell hypertrophy through inhibition of glycogen synthase kinase-3. J. Biol. Chem. 282, 19518–19525 [DOI] [PubMed] [Google Scholar]
- 38. Ayabe S., Kida T., Hori M., Ozaki H., Murata T. (2013) Prostaglandin D2 inhibits collagen secretion from lung fibroblasts by activating the DP receptor. J. Pharmacol. Sci. 121, 312–317 [DOI] [PubMed] [Google Scholar]
- 39. Landen J. W., Hau V., Wang M., Davis T., Ciliax B., Wainer B. H., Van Meir E. G., Glass J. D., Joshi H. C., Archer D. R. (2004) Noscapine crosses the blood-brain barrier and inhibits glioblastoma growth. Clin. Cancer Res. 10, 5187–5201 [DOI] [PubMed] [Google Scholar]
- 40. Zhou J., Gupta K., Yao J., Ye K., Panda D., Giannakakou P., Joshi H. C. (2002) Paclitaxel-resistant human ovarian cancer cells undergo c-Jun NH2-terminal kinase-mediated apoptosis in response to noscapine. J. Biol. Chem. 277, 39777–39785 [DOI] [PubMed] [Google Scholar]
- 41. Aneja R., Zhou J., Vangapandu S. N., Zhou B., Chandra R., Joshi H. C. (2006) Drug-resistant T-lymphoid tumors undergo apoptosis selectively in response to an antimicrotubule agent, EM011. Blood 107, 2486–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilborn J., Crofford L. J., Burdick M. D., Kunkel S. L., Strieter R. M., Peters-Golden M. (1995) Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J. Clin. Invest. 95, 1861–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keerthisingam C. B., Jenkins R. G., Harrison N. K., Hernandez-Rodriguez N. A., Booth H., Laurent G. J., Hart S. L., Foster M. L., McAnulty R. J. (2001) Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-β in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 158, 1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bonner J. C., Rice A. B., Ingram J. L., Moomaw C. R., Nyska A., Bradbury A., Sessoms A. R., Chulada P. C., Morgan D. L., Zeldin D. C., Langenbach R. (2002) Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am. J. Pathol. 161, 459–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petkova D. K., Clelland C. A., Ronan J. E., Lewis S., Knox A. J. (2003) Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology 43, 381–386 [DOI] [PubMed] [Google Scholar]
- 46. Xaubet A., Roca-Ferrer J., Pujols L., Ramírez J., Mullol J., Marin-Arguedas A., Torrego A., Gimferrer J. M., Picado C. (2004) Cyclooxygenase-2 is up-regulated in lung parenchyma of chronic obstructive pulmonary disease and down-regulated in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 21, 35–42 [PubMed] [Google Scholar]
- 47. Hodges R. J., Jenkins R. G., Wheeler-Jones C. P., Copeman D. M., Bottoms S. E., Bellingan G. J., Nanthakumar C. B., Laurent G. J., Hart S. L., Foster M. L., McAnulty R. J. (2004) Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E2 production. Am. J. Pathol. 165, 1663–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aneja R., Vangapandu S. N., Joshi H. C. (2006) Synthesis and biological evaluation of a cyclic ether fluorinated noscapine analog. Bioorg. Med. Chem. 14, 8352–8358 [DOI] [PubMed] [Google Scholar]
- 49. Aneja R., Vangapandu S. N., Lopus M., Chandra R., Panda D., Joshi H. C. (2006) Development of a novel nitro-derivative of noscapine for the potential treatment of drug-resistant ovarian cancer and T-cell lymphoma. Mol. Pharmacol. 69, 1801–1809 [DOI] [PubMed] [Google Scholar]
- 50. Aneja R., Vangapandu S. N., Lopus M., Viswesarappa V. G., Dhiman N., Verma A., Chandra R., Panda D., Joshi H. C. (2006) Synthesis of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Biochem. Pharmacol. 72, 415–426 [DOI] [PubMed] [Google Scholar]