

Background: The endoplasmic reticulum (ER) stress response is increasingly implicated in the pathogenesis of viral infections.
Results: Respiratory syncytial virus (RSV) infection induces the inositol-requiring enzyme 1 (IRE1) stress pathway. IRE1 activity inhibits RSV replication.
Conclusion: ER stress may be a novel cellular anti-RSV defense mechanism.
Significance: Identification of a novel regulator of RSV replication may have therapeutic implications.
Keywords: Cell Biology, Cell Signaling, ER Stress, Negative-strand RNA Viruses, Viral Replication, IRE1
Abstract
Despite being a major health problem, respiratory syncytial virus (RSV) infections remain without specific therapy. Identification of novel host cellular responses that play a role in the pathogenesis of RSV infection is needed for therapeutic development. The endoplasmic reticulum (ER) stress response is an evolutionarily conserved cellular signaling cascade that has been implicated in multiple biological phenomena, including the pathogenesis of some viral infections. In this study, we investigate the role of the ER stress response in RSV infection using an in vitro A549 cell culture model. We found that RSV infection induces a non-canonical ER stress response with preferential activation of the inositol-requiring enzyme 1 (IRE1) and activated transcription factor 6 (ATF6) pathways with no concomitant significant activation of the protein kinase R-like ER kinase (PERK) pathway. Furthermore, we discovered that IRE1 has an inhibitory effect on RSV replication. Our data characterize, for the first time, the nature of the ER stress response in the setting of RSV infection and identify the IRE1 stress pathway as a novel cellular anti-RSV defense mechanism.
Introduction
Respiratory syncytial virus (RSV)2 is a major cause of human respiratory infections (1). It is the leading cause of respiratory illness in young children. Almost all children acquire the infection sometime during their first 2 years of life (2, 3). Although protective immunity develops following an RSV infection, it is not long lasting. In fact, subjects at both ends of the age spectrum are most susceptible to infection, especially the ones with chronic cardiopulmonary diseases or immunosuppression (4). In the elderly population, studies have shown the prevalence and clinical significance of RSV infection to be comparable with that of influenza virus (5). To date, there are no effective pharmacologic therapies against RSV infection. Furthermore, and in contrast with influenza virus, attempts at developing an effective RSV vaccine have failed despite years of attempts (6, 7). Therefore, further understanding of molecular mechanisms involved in the pathogenesis of RSV infection is needed to guide the development of new therapeutic strategies.
The endoplasmic reticulum (ER) stress response is an evolutionarily conserved cellular signaling cascade with an increasingly recognized range of effects. Although it has been originally understood as a molecular rescue mechanism aimed at restoring ER homeostasis during stress conditions, the ER stress response is now known to play a role in diverse biological processes including apoptosis, inflammation, and metabolism (8–14). Furthermore, ER stress has been shown to be involved in the pathogenesis of certain viruses. For instance, cellular apoptosis has been shown to be mediated by the ER stress response in the setting of Japanese encephalitis virus, bovine diarrhea virus, Tula virus, severe acute respiratory syndrome coronavirus (SARS-CoV), and West Nile virus infections (15–20). Oxidative stress in the setting of hepatitis C infection has been shown to be mediated by ER stress (21, 22). We recently published a study showing that influenza A virus infection selectively activates the inositol-requiring enzyme 1 (IRE1) branch of the ER stress response and that this activation is essential for efficient viral replication (23). Surprisingly, the role of ER stress in RSV infection is unknown.
The ER stress response consists of three signaling pathways. The upstream mediators are three ER-resident transmembrane proteins: activating transcription factor 6 (ATF6), protein kinase R-like ER kinase (PERK), and inositol-requiring enzyme 1 (IRE1). Under normal conditions, these three upstream mediators are bound to a major ER chaperone, binding immunoglobulin protein (BiP), at their luminal N-terminal side, preventing their activation. During ER stress, BiP is released from ATF6, PERK, and IRE1 because of competitive binding to the increasing levels of misfolded proteins in the ER lumen. This allows the activation of the ER stress response (24). Recent work has found some systems in which not all arms of the ER stress response are activated equally. Alternative mechanisms by which some of these mediators, notably IRE1, sense ER stress and activate have been reported recently (25–27).
IRE1 has two catalytic domains, an endoribonuclease domain and a kinase domain. The endonuclease activity is responsible for the non-conventional cytosolic splicing of a 26-base intron in the X box binding protein 1 (XBP1) mRNA, leading to a translational reading frameshift. The spliced XBP1 mRNA translates into an active transcription factor that drives the expression of genes involved in the ER-associated degradation machinery (9, 28, 29). In contrast to the adaptive effects of its endonuclease function, the downstream effects of the IRE1 kinase function include phosphorylation of JNK and p38 MAP kinase, both of which are implicated in mediating some of the apoptotic effects of the ER stress response (9, 24, 28, 29).
When released from BiP, ATF6 translocates to the Golgi, where it is cleaved by resident proteases. Cleaved ATF6 functions as a transcription factor for chaperone genes, one of which is BiP, thus improving the ER protein folding capacity (9).
PERK homodimerizes when released from BiP, which induces its autophosphorylation and activation. PERK is a serine/threonine kinase that phosphorylates and inactivates eIF2α (eukaryotic translation initiation factor 2 α). Phosphorylation of eIF2α induces global shutdown of protein translation. Certain mRNAs, for example BiP mRNA, escape that inhibition and gain a translational advantage (9).
In this study, we investigate the effects of RSV infection on the three arms of the ER stress response and describe a novel role for IRE1 activity as an inhibitor of RSV replication.
EXPERIMENTAL PROCEDURES
Reagents
Tunicamycin (catalog no. 654380) was purchased from Calbiochem. 3,5-Dibromosalicylaldehyde (catalog no.122130) was purchased from Sigma-Aldrich. Abs used in this study were obtained from a variety of sources. BiP ab (catalog no. cs-3183) and PERK ab (cs-13073) were obtained from Cell Signaling Technology (Beverly, MA). HRP-conjugated anti-rabbit (catalog no. sc-2004) and anti-mouse (catalog no. sc-2005) abs were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). β-actin ab (catalog no. ab-8226) and HRP-conjugated GAPDH ab (catalog no. ab-9482) were obtained from Abcam (Cambridge, MA). RSV all-antigen ab (catalog no. B65860G) was obtained from Biodesign International (Saco, ME).
Epithelial Cell Culture and Viral Infection
A549 lung epithelial cells were obtained from the ATCC. A549 cells were used because they most closely mimic RSV observations in primary human airway cells (30–33). Cells were maintained in 75-cm2 tissue culture flasks (Corning) in minimal essential medium (Invitrogen) with 10% FBS and gentamicin. For infection, cells at 80% confluency were infected with human RSV strain A-2 (multiplicity of infection (MOI) 1). Viral stocks were obtained from Advanced Biotechnologies (Columbia, MD). The initial stock (2.2 × 107 pfu/ml) was placed in aliquots and kept frozen at −135 °C. The virus was never refrozen.
Other Cells
IRE1−/− mouse embryonic fibroblasts (MEFs) and XBP1−/− MEFs were prepared as described previously (34–36). IRE1−/−, XBP1−/−, and littermate wild-type control MEFs were maintained in 150-cm2 tissue culture flasks (Corning) in high-glucose Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS, l-glutamine, amino acids, non-essential amino acids, penicillin/streptomycin, and sodium pyruvate. Human tracheobronchial epithelial (HTBE) cells were obtained from Dr. Joseph Zabner and the Cell Culture Core under a protocol approved by the University of Iowa Institutional Review Board. Epithelial cells were isolated from tracheal and bronchial mucosa by enzymatic dissociation and cultured in Laboratory of Human Carcinogenesis 8e medium on plates coated with collagen/albumin for study up to passage 10 (37–40). Experiments were conducted using cells from at least three different donors.
Cellular Protein
Whole-cell protein extracts were prepared by lysis of cell monolayers in 200 ml of lysis buffer (50 mm Tris (pH 7.4), 150 mm NaCl, and 1% Nonidet P-40), a protease inhibitor mixture (Roche Applied Science), and a phosphatase inhibitor mixture (catalog no. 524625, Calbiochem). The lysates were sonicated for 20 s and kept at 4 °C for 30 min. After 5 min of centrifugation (14,000 rpm at 4 °C), the supernatant was saved as a whole-cell lysate. Protein determinations were made using a protein measurement kit (Bradford protein assay, catalog no. 500-0006) from Bio-Rad. Extracts were stored at −80 °C.
Immunoblot Analysis
Protein (25–30 mg) was mixed 1:1 with 23 sample buffer (20% glycerol, 4% SDS, 10% 2-mercaptophenol, 0.05% bromphenol blue, and 1.25 m Tris (pH 6.8)), loaded onto a 12 or 15% (ISG15) SDS-PAGE gel, and run at 150 V for 90 min. Cell proteins were transferred to an immunoblot polyvinylidene difluoride membrane (Bio-Rad) with a Bio-Rad semidry transfer system according to the instructions of the manufacturer. The polyvinylidene difluoride membrane was then incubated with primary ab (dilutions 1:500–1:1000) in 5% milk in Tris-buffered saline with 0.1% Tween 20 overnight. The blots were washed three times with Tris-buffered saline with 0.1% Tween 20 and incubated for 1 h with HRP-conjugated secondary anti-IgG ab (dilution 1:2000–1:20,000). The blots were washed again three times with Tris-buffered saline with 0.1% Tween 20, and immunoreactive bands were developed using the chemiluminescent substrate ECL Plus (Amersham Biosciences, Piscataway, NJ). An autoradiograph was obtained with exposure times of 10 s to 2 min.
Luciferase Activity Assay
Firefly and Renilla luciferase activity was measured using the Dual-Luciferase reporter assay system from Promega (Madison, WI) according to the instructions of the manufacturer.
RNA Isolation
RNA was isolated from cells using a reagents from the MirVana kit (Applied Biosystems, Austin, TX) according to the instructions of the manufacturer. After preparation, RNA samples were stored in a −80 °C freezer until use. RNA quality and quantity were assessed with an Experion automated electrophoresis system (Bio-Rad) using the Experion RNA StdSens analysis kit according to the protocol of the manufacturer. RNA quality was considered adequate for use if the 28 S/18 S ratio was >1.2 and the RNA quality indicator was >7.
Real-time RT-PCR for mRNA Quantitation
Total RNA (1 μg) was reverse-transcribed to cDNA using the iScript cDNA synthesis kit (Bio-Rad) following the instructions of the manufacturer. PCR reactions were performed using 2 μl of cDNA and 48 μl of master mix containing iQ SYBR Green Supermix (Bio-Rad), 15 pmol of forward primer, and 15 pmol of reverse primer in a CFX96 real-time PCR detection system (Bio-Rad) as follows: 3 min at 95 °C followed by 40 cycles of 10 s at 95 °C and 30 s at 55 °C and plate reading. The fluorescence signal generated with SYBR Green I DNA dye was measured during the annealing steps. The specificity of the amplification was confirmed using a melting curve analysis. Data were collected and recorded by CFX Manager Software (Bio-Rad) and expressed as a function of threshold cycle (CT). The relative quantity of the gene of interest was then normalized to the relative quantity of hypoxanthine phosphoribosyltransferase (ΔΔCT). The sample mRNA abundance was calculated by the equation 2− (ΔΔCT). Gene-specific primers were custom-synthesized and purchased from Integrated DNA Technologies (Iowa City, IA) on the basis of design using gene-specific nucleotide sequences from the National Center for Biotechnology Information sequence databases and the PrimerQuest Web interface (Integrated DNA Technologies).
XBP1 mRNA Splicing Assay
To determine the degree of XBP1 mRNA splicing, total cellular RNA was isolated as described above. Then, total XBP1 cDNA was synthesized and amplified using the Superscript III One Step RT PCR with platinum TaqDNA polymerase kit (catalog no.12574-018) (Invitrogen) following the instructions of the manufacturer. To amplify both the spliced and the unspliced variants of XBP1 cDNA, we used forward and reverse primers that circumvented the spliced segment. The PCR product was then separated by gel electrophoresis using a 10% acrylamide gel. Staining with a SYBR Green fluorescent dye was then performed for visualization of the separated DNA bands. The primer sequences used for the human and mouse XBP1 genes, respectively, were as follows: 5′-CCTTGTAGTTGAGAACCAGG-3′ (forward) and 5′-GGGCTTGGTATATATGTGG-3′ (reverse) and 5′-TTGTGGTTGAGAACCAGG-3′ (forward) and 5′-TCCATGGGAAGATGTTCTGG-3′ (reverse). We also used another method to quantitatively measure spliced XBP1 mRNA. This was achieved by quantitative real-time RT-PCR using a primer set that selectively amplifies the spliced variant of the human XBP1 cDNA (41). In this method, the forward primer is designed to span the 26-bp intron and, therefore, will only anneal to the spliced variant of the XBP1 cDNA. The primer sequences used for this method were 5′-GGTCTGCTGAGTCCGCAGCAGG-3′ (forward) and 5′-GGGCTTGGTATATATGTGG-3′ (reverse).
Plaque Assay
Viral titers of infected cells were measured by standard plaque assay using 90% confluent Vero cells. Briefly, the supernatant and adherent cells were removed, sonicated for 20 s on ice as described, and frozen at −70 °C to be assayed later by plaque assay. Vero cells were treated with serial 10-fold dilutions of the supernatant/lysed cell mixture. The cell cultures were incubated at 37 °C, 5% CO2 for 90 min with gentle rocking of the plates every 15 min. Overlay, consisting of Eagle's minimal essential medium (Cambrex), 10% FBS (JRH Biosciences), l-glutamine (Invitrogen), penicillin/streptomycin (Invitrogen), and 1% SeaKem ME-agarose (Cambrex) was prepared, and 4 ml of cooled overlay was added to each sample. Samples were gently swirled to mix and then allowed to cool in a laminar flow hood for 15 min. When the agar solidified, plates were incubated for 5 days at 37 °C, 5% CO2. Cells were stained by adding 2 ml of neutral red (Fisher Scientific) overlay and incubated for 24 h. The next day, plaques were counted in each well over a light box, and the concentration of virus was calculated.
Statistical Analysis
Two-tailed Student's t test was used for group comparisons. Statistical significance was determined on the basis of a p value of less than 0.05.
RESULTS
RSV Infection Induces ER Stress
To determine whether RSV infection leads to activation of the ER stress response, we infected A549 cells with RSV and measured the effect of the infection on expression of the downstream genes known to be transcriptionally activated during ER stress. To detect differences in ER stress markers, we set up our infection model to infect the majority of the cells in the system. Using immunofluorescent staining, we determined that infecting confluent A549 monolayer cell cultures with RSV at an MOI of 1 ensures 80% infectivity 12 h post-infection (data not shown). Therefore, an MOI of 1 was chosen for all subsequent experiments. For positive ER stress control, we treated cells with the N-linked glycosylation inhibitor tunicamycin (1 μg/ml for 4 h), a known ER stress-inducing agent. We found that RSV infection induced transcription of a number of conventional ER stress genes (Fig. 1A). The BiP gene, which encodes one of the major chaperones and is often considered a marker for ER stress, was induced at both the mRNA and protein levels (Fig. 1B). These data are consistent with induction of the ER stress response by RSV infection.
FIGURE 1.
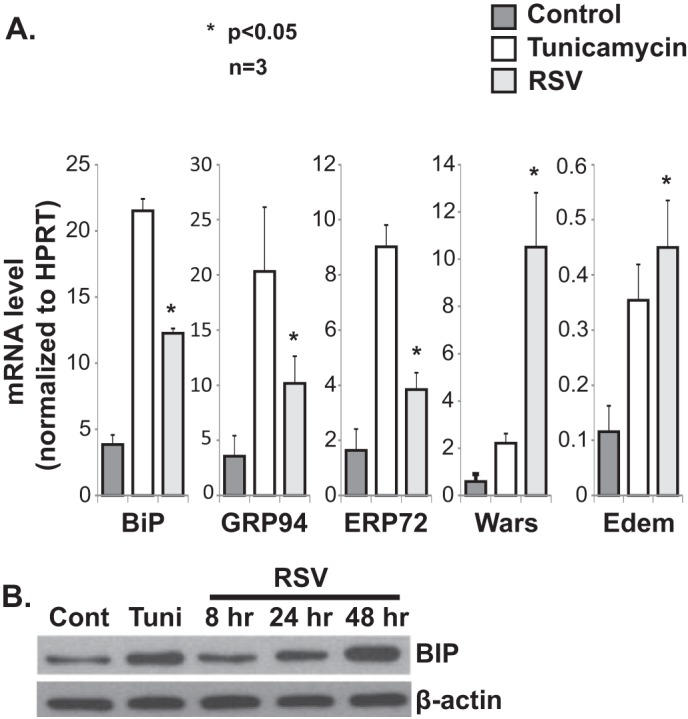
RSV infection induces the ER stress response. A, quantitative RT-PCR data showing induction of a number of ER stress genes by RSV infection in A549 cells. RNA was isolated from the indicated groups at 24 h post-infection. HPRT, hypoxanthine-guanine phosphoribosyltransferase. B, Western blot analysis showing a time-dependent increase in BiP expression post-RSV infection of A549 cells. Tunicamycin (Tuni) was used at 1 μg/ml for 4 h in both A and B. Cont, control.
RSV Infection Induces a Differential ER Stress Response
To better characterize the nature of the ER stress response induced by RSV, we examined the effect of infection on each of the three individual ER stress pathways: IRE1, ATF6, and PERK. One of the primary functions of IRE1 is splicing and removal of a 26-bp intron from an evolutionarily conserved ER stress transcription factor mRNA, XBP1. To determine the effect of RSV infection on IRE1 activity, we measured the degree of XBP1 mRNA splicing at 8 and 24 h post-RSV infection of A549 cells. This was done by isolating cellular RNA at the indicated time points and performing conventional PCR using primers that circumvent the spliced intron, thus allowing amplification of both the spliced and unspliced variants of XBP1 cDNA. The degree of splicing was then determined by separating the amplification product using gel electrophoresis followed by fluorescent staining (detailed under “Experimental Procedures”). Tunicamycin (1 μg/ml for 4 h) was used as a positive control. Our results show that RSV infection leads to a time-dependent, statistically significant increase in XBP1 mRNA splicing (Fig. 2A). We also measured the degree of splicing of XBP1 mRNA by quantitative RT-PCR using a set of primers validated previously to amplify only the spliced variant (41). The results also showed an increase in spliced XBP1 mRNA with RSV (Fig. 2B). These results demonstrate the activation of the IRE1 endonuclease by RSV infection.
FIGURE 2.

Effects of RSV infection on individual ER stress response pathways. A, an XBP1 PCR amplification product containing both spliced and unspliced variants was separated by DNA gel electrophoresis. PCR amplification of total XBP1 cDNA was performed using primers that circumvented the spliced intron. Cellular RNA was isolated from the indicated groups. A corresponding densitometry analysis from three independent experiments is shown. Cont, control; Tuni, tunicamycin. B, quantitative RT PCR data of spliced XBP1 mRNA. Cellular RNA was isolated from the indicated groups at 24 h post-infection. HPRT, hypoxanthine-guanine phosphoribosyltransferase. C, A549 cells transfected with p5xATF6-GL3 were treated as indicated. Firefly luciferase activity was measured 24 h post-infection and normalized to Renilla luciferase transfection control. Tunicamycin treatment at 1 μg/ml 4 h prior to harvest was used as a positive control. D, Western blot analysis showing the mobility shift of PERK indicating phosphorylation during ER stress. Total cellular proteins were isolated from the indicated groups at 24 h post-infection.
We next investigated the effect of RSV infection on the ATF6 pathway. Note that the RSV-induced transcription of ER stress genes suggests activation of ATF6 because these genes are at least partially dependent on ATF6 for their full up-regulation (42, 43). To detect ATF6 activity, we transfected cells with a plasmid encoding a firefly luciferase reporter gene under the control of five repeats of the ATF6 promoter. As a transfection control, cells were cotransfected with a Renilla luciferase reporter gene plasmid. We treated these cells with either tunicamycin (1 μg/ml for 4 h), as a positive control, or RSV. Firefly luciferase activity was measured 8 h post-infection and normalized to Renilla luciferase activity. The results show higher activity levels in the RSV-infected and the tunicamycin-treated groups compared with the control, indicating that RSV infection activates the ATF6 pathway (Fig. 2C).
To examine the PERK pathway, we performed a Western blot analysis using a PERK antibody on control cells and cells treated with tunicamycin (1 μg/ml for 4 h) or RSV (for 24 h). When active, PERK dimerizes and autophosphorylates. Phosphorylation of PERK can be detected as a mobility shift on SDS-PAGE. A shift in migration of the PERK band was seen only in the tunicamycin-treated but not in the RSV-infected cells, indicating that RSV infection does not induce significant activation of the PERK pathway (Fig. 2D). Taken together, these results indicate that RSV infection leads to a non-canonical ER stress response characterized by induction of the IRE1 and ATF6 pathways with no concomitant significant activation of the PERK pathway.
IRE1 Inhibits RSV Replication
We recently published a study showing that IRE1 activation is necessary for efficient replication of influenza A virus, another RNA respiratory virus (23). Therefore, and in light of the pathway-specific activation of the ER stress response during RSV infection, we wanted to investigate whether IRE1 also plays a role in RSV replication. Surprisingly, our findings indicate a different role for IRE1 in RSV replication compared with its effect on influenza A virus.
To modulate the IRE1 endonuclease activity, we made use of a selective IRE1 endonuclease inhibitor, 3,5-dibromosalicylaldehide (IRE1 inhibitor) (23, 44). A549 cells were infected with RSV and cotreated with the IRE1 inhibitor or vehicle as a control. Using Western blot analysis with a polyclonal all-antigen anti-RSV antibody, we found that viral protein expression is higher in the IRE1 inhibitor-treated cells in a dose-dependent manner (Fig. 3A). Consistent with this finding, viral titers were also found to be higher in the supernatant of infected cells that are cotreated with the IRE1 inhibitor compared with vehicle (Fig. 3B). This suggests an inhibitory effect of the IRE1 endonuclease on RSV replication.
FIGURE 3.

IRE1 inhibits RSV replication. A, Western blot analysis using an all-antigen polyclonal RSV antibody, detecting G, F, P, and M2-1 viral proteins, 8 h post-infection of A549 cells with or without cotreatment with various doses of the IRE1 endonuclease inhibitor 3,5-dibromosalicylaldehide. B, viral titers measured from supernatant of RSV-infected A549 cells with or without cotreatment, as indicated, 8 h post-infection. C, Western blot for RSV proteins performed at different time points on WT versus IRE1 KO MEFs infected with RSV at an MOI of 1 with corresponding densitometry obtained from three independent experiments shown at the bottom. D, viral titers measured from supernatant of RSV-infected wild-type versus IRE1 knockout MEFs 24 h post-infection. E, quantitative RT-PCR data for the G and N RSV genes at 4 and 24 h post-infection of wild-type versus IRE1 knockout MEFs. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
To test this hypothesis further, we conducted experiments using IRE1 KO MEFs along with wild-type littermate control MEFs. Cells were cultured in monolayers on tissue culture plates and infected with RSV at an MOI of 1. Consistent with our IRE1 pharmacologic inhibitor results, IRE1 knockout led to a significant increase in both RSV protein expression and viral titers (Fig. 3, C and D). We also measured mRNA levels of two RSV genes, the G and the N genes, at 4 and 24 h post-infection. We found that mRNA levels were also increased in the IRE1 knockout cells (Fig. 3E). Taken together, these results indicate that IRE1 inhibits RSV replication at an early stage of the replication cycle of the virus.
RSV Replication Is Not Affected by Altering the Downstream Signaling Effects of the IRE1 Stress Pathway
IRE1 endonuclease signaling is mediated by splicing of the XBP1 mRNA, allowing it to translate into an active transcription factor, thus resulting in gene expression changes. To investigate whether the IRE1-mediated inhibition of RSV replication is a downstream signaling effect of the IRE1 pathway or an upstream effect mediated directly by the IRE1 enzyme, we compared RSV replication in XBP1 knockout MEFs versus wild-type littermate control MEFs. Cells were infected with RSV at an MOI of 1. Using Western blot analysis, viral proteins were measured and found to be similar between the two groups of cells at different time points (Fig. 4A). RSV G and N mRNA levels were measured at 4 and 24 h post-infection. A trend for slightly lower levels in the XBP1 knockout group was noted. However, the differences were not statistically significant (Fig. 4B). Viral titers in the supernatant of the infected cells were measured at 24 h post-infection and were found to be similar (Fig. 4C). To ensure that XBP1 was actually activated in the wild-type MEFs following RSV infection, we measured XBP1 mRNA splicing in infected wild-type MEFs at 8 and 24 h post-infection. This was done by total XBP1 PCR followed by gel electrophoresis, as described earlier. Tunicamycin (1 μg/ml for 4 h) was used as a positive control. IRE1 knockout cells with or without tunicamycin treatment were also included as a negative control. The results show a time-dependent increase in the spliced XBP1 variant in response to RSV infection (Fig. 4D). Taken together, these results indicate that IRE1 stress pathway signaling downstream of the XBP1 mRNA splicing has no effect on RSV replication.
FIGURE 4.

RSV replication is not affected by XBP1 knockout. A, Western blot analysis for RSV G, F, P, and M2-1 proteins performed at different time points on WT versus XBP1 KO MEFs infected with RSV at an MOI of 1. B, quantitative RT-PCR data for the G and N RSV genes at 4 and 24 h post-infection of wild-type versus XBP1 knockout MEFs. ns, not significant. C, viral titers measured from supernatant of RSV-infected wild-type versus XBP1 knockout MEFs 24 h post-infection. D, an XBP1 PCR amplification product containing both spliced and unspliced variants was separated by DNA gel electrophoresis. PCR amplification of total XBP1 cDNA was performed using primers that circumvented the spliced intron. Cellular RNA was isolated from the indicated groups. Tunicamycin (Tuni) was used at 1 μg/ml for 4 h. A representative gel from three independent experiments is shown. Cont, control.
RSV and ER Stress in Primary HTBE Cells
A549 cells are a transformed cell line, hence their response to RSV infection may be different from that of primary respiratory epithelial cells. Therefore, we wanted to investigate whether ER stress also plays a role in RSV infection of primary HTBE cells. We reproduced a selected number of our findings in an in vitro primary HTBE cell culture model. HTBE cells were cultured in monolayers on collagen-coated plates and infected with RSV at an MOI of 1. Tunicamycin (1 μg/ml for 4 h) was used as an ER stress positive control. Using quantitative real-time RT-PCR, we found that RSV infection leads to an increase in the expression level of BiP mRNA consistent with induction of the ER stress response (Fig. 5A). We also found that RSV infection causes an increase in the level of spliced XBP1 mRNA consistent with activation of IRE1 endonuclease (Fig. 5B). Furthermore, we found that cotreatment of RSV-infected HTBE cells with the selective IRE1 endonuclease inhibitor 3,5-dibromosalicylaldehide leads to an increase in RSV protein expression, suggesting that IRE1 endonuclease activity inhibits RSV replication (Fig. 5C). Knowing that the IRE1 pathway is implicated in the ER-associated degradation machinery, we wanted to rule out the possibility of a generalized posttranslational glycoprotein stability effect of the IRE1 inhibitor. Therefore, we probed for the EGF receptor as a control cellular transmembrane glycoprotein and found it to be unchanged (Fig. 5C). Taken together, and consistent with our previous A549 cell data, these results indicate that the ER stress response, and in particular IRE1, is induced by RSV infection and may modulate viral replication in primary HTBE cells.
FIGURE 5.

ER stress and RSV infection in HTBE cells. A, quantitative RT-PCR data showing induction of BiP mRNA levels by RSV infection in HTBE cells. RNA was isolated from the indicated groups (of four donors) at 48 h post infection. HPRT, hypoxanthine-guanine phosphoribosyltransferase. B, quantitative RT PCR data of spliced XBP1 mRNA. Cellular RNA was isolated from the indicated groups (of three donors) at 24 h post-infection. Tunicamycin was used at 1 μg/ml for 4 h in both A and B. C, Western blot analysis using an all-antigen polyclonal RSV antibody, detecting G, F, P, and M2-1 viral proteins, 8 h post-infection of HTBE cells with or without cotreatment with the selective IRE1 endonuclease inhibitor (inh) 3,5-dibromosalicylaldehide at 20 μm. Three representative blots are shown along with corresponding densitometry analysis from three independent experiments using cells from three different donors. The EGF receptor was probed as a control cellular transmembrane glycoprotein.
DISCUSSION
In this work, we characterized, for the first time, the nature of the ER stress response induced by RSV using an in vitro A549 cell culture model. We showed that the infection leads to a non-canonical, preferential activation of the IRE1 and ATF6 pathways with no concomitant significant activation of the PERK pathway. This conclusion was reached on the basis of experimental data showing RSV-induced splicing of the XBP1 mRNA, transcriptional activation of ATF6-driven genes, and lack of significant PERK phosphorylation. Using a pharmacologically selective IRE1 endonuclease inhibitor as well as genetic IRE1 knockout as methods to modulate IRE1 activity, we also discovered that the IRE1 enzyme has an inhibitory effect on RSV replication. Then, using an XBP1 knockout cell line, we showed that the IRE1-mediated inhibition of RSV replication cannot be entirely the result of downstream signaling through XBP1 mRNA splicing. Finally, we reproduced a selected number of findings in HTBE cells, suggesting that IRE1 may also play a role in RSV replication in the primary cell line.
During viral infection, viral protein synthesis will overload the protein translation and modification systems of the cell (15, 45). Therefore, it makes sense that a viral infection will activate the ER stress response. Over the last decade, the ER stress response has been increasingly recognized to be activated in the setting of certain viral infections (15, 16, 19, 46–49). Interestingly, and through unknown mechanisms, different viruses have been shown to selectively induce different branches of the ER stress response. It has become clear lately that different conditions can result in very different ER stress responses. The mechanisms by which the different arms of the ER stress response are modulated are not fully understood. Along these lines, investigators have recently discovered novel modes of IRE1 activation through direct interaction with ligands at specific binding sites on both its luminal and cytosolic domains (25, 50–52). Viruses are a very diverse group of intracellular pathogens. Therefore, it is conceivable that different viruses lead to various ER stress response outputs. For instance, positive-sense RNA viruses require the formation of ER-derived vesicles during their replication, whereas negative-sense RNA viruses do not cause similar ER perturbations. Although such properties may account for the observed differences in virus-induced ER stress responses, it is clearly not the only factor. For instance, hepatitis C virus and West Nile virus are both positive-sense, single-stranded RNA viruses. However, they exert opposite effects on the IRE1 stress pathway (19, 22). Furthermore, RSV and influenza A virus, both negative-sense RNA viruses, exert opposite effects on the ATF6 pathway. Here we show that RSV infection activates ATF6, whereas our group has published data previously showing that influenza A virus inhibits ATF6 activation (23). On the other hand, the properties of the host cells different viruses infect may also have a role in determining the ER stress response output. Hepatocytes, for example, are specialized secretory cells that are actively synthesizing and secreting large amounts of proteins and might be more sensitive to ER stress than other cell types. It is clear that the interaction between viruses and the ER stress pathways is complex and far from being completely understood.
In this work, we showed that IRE1 inhibits RSV replication. However, the complete mechanism of this interaction is still unclear. Investigators have recently discovered a novel function of IRE1 endonuclease, which is the regulated IRE1-dependent decay of messenger RNAs. In addition to XBP1 mRNA, IRE1 targets a variety of other mRNAs, contributing to their decay and, thus, a decrease in the translocation of nascent proteins to the ER. It is thought that the ER membrane-targeting sequence of the nascent translated protein is responsible for bringing the target mRNA into close proximity with the IRE1 enzyme. It is unclear whether IRE1 nonspecifically targets all mRNAs encoding transmembrane and secretory proteins or whether it specifically recognizes certain mRNA sequences for its regulated IRE1-dependent decay function (53). Our results indicate that, in the presence of IRE1, RSV mRNA levels are lower and that this effect is not a downstream effect of XBP1 splicing. This suggests that RSV mRNA could be another regulated IRE1-dependent decay target for the IRE1 endonuclease.
We believe it is unlikely that the IRE1 kinase domain is responsible for RSV inhibition. This is because the downstream signaling of the IRE1 kinase, through activation of MAPKs, is not specific to IRE1. MAPKs have multiple common upstream mediators, notably TLR3 and TLR4, both of which are also known to be activated during RSV infection (54, 55). Therefore, knocking out IRE1 would not necessarily abrogate MAPK signaling. The important role of the endonuclease domain, not the kinase domain, of IRE1 in RSV infection is also supported by our results showing enhanced RSV replication in response to the selective IRE1 endonuclease inhibitor.
Although more investigation is clearly needed to elucidate the mechanism of interaction between IRE1 and RSV, identification of the IRE1 stress pathway as a cellular anti-RSV defense mechanism is completely novel. Even though a number of our observations were reproduced in primary HTBE cells, this work does not comprise an in-depth mechanistic investigation in these cells. Therefore, and despite the shared similarities with the A549 data, we cannot formally exclude a different mechanism leading to the results observed in the primary HTBE cells. Furthermore, the in vivo relevance of these findings still needs to be investigated because this could pave the way to the development of novel therapeutic approaches against human RSV infection.
This project was supported, in whole or in part, by National Institutes of Health Grants R01 HL079901, R01 HL096625, and R21 HL109589 (to M. M. M.); R01 DK084058 (to D. T. R.); NIEHS, National Institutes of Health Grant P30 ES005605; and National Center for Research Resources (NCRR), National Institutes of Health Grant UL1RR024979. This work was also supported by American Thoracic Society Foundation Research Grant 2012-04 (to I. H.) and by the National Institute for Environmental Health Sciences through the University of Iowa Environmental Health Sciences Research Center.
- RSV
- respiratory syncytial virus
- ER
- endoplasmic reticulum
- PERK
- protein kinase R-like ER kinase
- ATF
- activated transcription factor
- BiP
- binding immunoglobulin protein
- XBP
- X box binding protein
- ab
- antibody
- MOI
- multiplicity of infection
- MEF
- mouse embryonic fibroblast
- HTBE
- human tracheobronchial epithelial.
REFERENCES
- 1. Nair H., Nokes D. J., Gessner B. D., Dherani M., Madhi S. A., Singleton R. J., O'Brien K. L., Roca A., Wright P. F., Bruce N., Chandran A., Theodoratou E., Sutanto A., Sedyaningsih E. R., Ngama M., Munywoki P. K., Kartasasmita C., Simões E. A., Rudan I., Weber M. W., Campbell H. (2010) Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children. A systematic review and meta-analysis. Lancet 375, 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glezen W. P., Taber L. H., Frank A. L., Kasel J. A. (1986) Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child 140, 543–546 [DOI] [PubMed] [Google Scholar]
- 3. Shay D. K., Holman R. C., Newman R. D., Liu L. L., Stout J. W., Anderson L. J. (1999) Bronchiolitis-associated hospitalizations among US children, 1980–1996. JAMA 282, 1440–1446 [DOI] [PubMed] [Google Scholar]
- 4. Walsh E. E., Falsey A. R., Hennessey P. A. (1999) Respiratory syncytial and other virus infections in persons with chronic cardiopulmonary disease. Am. J. Respir. Crit. Care Med. 160, 791–795 [DOI] [PubMed] [Google Scholar]
- 5. Falsey A. R., Hennessey P. A., Formica M. A., Cox C., Walsh E. E. (2005) Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352, 1749–1759 [DOI] [PubMed] [Google Scholar]
- 6. Beeler J. A., Eichelberger M. C. (2013) Influenza and respiratory syncytial virus (RSV) vaccines for infants. Safety, immunogenicity, and efficacy. Microb. Pathog. 55, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim H. W., Canchola J. G., Brandt C. D., Pyles G., Chanock R. M., Jensen K., Parrott R. H. (1969) Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89, 422–434 [DOI] [PubMed] [Google Scholar]
- 8. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu C., Bailly-Maitre B., Reed J. C. (2005) Endoplasmic reticulum stress. Cell life and death decisions. J. Clin. Invest. 115, 2656–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D. T., Back S. H., Kaufman R. J. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599 [DOI] [PubMed] [Google Scholar]
- 11. Gupta S., Cuffe L., Szegezdi E., Logue S. E., Neary C., Healy S., Samali A. (2010) Mechanisms of ER stress-mediated mitochondrial membrane permeabilization. Int. J. Cell Biol. 2010, 170215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakajima S., Hiramatsu N., Hayakawa K., Saito Y., Kato H., Huang T., Yao J., Paton A. W., Paton J. C., Kitamura M. (2011) Selective abrogation of BiP/GRP78 blunts activation of NF-κB through the ATF6 branch of the UPR. Involvement of C/EBPβ and mTOR-dependent dephosphorylation of Akt. Mol. Cell. Biol. 31, 1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pahl H. L., Baeuerle P. A. (1996) Activation of NF-κB by ER stress requires both Ca2+ and reactive oxygen intermediates as messengers. FEBS Lett. 392, 129–136 [DOI] [PubMed] [Google Scholar]
- 15. He B. (2006) Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13, 393–403 [DOI] [PubMed] [Google Scholar]
- 16. Su H. L., Liao C. L., Lin Y. L. (2002) Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76, 4162–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X. D., Lankinen H., Putkuri N., Vapalahti O., Vaheri A. (2005) Tula hantavirus triggers pro-apoptotic signals of ER stress in Vero E6 cells. Virology 333, 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye Z., Wong C. K., Li P., Xie Y. (2008) A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 1780, 1383–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Medigeshi G. R., Lancaster A. M., Hirsch A. J., Briese T., Lipkin W. I., Defilippis V., Früh K., Mason P. W., Nikolich-Zugich J., Nelson J. A. (2007) West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ambrose R. L., Mackenzie J. M. (2011) West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 85, 2723–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tardif K. D., Waris G., Siddiqui A. (2005) Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13, 159–163 [DOI] [PubMed] [Google Scholar]
- 22. Tardif K. D., Mori K., Kaufman R. J., Siddiqui A. (2004) Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164 [DOI] [PubMed] [Google Scholar]
- 23. Hassan I. H., Zhang M. S., Powers L. S., Shao J. Q., Baltrusaitis J., Rutkowski D. T., Legge K., Monick M. M. (2012) Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 287, 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rutkowski D. T., Arnold S. M., Miller C. N., Wu J., Li J., Gunnison K. M., Mori K., Sadighi Akha A. A., Raden D., Kaufman R. J. (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gardner B. M., Walter P. (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hetz C., Bernasconi P., Fisher J., Lee A. H., Bassik M. C., Antonsson B., Brandt G. S., Iwakoshi N. N., Schinzel A., Glimcher L. H., Korsmeyer S. J. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312, 572–576 [DOI] [PubMed] [Google Scholar]
- 27. Papa F. R., Zhang C., Shokat K., Walter P. (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 28. Rutkowski D. T., Kaufman R. J. (2004) A trip to the ER. Coping with stress. Trends Cell Biol. 14, 20–28 [DOI] [PubMed] [Google Scholar]
- 29. Naidoo N. (2009) ER and aging-protein folding and the ER stress response. Ageing Res. Rev. 8, 150–159 [DOI] [PubMed] [Google Scholar]
- 30. Monick M. M., Yarovinsky T. O., Powers L. S., Butler N. S., Carter A. B., Gudmundsson G., Hunninghake G. W. (2003) Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol. Chem. 278, 53035–53044 [DOI] [PubMed] [Google Scholar]
- 31. Monick M. M., Powers L. S., Hassan I., Groskreutz D., Yarovinsky T. O., Barrett C. W., Castilow E. M., Tifrea D., Varga S. M., Hunninghake G. W. (2007) Respiratory syncytial virus synergizes with Th2 cytokines to induce optimal levels of TARC/CCL17. J. Immunol. 179, 1648–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Monick M. M., Cameron K., Staber J., Powers L. S., Yarovinsky T. O., Koland J. G., Hunninghake G. W. (2005) Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 280, 2147–2158 [DOI] [PubMed] [Google Scholar]
- 33. Groskreutz D. J., Monick M. M., Powers L. S., Yarovinsky T. O., Look D. C., Hunninghake G. W. (2006) Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 176, 1733–1740 [DOI] [PubMed] [Google Scholar]
- 34. Arensdorf A. M., Rutkowski D. T. (2013) Endoplasmic reticulum stress impairs IL-4/IL-13 signaling through C/EBPβ-mediated transcriptional suppression. J. Cell Sci. 126, 4026–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., Kaufman R. J. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee A. H., Iwakoshi N. N., Glimcher L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Look D. C., Rapp S. R., Keller B. T., Holtzman M. J. (1992) Selective induction of intercellular adhesion molecule-1 by interferon-γ in human airway epithelial cells. Am. J. Physiol. 263, L79–87 [DOI] [PubMed] [Google Scholar]
- 38. Hunter J. A., Finkbeiner W. E., Nadel J. A., Goetzl E. J., Holtzman M. J. (1985) Predominant generation of 15-lipoxygenase metabolites of arachidonic acid by epithelial cells from human trachea. Proc. Natl. Acad. Sci. U.S.A. 82, 4633–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dickey D. D., Excoffon K. J., Young K. R., Parekh K. R., Zabner J. (2012) Hoechst increases adeno-associated virus-mediated transgene expression in airway epithelia by inducing the cytomegalovirus promoter. J. Gene Med. 14, 366–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karp P. H., Moninger T. O., Weber S. P., Nesselhauf T. S., Launspach J. L., Zabner J., Welsh M. J. (2002) An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol. Biol. 188, 115–137 [DOI] [PubMed] [Google Scholar]
- 41. Hirota M., Kitagaki M., Itagaki H., Aiba S. (2006) Quantitative measurement of spliced XBP1 mRNA as an indicator of endoplasmic reticulum stress. J. Toxicol. Sci. 31, 149–156 [DOI] [PubMed] [Google Scholar]
- 42. Wu J., Rutkowski D. T., Dubois M., Swathirajan J., Saunders T., Wang J., Song B., Yau G. D., Kaufman R. J. (2007) ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364 [DOI] [PubMed] [Google Scholar]
- 43. Adachi Y., Yamamoto K., Okada T., Yoshida H., Harada A., Mori K. (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 33, 75–89 [DOI] [PubMed] [Google Scholar]
- 44. Volkmann K., Lucas J. L., Vuga D., Wang X., Brumm D., Stiles C., Kriebel D., Der-Sarkissian A., Krishnan K., Schweitzer C., Liu Z., Malyankar U. M., Chiovitti D., Canny M., Durocher D., Sicheri F., Patterson J. B. (2011) Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 286, 12743–12755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dimcheff D. E., Faasse M. A., McAtee F. J., Portis J. L. (2004) Endoplasmic reticulum (ER) stress induced by a neurovirulent mouse retrovirus is associated with prolonged BiP binding and retention of a viral protein in the ER. J. Biol. Chem. 279, 33782–33790 [DOI] [PubMed] [Google Scholar]
- 46. Li B., Gao B., Ye L., Han X., Wang W., Kong L., Fang X., Zeng Y., Zheng H., Li S., Wu Z., Ye L. (2007) Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 124, 44–49 [DOI] [PubMed] [Google Scholar]
- 47. Jheng J. R., Lau K. S., Tang W. F., Wu M. S., Horng J. T. (2010) Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell. Microbiol. 12, 796–813 [DOI] [PubMed] [Google Scholar]
- 48. Kim H. T., Waters K., Stoica G., Qiang W., Liu N., Scofield V. L., Wong P. K. (2004) Activation of endoplasmic reticulum stress signaling pathway is associated with neuronal degeneration in MoMuLV-ts1-induced spongiform encephalomyelopathy. Lab. Invest. 84, 816–827 [DOI] [PubMed] [Google Scholar]
- 49. Bechill J., Chen Z., Brewer J. W., Baker S. C. (2008) Coronavirus infection modulates the unfolded protein response and mediates sustained translational repression. J. Virol. 82, 4492–4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wiseman R. L., Zhang Y., Lee K. P., Harding H. P., Haynes C. M., Price J., Sicheri F., Ron D. (2010) Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell 38, 291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Onn A., Ron D. (2010) Modeling the endoplasmic reticulum unfolded protein response. Nat. Struct. Mol. Biol. 17, 924–925 [DOI] [PubMed] [Google Scholar]
- 52. Promlek T., Ishiwata-Kimata Y., Shido M., Sakuramoto M., Kohno K., Kimata Y. (2011) Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 22, 3520–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hollien J., Lin J. H., Li H., Stevens N., Walter P., Weissman J. S. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stewart M. J., Kulkarni S. B., Meusel T. R., Imani F. (2006) c-Jun N-terminal kinase negatively regulates dsRNA and RSV induction of tumor necrosis factor-α transcription in human epithelial cells. J. Interferon Cytokine Res. 26, 521–533 [DOI] [PubMed] [Google Scholar]
- 55. Rallabhandi P., Phillips R. L., Boukhvalova M. S., Pletneva L. M., Shirey K. A., Gioannini T. L., Weiss J. P., Chow J. C., Hawkins L. D., Vogel S. N., Blanco J. C. (2012) Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. mBio 3, e00218–12 [DOI] [PMC free article] [PubMed] [Google Scholar]