

Background: The SREBP-SCAP pathway controls lipid homeostasis.
Results: SCAP Golgi-to-ER transport requires cleavage of SREBP precursor, reductions in SREBP cleavage at site-1 lead to SCAP degradation in lysosomes, and SREBP control of SCAP expression is conserved in yeast.
Conclusion: Site-1 protease activity and SREBP binding regulate SCAP expression through degradation.
Keywords: Cholesterol, Lipids, Lysosomes, Protein Degradation, Transcription Factors, SCAP, SREBP, Recycling
Abstract
Sterol regulatory element-binding protein (SREBP) transcription factors are central regulators of cellular lipogenesis. Release of membrane-bound SREBP requires SREBP cleavage-activating protein (SCAP) to escort SREBP from the endoplasmic reticulum (ER) to the Golgi for cleavage by site-1 and site-2 proteases. SCAP then recycles to the ER for additional rounds of SREBP binding and transport. Mechanisms regulating ER-to-Golgi transport of SCAP-SREBP are understood in molecular detail, but little is known about SCAP recycling. Here, we have demonstrated that SCAP Golgi-to-ER transport requires cleavage of SREBP at site-1. Reductions in SREBP cleavage lead to SCAP degradation in lysosomes, providing additional negative feedback control to the SREBP pathway. Current models suggest that SREBP plays a passive role prior to cleavage. However, we show that SREBP actively prevents premature recycling of SCAP-SREBP until initiation of SREBP cleavage. SREBP regulates SCAP in human cells and yeast, indicating that this is an ancient regulatory mechanism.
Introduction
Maintenance of cellular lipid homeostasis is essential for cell survival and disruptions to homeostasis lead to diseases (1–3). Sterol regulatory element-binding proteins (SREBPs)2 are membrane-bound basic helix-loop-helix leucine zipper transcription factors that serve as master regulators of lipid homeostasis by regulating synthesis of cholesterol, fatty acids, and triglycerides (4). Two genes, SREBF1 and SREBF2, code for three mammalian SREBP isoforms (SREBP-1a, SREBP-1c, and SREBP-2). SREBP cleavage-activating protein (SCAP) is a central regulator of lipogenesis that controls the activity of all three SREBP isoforms. SREBP activation is completely blocked in cultured cells lacking SCAP (5) and in livers of Scap knock-out mice (6). Consistent with this central role for SCAP in lipid synthesis, inhibition of liver SCAP blocks hepatic steatosis in genetic and dietary rodent models of obesity-induced diabetes (7). Accumulating evidence suggests additional roles for SREBPs in diabetes, immune responses, and cancer (8), necessitating a complete understanding of SREBP pathway regulation.
Current models provide a clear understanding of how SCAP regulates SREBP activity in response to lipid supply (4). Newly synthesized SREBP binds SCAP in the ER (Fig. 1A). Cholesterol and oxysterols regulate ER exit of the SCAP-SREBP complex by controlling binding of SCAP to the ER-resident Insig proteins. In the presence of excess sterols, SCAP binds Insig, preventing recognition of a cytosolic MELADL sequence by COPII coat proteins and blocking SCAP-SREBP entry into ER transport vesicles (Fig. 1A). In sterol-depleted cells, SCAP-SREBP exits the ER and travels to the Golgi, where SREBP is proteolytically activated. First, site-1 protease (S1P) cleaves SREBP in the luminal loop, producing an intermediate in which the N-terminal transcription factor remains attached to the membrane and a C-terminal fragment attached to SCAP. Site-2 protease (S2P) then cleaves the intermediate, releasing the transcription factor from the membrane. This ordered cleavage is obligatory insomuch as full-length SREBP is not cleaved by S2P in the absence of S1P (9). SCAP then recycles from the Golgi to the ER for additional rounds of SREBP transport and cleavage.
FIGURE 1.

Sterols regulate SCAP post-transcriptionally in cells lacking S1P. A, diagram of SREBP-SCAP complex showing sites of S1P and S2P cleavage in SREBP and the MELADL ER exit sequence in SCAP. B, wild-type CHO cells (CHO/pS2P), S1P-deficient CHO cells (SRD-12B), and SCAP-deficient cells (SRD-13A) were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS (for CHO/pS2P) or medium B (for SRD-12B and SRD-13A). On day 1, cells were refed medium C in the absence or presence of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol). After 16 h, ALLN was added to a final concentration of 25 μg/ml, and cells were harvested 1 h later. C, CHO-7 cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS. On day 1, the cells were refed medium C with the addition of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol) and S1P inhibitor PF-429242 (50 μm) as indicated. After 16 h, ALLN was added to a final concentration of 25 μg/ml, and cells were harvested 1 h later. For B and C, aliquots of membrane (25 μg protein/lane for SCAP, S1P, calnexin, and SREBP1/2-P) and nuclear extract (40 μg protein/lane for SREBP1/2-N) fractions were immunoblotted as indicated. D, CHO-7 cells were set up on day 0 at 2 × 105 cells/well (6-well plate) in medium A supplemented with 5% (v/v) FCS. Cells were treated as in described in C. After 16 h, cells were harvested and subjected to RNA preparation and RT-qPCR analysis as described under “Experimental Procedures.” Error bars represent the standard deviation of fold changes from three biological replicates (mean ± S.D.). FASN and LDLR are targets of SREBP1 and SREBP2, respectively. ABCA1 is the target of nuclear receptor LXR.
Despite understanding the mechanisms controlling the ER-to-Golgi transport of SCAP-SREBP in molecular detail, little is known about regulation of SCAP Golgi-to-ER recycling. A single study has demonstrated that SCAP cycles between the ER and Golgi (10). In sterol-depleted cells, SCAP acquires Golgi carbohydrate modifications, but localizes to the ER at steady state, indicating that SCAP recycles from the Golgi to the ER. Here, we present genetic and pharmacologic evidence demonstrating that SREBP cleavage regulates SCAP Golgi-to-ER recycling. In the absence of S1P cleavage, SCAP fails to recycle to the ER and is degraded in lysosomes. Binding of uncleaved SREBP actively blocks SCAP recycling, because SCAP cycles normally when binding to SREBP is prevented. Indeed, SREBP regulation of SCAP recycling is a fundamental mechanism as it is conserved in the fission yeast Schizosaccharomyces pombe where SREBPs are proteolytically activated by a divergent mechanism that does not involve S1P and S2P. This study outlines a new negative feedback mechanism in lipogenesis, identifies the first pathway for SCAP degradation, and defines a regulatory role for SREBP prior to proteolytic activation.
EXPERIMENTAL PROCEDURES
Reagents
We obtained yeast extract, peptone, and agar from BD Biosciences; S1P inhibitor PF-429242 from Shanghai APIs Chemical Co.; proteasome inhibitor MG132 (C2211), lysosome inhibitor ammonium chloride (A9434), mevalonolactone (M4667, for sodium mevalonate preparation), puromycin dihydrochloride (P8833), oleic acid-albumin (O3008), doxycycline (D9891), crystal violet (C3886), soybean trypsin inhibitor (T9003), glass beads (G8772, for yeast cell lysis), trypsin (T8003), and lipoprotein-deficient serum (LPDS; S5394) from Sigma-Aldrich (catalogue numbers in parentheses); cell culture media DMEM (10-013), DMEM/F12 (10-092), and penicillin-streptomycin (30-002) from Corning Cellgro; FuGENE 6 and RNase-free DNase I (10104159001) from Roche Applied Science; random primer mix (S1330), M-MuLV reverse transcriptase (M0253L), murine RNase inhibitor (M0314L), oligo d(T)23VN (S1327S), and endoglycosidase Hf (P0703) from New England Biolabs; GoTaq real-time PCR mix (A6002) from Promega; SCAP trafficking inhibitor fatostatin (341329) and compactin (mevastatin, 474705) from Millipore; and BioCoatTM collagen-coated culture dish (VWR 62405-617) from BD Biosciences.
S. pombe Strains and Culture
We obtained wild-type haploid S. pombe KGY425 from ATCC. Strains scp1Δ, dsc1Δ, dsc2Δ, dsc3Δ, and dsc4Δ have been described previously (11, 12). Strain dsc2Δ sre1Δ was generated in this study by standard mating procedure. All strains were grown to log phase at 30 °C in YES medium (5 g/liter yeast extract plus 30 g/liter glucose and supplements consisting of 225 mg/liter each uracil, adenine, leucine, histidine, and lysine). On day 0, wild-type and mutant yeast cells were inoculated from saturated cultures into 10 ml of fresh YES medium and grown overnight at 30 °C. On day 1, exponential phase cells were inoculated into 10 ml of fresh YES medium under aerobic or anaerobic conditions for 2 h. For hypoxic treatment, cells were grown in YES medium and collected by centrifugation, and the oxygenated medium was removed by aspiration. Cells were resuspended in deoxygenated YES medium inside an Invivo2 400 Hypoxia Workstation (Biotrace, Inc.). Anaerobic conditions were maintained in the work station using 10% hydrogen gas with balanced nitrogen in the presence of palladium catalyst. Deoxygenated medium was prepared by preincubation for >24 h in the work station. After resuspension, the cultures were agitated at 30 °C for the indicated times, and 5 × 107 cells of exponential phase culture were harvested for microsome extraction or RNA preparation.
Antibodies
We obtained the following antibodies: rabbit polyclonal anti-calnexin (208880), HRP-conjugated mouse monoclonal anti-T7 tag (69048), and mouse monoclonal anti-HSV Tag (69171) from EMD Millipore Chemicals; rabbit polyclonal anti-LC3 (PM036) from MBL International Corp.; and HRP-conjugated donkey anti-mouse and anti-rabbit IgG from Jackson ImmunoResearch Laboratories. Antisera to S. pombe Sre1 (11), Scp1 (13), Dsc1, Dsc2, Dsc3, and Dsc4 (12), Dsc5 (14), hamster S1P (U1683 (15)), hamster SCAP (R139 or 9D5) (16), hamster SREBP1 (2A4) (17), and hamster SREBP2 (7D4) (18) have been described previously.
Construction of Inducible SCAP and SREBP2 Expression Vectors
The expression vector pTetOn_CMV_2C1-SCAP C-terminal domain (CTD) encodes amino acids 1–29 of cytochrome P450–2C1 followed by amino acids 731–1276 of hamster SCAP and three tandem copies of the T7 epitope tag (MASMTGGQQMG). The expression vectors pTetOn_CMV_HSV-SREBP2 (WT and R519A) encode two copies of the HSV epitope tag (QPELAPEDPEDC) followed by amino acids 14–1141 of human SREBP2. To generate these plasmids, we first removed the TurboRFP-shRNA cassette from the shRNA plasmid pTRIPz (Open Biosystems) by AgeI/MluI digestion and then ligated into a 250-bp fragment flanked by AgeI and MluI sites containing multiple cloning sites (AgeI/HpaI/ClaI/XhoI/PacI/AscI) and bovine growth hormone poly(A) signal from pcDNA3.1-Myc-His A (Invitrogen) to generate the intermediate doxycycline-inducible protein expression vector pTetOn_CMV. Fragments flanked by AgeI and XhoI sites encoding 2×HSV-human SREBP (WT/R519A) were amplified from vectors pTK-HSV-BP2 (WT/R519A) (19), digested, and inserted into the same sites of pTetOn_CMV vector to generate pTetOn_CMV_HSV-SREBP2 (WT/R519A). Fragments flanked by AgeI and XhoI sites encoding 2C1-SCAP CTD were amplified from vectors P450 TM/SCAP-(731–1276) (20) and pCMV-SCAP-(732–1276)-T7 (16) respectively, digested, and inserted into the same sites of pTetOn_CMV vector to generate pTetOn CMV-2C1-SCAP CTD.
Mammalian Cell Culture
Cells were maintained in monolayer culture at 37 °C in 5% CO2. CHO-7 is a Chinese hamster ovary (CHO) line derived from CHO-K1 selected for growth in lipoprotein-deficient serum (21). CHO/pS2P cells (22) are a clone of CHO-7 cells stably expressing human S2P. CHO-7 and CHO/pS2P cells were maintained in medium A (DMEM/F-12 (1:1) containing 100 units/ml penicillin and 100 μg/ml streptomycin sulfate) supplemented with 5% (v/v) fetal calf serum. S1P-deficient SRD-12B cells and SCAP-deficient SRD-13A cells were selected from γ-irradiated CHO/pS2P cells (5, 22). In experiments using SRD-12B or SRD-13A cells, the parental line, CHO/pS2P, was used as a control. M19 cells are CHO-K1-derived mutant cells lacking the gene encoding the S2P (23, 24). SRD12B, SRD-13A, and M19 cells were maintained in medium B (medium A, 5% (v/v) FCS, 5 μg/ml cholesterol, 1 mm sodium mevalonate, 20 mm sodium oleate). WSC17 and WSC18 cells are CHO-7 cells stably transfected with pTetOn_CMV_HSV-SREBP2 (WT and R519A mutant, respectively). 2C1-SCAP CTD cells are CHO-7 cells stably transfected with pTetOn_CMV_2C1-SCAP CTD. These stable lines were maintained in medium A supplemented with 5% FCS and 9 μg/ml puromycin. NIH3T3 is a mouse fibroblast cell line (25). COS-7 is a cell line derived from monkey kidney tissue (26). HEK293 is a cell line derived from human embryonic kidney (27). Pa03c is a human pancreatic cancer cell line generously provided by Dr. Anirban Maitra at Johns Hopkins University (28). NIH3T3, COS-7, Pa03c, and HEK293 were maintained in medium D (DMEM containing 100 units/ml penicillin and 100 μg/ml streptomycin sulfate) supplemented with 10% fetal calf serum. Atg7−/− mouse embryonic fibroblasts (MEFs) and corresponding control wild-type MEFs were kindly provided by Dr. Masaaki Komatsu at Tokyo Metropolitan Institute of Medical Science, Japan (29). We obtained primary MEFs from Millipore, and primary mouse hepatocytes were isolated from C57BL/6J mice as described (30). Animal care and procedures were approved by the Animal Care and Use Committee of Johns Hopkins University. For sterol depletion treatment, CHO-derived cells were cultured in medium C (medium A, 5% (v/v) LPDS, 50 μm sodium compactin, 50 μm sodium mevalonate), and all other cells were cultured in medium E (medium D, 10% (v/v) LPDS, 50 μm sodium compactin, 50 μm sodium mevalonate). Chemical inhibitors were dissolved in the following solvents, and control cells were treated with an equal concentration of vehicle: PF-429242 in DMSO, fatostatin in DMSO, and MG132 in DMSO.
RNA Preparation and RT-qPCR Analysis
Total RNA was isolated from yeast and mammalian cells using RNA STAT-60. For RT-qPCR analysis of transcript abundance, total RNA (2 μg/sample) was treated with RNase-free DNase I in a total volume of 10 μl at 20–25 °C for 15 min. Reactions were stopped by the addition of 1 μl of 25 mm EDTA. After heating at 65 °C for 10 min, each sample received 4 μl of dNTPs (2.5 mm), 2 μl of 10× RT buffer, 2 μl of primers (oligo d(T)23VN for S. pombe samples and random primer mix for CHO samples), 1 μl of RNase inhibitor, and 1 μl of M-MuLV reverse transcriptase. Reverse transcription was carried out at 25 °C for 5 min followed by 42 °C for 60 min and then 80 °C for 10 min. cDNAs of the tested genes were quantified by real-time PCR using SYBR Green qPCR master mix. β-Actin (for CHO cells samples) or tub1+ (for S. pombe samples) served as the internal control to calculate the relative expression across different samples. Expression was normalized to sterol-depleted CHO-7 cells or wild-type S. pombe cells cultured in the presence of oxygen. Error bars represent standard deviation of fold changes from three biological replicates (mean ± S.D.).
Immunoblotting
Protein concentration in nuclear extracts and membrane fractions was measured using the BCA kit (Pierce), and the samples were mixed with 5× SDS loading buffer (150 mm Tris-HCl, pH 6.8, 15% SDS, 25% glycerol, 0.2% bromphenol blue, and 12.5% β-mercaptoethanol). After heating at 37 °C for 30 min (membrane fraction) or boiling at 100 °C for 5 min (nuclear extracts), proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences). The filters were incubated with the antibodies described in the legends for Figs. 1–6. Bound antibodies were visualized with peroxidase-conjugated affinity-purified donkey anti-mouse or anti-rabbit IgG using the ECL substrate system (Pierce) according to the manufacturer's instructions. Filters were exposed to film at room temperature for 2–120 s. Gels were calibrated with prestained molecular mass markers (Bio-Rad). Working concentrations of primary antibodies were: 5 μg/ml rabbit polyclonal R139 IgG (against SCAP), 10 μg/ml mouse monoclonal 9D5 (against hamster SCAP for SCAP ER-exit assay), 1:250 diluted rabbit anti-serum U1683 (against S1P), 0.5 μg/ml rabbit polyclonal 208880 (against calnexin), 5 μg/ml mouse monoclonal 2A4 (against SREBP1), 5 μg/ml mouse monoclonal 7D4 (against SREBP2), 1:5000 diluted HRP-conjugated mouse monoclonal 69048 (against T7 tag), 1 μg/ml mouse monoclonal 69171 (against HSV tag), 1:500 diluted mouse monoclonals 8G4C11, 1G1D6, and 7B4A3 (against Scp1), 1:5,000 diluted rabbit polyclonal anti-Dsc5 anti-sera, and 1:1000 diluted rabbit polyclonal PM036 (against LC-3).
FIGURE 2.
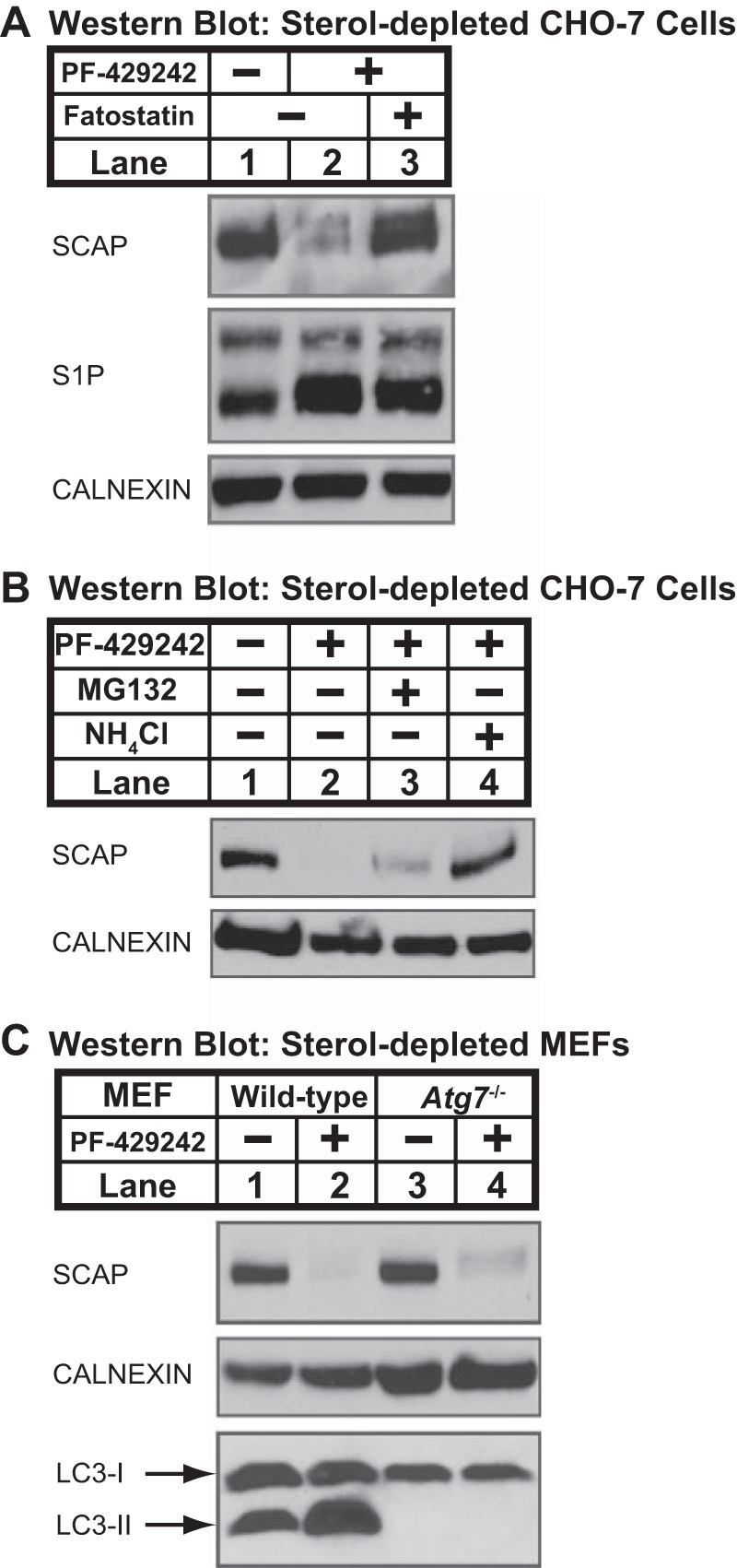
Loss of SCAP requires ER exit and is blocked by ammonium chloride. A, CHO-7 cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS. On day 1, cells were refed medium C containing PF-429242 (50 μm) and fatostatin (10 μm) as indicated. B, CHO-7 cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS. On day 1, cells were refed medium C containing vehicle, PF-429242 (50 μm), MG132 (50 μm), and NH4Cl (50 mm) as indicated. C, MEFs (wild-type control and Atg7−/−) were set up for experiments on day 0 at 1.5 × 106 cells/100-mm dish in medium D supplemented with 10% (v/v) FCS. On day 1, the cells were refed medium E in the absence or presence of PF-429242 (50 μm). A–C, after 16 h, the cells were fractionated, and membrane fractions (25 μg protein/lane) were subjected to immunoblot analysis using the indicated antibodies.
FIGURE 3.
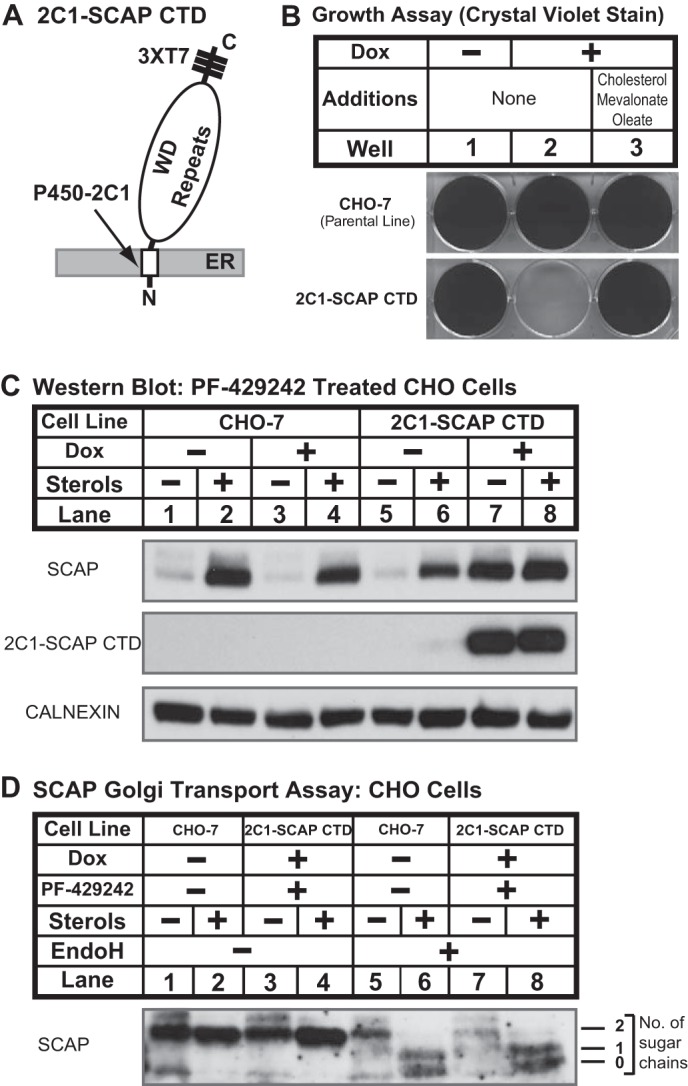
SCAP degradation requires binding to SREBP. A, diagram shows structure of 2C1-SCAP CTD fusion protein in which the N-terminal 1–29 amino acids of cytochrome P450–2C1 are fused to the C-terminal domain of hamster SCAP (amino acids 731–1276) with three repeats of T7 epitope. B, CHO-7 and 2C1-SCAP CTD cells were seeded on day 0 at a density of 3 × 104 cells/well (6-well plate) in medium A supplemented with 5% (v/v) FCS. On day 1, cells were refed with medium A supplemented with 5% (v/v) LPDS in the absence or presence of doxycycline (Dox) (1 μg/ml) or medium B plus doxycycline (1 μg/ml). Cells were refed every 2 days. On day 14, cells were fixed in cold methanol and stained with 0.05% crystal violet. C, CHO-7 and 2C1-SCAP CTD cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS. On day 1, cells were refed medium C in the absence or presence of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol), PF-429242 (50 μm), or doxycycline (1 μg/ml) as indicated. After 16 h, cells were fractionated, and membrane fractions (25 μg protein/lane) were subjected to immunoblot analysis with the indicated antibodies and anti-T7 to detect 2C1-SCAP CTD. D, CHO-7 and 2C1-SCAP CTD cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% FCS plus 25-hydroxycholesterol (1 μg/ml). On day 1, cells were refed medium C in the absence or presence of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol), PF-429242 (50 μm), or doxycycline (1 μg/ml) as indicated. After 16 h, cells were harvested (4 dishes/condition) and subjected to cell fractionation. Aliquots of membrane fractions (50 μg) were analyzed using the SCAP endoglycosidase H (EndoH) assay described under “Experimental Procedures” and then subjected to immunoblot analysis with 10 μg/ml anti-SCAP mouse monoclonal 9D5. Filters were exposed to film at room temperature for 60 s.
FIGURE 4.

SCAP recycling requires SREBP cleavage at site-1. A, S2P-deficient (M19) cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium B. On day 1, the cells were refed medium C in the absence or presence of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol) and PF-429242 (50 μm). After 16 h, cells were fractionated, and membrane fractions (25 μg protein/lane) were subjected to immunoblot analysis as indicated. B, CHO-7, WSC17 (HSV-SREBP2_WT), and WSC18 (HSV-SREBP2_R519A) cells were set up on day 0 at 3 × 104 cells/well (6-well plate) in medium A supplemented with 5% (v/v) FCS. On day 1, cells were refed with medium A supplemented with 5% (v/v) LPDS in the absence or presence of doxycycline (Dox) (1 μg/ml) or medium B plus doxycycline (1 μg/ml). Cells were refed every 2 days. On day 14, cells were fixed in cold methanol and stained with 0.05% crystal violet. C, CHO-7 cells, WSC17 (HSV-SREBP2_WT), and WSC18 (HSV-SREBP2_R519A) cells were set up on day 0 at 1.5 × 106 cells/100-mm dish in medium A supplemented with 5% (v/v) FCS. On day 1, the cells were refed medium C in the absence or presence of sterols (1 μg/ml 25-HC, 10 μg/ml cholesterol) and doxycycline (1 μg/ml) as indicated. After 16 h, cells were fractionated, and membrane fractions (25 μg protein/lane) were subjected to immunoblot analysis as noted.
FIGURE 5.
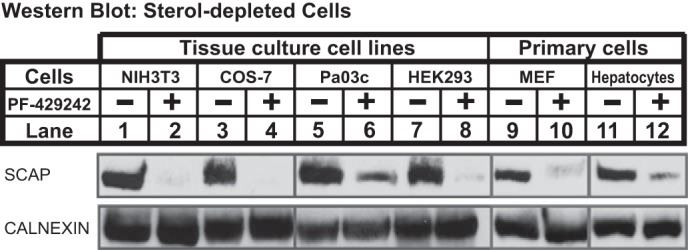
Mammalian SREBP regulates SCAP recycling through a common mechanism. NIH3T3, COS-7, Pa03c, HEK293, and primary MEFs (MEF-P3, isolated from CF1 strain, passage 3) were set on day 0 at 1 × 106 cells/100-mm dish in medium D supplemented with 10% (v/v) FCS. On day 1, the cells were refed medium E in the absence or presence of PF-429242 (50 μm). Primary mouse hepatocytes were isolated from C57BL/6J mice and set up for experiments on day 0 at 3 × 106 cells/100-mm dish (BD BioCoat collagen-coated plates) in medium D supplemented with 10% (v/v) FCS. On day 1, the cells were refed medium E in the absence or presence of PF-429242 (50 μm). After 16 h, cells were fractionated, and membrane fractions (25 μg protein/lane) were subjected to immunoblot analysis using the indicated antibodies.
FIGURE 6.

Yeast SREBP regulates SCAP recycling. A, Western blot of membrane fractions from wild-type and the indicated mutant strains probed with anti-Scp1 IgGs or anti-Dsc5 serum. Expression of scp1+ was analyzed by RT-qPCR. B, Western blot of membrane fractions from wild-type and dsc2Δ mutant cells grown for 2 h in the presence or absence of oxygen probed with anti-Scp1 IgGs. Expression of scp1+ was analyzed by RT-qPCR. C, Western blot of membrane fractions from wild-type, dsc2Δ, and dsc2Δsre1Δ mutant strains probed with anti-Scp1 IgGs and anti-Dsc5 serum. Expression of scp1+ was analyzed by RT-qPCR. Error bars represented the standard deviation of fold changes from three biological replicates (mean ± S.D.). ND, not detected.
Mammalian Cell Growth Assay
The growth assay used for CHO-7 and other stable cell lines has been described previously (31). Briefly, cells were seeded on day 0 at a density of 3 × 104 cells/well (6-well plate) in medium A supplemented with 5% (v/v) fetal calf serum. On day 1, cells were refed as indicated in the figure legends for Figs. 3 and 4. Cells were refed every 2 days. On day 14, cells were washed with PBS once, fixed in cold methanol at −20 °C for 10 min, and stained with 0.05% crystal violet at room temperature for 10 min. Plate images were scanned in transmitted light mode at a resolution of 300 dpi.
Cell Fractionation
Harvested yeast (2.5 × 109) were resuspended in 50 μl of B88 buffer (20 mm HEPES-KOH, pH 7.2, 150 mm KOAc, 5 mm MgOAc, and 250 mm sorbitol) with a mixture of protease inhibitors (10 μg/ml pepstatin A, 20 μg/ml leupeptin, and 1 mm PMSF); glass beads were added, and the mixture was lysed for 10 min at 4 °C using Disruptor Genie. Then, 0.5 ml of B88 buffer with a mixture of protease inhibitors (5 μg/ml pepstatin A, 10 μg/ml leupeptin, and 0.5 mm PMSF) was added to the cell lysates, and all liquid was transferred to a new Eppendorf tube and then centrifuged at 500 × g at 4 °C for 5 min. The recovered supernatant was then centrifuged at 20,000 × g for 10 min at 4 °C, and the pellet was dissolved in 50 μl of SDS lysis buffer (10 mm Tris-HCl, pH 6.8, 100 mm NaCl, 1% (v/v) SDS, 1 mm sodium EDTA, and 1 mm sodium EGTA) and designated as the membrane fraction.
Mammalian cell fractionation has been described previously (32). Briefly, harvested mammalian cells were allowed to swell in 0.5 ml of hypotonic buffer A (10 mm HEPES-KOH, pH 7.6, 10 mm KCl, 1.5 mm MgCl2, 1 mm sodium EDTA, 1 mm sodium EGTA, 250 mm sucrose, and a mixture of protease inhibitors (5 μg/ml pepstatin A, 10 μg/ml leupeptin, 0.5 mm PMSF, 1 mm DTT, and 25 μg/ml ALLN)) for 30 min on ice, passed 30 times through a 22G1/2-gauge needle, and centrifuged at 890 × g at 4 °C for 5 min to collect the nuclei. The nuclei pellet was resuspended in 0.1 ml of buffer C (20 mm HEPES-KOH, pH 7.6, 0.42 m NaCl, 2.5% (v/v) glycerol, 1.5 mm MgCl2, 1 mm sodium EDTA, 1 mm sodium EGTA, and a mixture of protease inhibitors (5 μg/ml pepstatin A, 10 μg/ml leupeptin, 0.5 mm PMSF, 1 mm DTT, and 25 μg/ml ALLN)). The suspension was rotated at 4 °C for 60 min and centrifuged at 20,000 × g in a microcentrifuge for 20 min at 4 °C. The supernatant was designated as nuclear extract. The supernatant from the original 890 × g spin was centrifuged at 20,000 × g for 20 min at 4 °C to collect membranes. For subsequent treatment with trypsin/endoglycosidase Hf, this membrane pellet was resuspended in 0.1 ml of buffer A containing 100 mm NaCl. For subsequent immunoblot analysis, the pellet was dissolved in 0.1 ml of SDS lysis buffer and designated the membrane fraction.
SCAP Golgi Transport Assay
ER-to-Golgi transport of SCAP was monitored by the presence of Golgi-specific oligosaccharide modifications on SCAP (10). Briefly, cell membranes (50 μg) were incubated in the presence of 0.5 μg of trypsin in a total volume of 29 μl of buffer A containing 100 mm NaCl for 30 min at 30 °C. Reactions were stopped by the addition of 1 μl of soybean trypsin inhibitor (200 units). Then individual samples received 5 μl of 3.5% (w/v) SDS, 7% (v/v) β-mercaptoethanol. After heating at 100 °C for 10 min, each sample received sequential additions of 4.5 μl of 0.5 m sodium citrate, pH 5.5, and 2.5 μl of 17× protease inhibitors (with 1× corresponding to 10 μg/ml leupeptin, 5 μg/ml pepstatin A, and 2 μg/ml aprotinin) followed by 1 μl of endoglycosidase Hf. The reaction was carried out at 37 °C for 16 h and then stopped by the addition of 10 μl of 5× SDS loading buffer. The mixtures were heated at 100 °C for 5 min and subjected to 12% SDS-PAGE.
RESULTS
Sterols Regulate SCAP in S1P-deficient CHO Cells
SCAP plays a central role in the regulation of SREBP activation, but little is known about the regulation of SCAP expression, either mRNA or protein. Rawson et al. (5) observed loss of SCAP when S1P-deficient CHO cells (SRD-12B) were sterol-depleted. To investigate the mechanism for SCAP loss, we conducted a similar experiment. Membranes from the parental cell line CHO/pS2P contained SCAP that was unaffected by the absence or presence of sterols (Fig. 1B, lanes 1and 2). S1P-deficient SRD-12B cells had similar amounts of SCAP when incubated in the presence of sterols, but SCAP was reduced when cells were depleted of sterols (Fig. 1B, lanes 3 and 4). SCAP-deficient SRD-13A cells served as a specificity control for SCAP antibody (5). As expected, SREBP2 proteolytic activation was induced in sterol-depleted CHO/pS2P cells leading to the accumulation of nuclear SREBP2-N, and S1P-deficient cells failed to activate SREBP2 (Fig. 1B, lanes 3 and 4). Thus, consistent with the previous report (5), sterols regulated SCAP expression in S1P-deficient CHO cells.
Sterols Regulate SCAP Post-transcriptionally in Cells Lacking S1P Activity
SRD-12B cells were generated by γ-irradiation and may contain multiple mutations (22). To test independently whether the sterol-dependent loss of SCAP resulted from lack of S1P activity, we treated wild-type CHO cells with the S1P chemical inhibitor PF-429242 (33). In the absence of PF-429242, sterol depletion induced cleavage of both SREBP1 and SREBP2 in CHO-7 cells (Fig. 1C, lanes 1 and 2). The addition of PF-429242 blocked SREBP activation, demonstrating that PF-429242 inhibits S1P activity (Fig. 1C, lanes 3 and 4). In vehicle-treated cells, sterols had no effect on SCAP, but PF-429242 treatment led to a dramatic loss of SCAP in sterol-depleted cells (Fig. 1C, lanes 1–3). The addition of sterols prevented SCAP loss in PF-429242-treated cells (Fig. 1C, lane 4), suggesting that SCAP loss required ER exit. Interestingly, PF-429242 treatment increased the levels of both S1P proenzyme and the faster migrating active S1P (S1P blot, Fig. 1C), demonstrating the existence of an uncharacterized S1P feedback mechanism (15). Together, these genetic and pharmacologic data demonstrate that sterols regulate SCAP in cells lacking S1P activity.
To test whether sterols regulate SCAP through changes in transcription, we quantified SCAP mRNA in CHO-7 cells under identical conditions. Consistent with the Western results for SREBP activation in Fig. 1C, expression of FASN (fatty acid synthase, a SREBP1 target gene) and LDLR (LDL receptor, a SREBP2 target gene) was induced upon sterol depletion and was blocked by sterol addition or treatment with PF-429242 (Fig. 1D). Expression of ABCA1, a target of the LXR nuclear receptor, was stimulated by sterol treatment as expected. SCAP mRNA expression was unaffected by treatment with either sterols or the S1P inhibitor PF-429242 (Fig. 1D), indicating that sterols regulate SCAP expression post-transcriptionally.
SCAP Is Degraded in the Lysosome in the Absence of S1P Activity
Sterol depletion stimulates ER-to-Golgi transport of SCAP (34). To test whether sterol-dependent loss of SCAP required its ER exit, we treated cells with fatostatin, a chemical inhibitor that blocks ER exit of SCAP (35). In sterol-depleted CHO-7 cells, inhibition of S1P with PF-429242 resulted in SCAP loss (Fig. 2A, lanes 1 and 2). Treatment with fatostatin blocked SCAP loss, demonstrating that SCAP loss requires exit from the ER (Fig. 2A, lane 3). S1P accumulated upon PF-429242 treatment (Fig. 2A, lanes 2 and 3), and fatostatin did not block S1P accumulation, indicating that fatostatin did not interfere with S1P inhibition by PF-429242. Fatostatin also prevented SCAP loss in sterol-depleted SRD-12B cells (data not shown). These data demonstrate that sterols regulate SCAP degradation in cells lacking S1P and that SCAP degradation requires ER-to-Golgi transport.
Cellular protein degradation occurs primarily in the lysosome or through the ubiquitin-proteasome pathway (36). We next tested whether chemical inhibition of these degradative pathways prevented SCAP loss (Fig. 2B). Ammonium chloride (NH4Cl) inhibits lysosomal hydrolases by preventing acidification of the endosomal/lysosomal compartments (37), and MG132 blocks the proteasome (38). Treatment of CHO-7 cells with ammonium chloride, but not MG132, completely prevented sterol-regulated SCAP degradation (Fig. 2B). Peptide aldehydes, such as MG132, can also inhibit lysosomal proteases and calpains (39), suggesting that the mild inhibitory effect of MG132 was due to non-proteasomal effects of this inhibitor. A second lysosomal inhibitor, monensin, also prevented SCAP degradation in CHO-7 cells (data not shown) (37). Finally, ammonium chloride, but not MG132, blocked SCAP degradation in sterol-depleted, S1P-deficient SRD-12B cells (data not shown). Collectively, these data demonstrate that in the absence of S1P activity, SCAP is degraded in the lysosome following ER-to-Golgi transport.
SCAP lysosomal degradation could occur either because of inappropriate trafficking of SCAP to the lysosome or through basal autophagy. To test whether SCAP degradation requires autophagy, we examined SCAP degradation in Atg7−/− MEFs, which are defective for Atg12 conjugation and the LC3 lipidation required for autophagosome formation (29, 40). Similar to CHO-7 cells (Fig. 1C), in sterol-depleted wild-type MEFs, SCAP was degraded upon inhibition of S1P by PF-429242 treatment (Fig. 2C, lanes 1 and 2). In Atg7−/− MEFs, SCAP was also degraded upon PF-429242 treatment (Fig. 2C, lanes 3 and 4), indicating that S1P-dependent SCAP degradation does not require autophagy. Production of lipidated LC3-II was blocked in Atg7−/− MEFs as expected (Fig. 2C, lanes 1–4) (40). Based on our results up to this point, we concluded that in the absence of S1P activity, SCAP fails to recycle from the Golgi to the ER and trafficks to the lysosome where it is degraded. SCAP lysosomal degradation therefore provides an assay for Golgi-to-ER recycling.
SREBP Binding Prevents SCAP Golgi-to-ER Recycling in the Absence of S1P
S1P has multiple substrates in addition to SREBPs, including viral proteins and CREB family transcription factors such as ATF6 and OASIS (41). To investigate whether the effects of S1P inhibition on SCAP recycling are direct, we tested whether SCAP degradation requires binding to SREBP. Cells lacking all three SREBP isoforms were not available. As an alternative, we generated 2C1-SCAP CTD CHO cells in which binding of SCAP to SREBPs is blocked by the doxycycline-regulated overexpression of the SCAP CTD (amino acids 731–1276) that mediates SREBP binding (16). Overexpression of SCAP CTD anchored to the ER membrane by the transmembrane segment from cytochrome P450–2C1 blocks SREBP cleavage by competing for SREBP binding to full-length SCAP (42). The SCAP ER exit signal (MELADL) is absent from the SCAP CTD (Fig. 1A), and thus SREBP bound to SCAP CTD will be retained in the ER resulting in cholesterol and fatty acid auxotrophy. Conversely, full-length SCAP not bound to SREBP will traffick to the Golgi. Consistent with this design, 2C1-SCAP CTD cells failed to grow in the absence of exogenous lipids when 2C1-SCAP CTD expression was induced by doxycycline (Fig. 3B). The addition of the SREBP-dependent pathway products cholesterol, mevalonate, and oleate rescued growth, indicating that SCAP CTD blocked cell growth by inhibiting the SREBP pathway.
To test whether the effects of S1P inhibition on SCAP recycling are due to its role in SREBP cleavage, we examined SCAP degradation in 2C1-SCAP CTD cells. In this experiment, all cells were treated with PF-429242. In parental CHO-7 cells, sterol depletion led to SCAP degradation, and doxycycline had no effect on sterol-regulated SCAP degradation (Fig. 3C, lanes 1–4). In 2C1-SCAP CTD cells, sterol depletion caused SCAP degradation in the absence of doxycycline (Fig. 3C, lanes 5 and 6). In the presence of doxycycline, overexpression of 2C1-SCAP CTD prevented SCAP degradation in sterol-depleted cells (Fig. 3C, lanes 7 and 8). We confirmed that full-length SCAP trafficked to the Golgi normally in 2C1-SCAP CTD cells using an established assay (10). Under sterol-depleted conditions, SCAP in both cell lines acquired endoglycosidase H-resistant oligosaccharides due to the action of Golgi mannosidase II (Fig. 3D, lanes 5–8). Thus, binding of SREBP to SCAP in the absence of S1P activity prevents SCAP recycling, resulting in its degradation. These data demonstrate that SREBP controls SCAP recycling and that the effects of S1P inhibition are mediated by its role in SREBP cleavage.
SCAP Recycling Requires SREBP Cleavage at Site-1
Our results indicate that inhibition of SREBP cleavage prevents SCAP Golgi-to-ER recycling and leads to SCAP degradation in the lysosome. Next, we investigated the mechanism by which SREBP controls SCAP recycling. The failure of SCAP to recycle in the absence of S1P could result from either (1) the absence of active SREBP transcription factors and downstream gene expression or (2) a failure to cleave the SREBP precursor at site-1. To test whether a lack of active SREBP transcription factors prevents SCAP recycling, we examined SCAP regulation in M19 CHO cells that lack site-2 protease (23, 24). In M19 cells, S1P cleaves the SREBP precursor to produce a membrane-tethered N-terminal SREBP intermediate, but no active SREBP transcription factor is made. Unlike S1P-deficient SRD-12B cells (Fig. 1B), sterol depletion of M19 cells had no effect on SCAP expression (Fig. 4A, lanes 1 and 2). However, inhibition of S1P with PF-429242 in M19 cells led to sterol-regulated SCAP degradation as in wild-type CHO cells (Fig. 4A, lanes 3 and 4). Thus, the effects of S1P inhibition on SCAP recycling are not due to the absence of active SREBP transcription factors. Rather, SCAP failed to recycle when SREBPs were not cleaved by S1P.
To test directly whether SCAP recycling requires SREBP cleavage at site-1, we asked whether binding of non-cleavable SREBP targets SCAP for degradation in cells with active S1P. Mutation of SREBP2 in the protease recognition sequence (R519A) blocks cleavage by S1P (19, 43). We generated inducible CHO-7 lines overexpressing either wild-type HSV-tagged SREBP2 or S1P-resistant SREBP2 (R519A). Overexpressed HSV-SREBP2 will compete with endogenous SREBPs for SCAP binding, causing the majority of SCAP to bind to HSV-tagged SREBP2. Thus, we expected that overexpression of HSV-SREBP2 (R519A), but not wild-type HSV-SREBP2, would inhibit SREBP activation. Wild-type CHO cells and cells overexpressing HSV-SREBP2 grew normally in the absence of exogenous lipids, and doxycycline had no effect on cell growth (Fig. 4B, wells 1 and 2). As expected, overexpression of HSV-SREBP2 (R519A) inhibited growth in lipoprotein-deficient medium, and the addition of excess cholesterol, mevalonate, and oleate rescued growth (Fig. 4B, well 3). Thus, overexpression of HSV-SREBP2 (R519A) competes with endogenous SREBPs for SCAP binding and prevents SREBP activation.
Having established conditions in which SCAP binds either HSV-SREBP2 or non-cleavable SREBP2 (R519A), we next tested the effect on SCAP recycling. In parental CHO-7 cells and cells overexpressing HSV-SREBP2, sterol depletion and doxycycline treatment had no effect on SCAP expression (Fig. 4C, lanes 1–8). Importantly, overexpression of non-cleavable HSV-SREBP2 (R519A) led to sterol-regulated degradation of SCAP (Fig. 4C, lanes 11 and 12). SCAP degradation in sterol-depleted cells required HSV-SREBP2 (R519A) expression, because SCAP was stable in the absence of doxycycline treatment. Thus, uncleaved SREBP is sufficient to prevent SCAP Golgi-to-ER recycling, leading to its degradation in the lysosome. These data also demonstrate that the effects observed in cells lacking S1P activity result from the failure to cleave SREBPs at site-1 and not through the action of another S1P substrate.
Mammalian SREBP Regulates SCAP Recycling through a Common Mechanism
We next tested whether regulation of SCAP recycling by SREBP is limited to CHO cells or a general mechanism. We analyzed a range of established tissue culture cell lines, including mouse embryonic fibroblasts (NIH3T3), African green monkey kidney fibroblasts (COS-7), human pancreatic cancer cells (Pa03c), and human embryonic kidney cells (HEK293). Like CHO-7 cells (Fig. 1C), SCAP was degraded in each cell line upon inhibition of S1P by PF-429242 treatment (Fig. 5, lanes 1–8). We observed a similar regulation of SCAP degradation in primary mouse embryonic fibroblasts and primary mouse hepatocytes (Fig. 5, lanes 9–12). These data indicate that regulation of SCAP recycling by SREBP cleavage is a common mechanism conserved across mammals.
Yeast SREBP Regulates SCAP Recycling
Sterol regulation of the SREBP pathway is conserved in fission yeast S. pombe, where low oxygen activates SREBP to promote hypoxic adaptation (45). Scp1 and Sre1 are the yeast orthologs of mammalian SCAP and SREBP, respectively (11). Scp1 forms a complex with Sre1 and transports Sre1 from the ER to the Golgi at a basal rate in the presence of oxygen (46). Low oxygen inhibits sterol synthesis, increasing Sre1 cleavage and activating hypoxic gene expression. However, unlike in mammals, Sre1 cleavage does not require S1P and S2P, which are absent from fission yeast (47). Rather, Sre1 proteolytic cleavage occurs through a distinct mechanism that requires the Golgi Dsc E3 ubiquitin ligase complex and uncharacterized protease(s) (12). To test whether SREBP regulation of SCAP recycling is conserved in fission yeast, we examined Scp1 expression in dsc mutants in which Sre1 cleavage is blocked. Cells lacking each of the four core subunits of the Dsc E3 ligase (Dsc1–Dsc4) showed a dramatic reduction in Scp1 (Fig. 6A). Reduced Scp1 expression resulted from Scp1 degradation in dsc mutants as scp1+ mRNA was unchanged (Fig. 6A, bottom panel). As in mammals, Scp1 degradation was sterol-regulated insomuch as growth under low oxygen further reduced Scp1 expression with no effect on scp1+ mRNA (Fig. 6B). Finally, SCAP degradation requires binding to SREBP in mammals (Fig. 3C), and likewise dsc-dependent Scp1 degradation required Sre1. Deletion of dsc2+ decreased Scp1 expression, and deletion of sre1+ in dsc2Δ cells prevented Scp1 degradation (Fig. 6C). scp1+ mRNA was unchanged in these strains, indicating that changes in Scp1 expression did not result from transcriptional regulation (Fig. 6C, bottom panel). Collectively, these results demonstrated that despite distinct mechanisms for SREBP cleavage between yeast and mammals, SREBP regulation of SCAP recycling is conserved.
DISCUSSION
Mechanisms controlling ER-to-Golgi transport of the SCAP-SREBP complex and subsequent cleavage of SREBP are understood in detail (48). After transport to the Golgi, SCAP recycles to the ER for additional rounds of SREBP binding and transport, but nothing is known about how recycling is regulated (10). In this study, we identified requirements for SCAP recycling and revealed the first mechanism controlling SCAP expression. Upon arrival in the Golgi, S1P initiates SREBP activation by cleaving it in half at site-1 (Fig. 1A). The N-terminal SREBP product is subsequently cleaved by S2P to release the active N-terminal transcription factor from the membrane. We demonstrated that SCAP Golgi-to-ER recycling requires SREBP cleavage at site-1. If SREBP fails to be cleaved by S1P, SCAP does not recycle and is ultimately degraded in the lysosome. Importantly, this mechanism prevents premature return of the SCAP-SREBP complex to the ER, ensuring that SREBP cleavage initiates before SCAP recycles for another round of transport. Conservation of this mechanism in cells from human, monkey, mouse, and yeast underscores its significance for the fidelity of the SREBP pathway.
Studies of SCAP ER-to-Golgi transport revealed that sterol-dependent conformational changes in SCAP control entry into COPII-coated ER transport vesicles (49, 50). In the absence of sterols, COPII proteins bind the cytosolic MELADL sequence and recruit SCAP-SREBP into forming vesicles for Golgi transport (51). Examining the other half of the transport cycle, we found that SCAP Golgi-to-ER transport requires SREBP cleavage at site-1 and that uncleaved SREBP actively blocks recycling (Fig. 4). SCAP not bound to SREBP cycled normally between the ER and Golgi (Fig. 3), indicating that SCAP contains an ER retrieval signal. The best characterized ER retrieval signals are a dilysine -KKXX sequence at the cytosolic C terminus of ER membrane proteins and a luminal -KDEL sequence at the C terminus of both soluble and membrane-bound ER-resident proteins (52). Neither signal is present in SCAP, suggesting the existence of a novel ER retrieval signal. Alternatively, SCAP could bind to an unidentified adaptor protein that contains a canonical retrieval signal. Current models depict SREBPs as playing an inert or passive role prior to their proteolytic activation and downstream transcriptional activity. The discovery that uncleaved SREBP precursor regulates SCAP recycling assigns a function to SREBP prior to cleavage.
Understanding exactly how SREBP prevents SCAP recycling will require additional studies. However, the SCAP membrane domain forms tetramers, and the SREBP N-terminal basic helix-loop-helix leucine zipper domains dimerize (53, 54), suggesting that SCAP-SREBP exists as a large hetero-oligomer. One possibility is that S1P cleavage disrupts SREBP dimers, altering the oligomeric state of the SCAP-SREBP complex and revealing an ER retrieval signal. SCAP likely returns to the ER in COPI-coated vesicles that transport other recycled cargo (55).
The path that SCAP takes to the lysosome and the signals involved in this transport step are less clear. Notably, lysosomal transport does not require autophagy because SCAP is degraded normally in Atg7−/− embryonic fibroblasts blocked for autophagosome formation (Fig. 2C). Sorting of transmembrane proteins from the Golgi to the endosomes-lysosomes is mediated by signals present within the cytosolic domains of proteins (44). Well studied signals are the tyrosine-based sorting motif YXXΦ and dileucine-based motifs (D/E)XXXL(L/I) and DXXLL. Both dileucine motifs are present in the C-terminal cytosolic domain of SCAP. In addition to peptide motifs, membrane proteins can be ubiquitinated and sorted to the endosomal-lysosomal system. Future studies will determine the signals and sorting machinery required to direct SCAP from the Golgi to the lysosome.
Synthesis of SCAP is not subject to feedback regulation at transcription or translation (4). Consequently, whether SCAP expression can be regulated has been unknown. Here, we show that S1P activity regulates SCAP expression. In cells with limiting S1P activity, SREBP is not cleaved at site-1, and SCAP is degraded in the lysosome. This degradative pathway adds new negative feedback regulation to the SREBP system. Changes in S1P activity will control the balance between SCAP recycling and degradation, thereby dictating the amount of SCAP available for ER-to-Golgi transport and activation of SREBP. In this way, the flow of SREBP substrate to S1P can be tuned to S1P activity. S1P has multiple substrates, including viral proteins and cAMP-responsive element-binding protein (CREB) family transcription factors. Substrate competition may limit S1P activity toward SREBPs, providing a mechanism for cross-talk between the SREBP pathway and other pathways.
Our finding that SREBP cleavage at site-1 regulates SCAP Golgi-to-ER recycling identifies a new point of regulation in the SREBP pathway. Molecular understanding of the signals and proteins involved in this process will require the development of a direct assay for Golgi-to-ER recycling as was the case for ER-to-Golgi transport of SCAP. A detailed view of how SCAP either recycles to the ER or trafficks to the lysosome for degradation may reveal new opportunities for chemical control of the pathway and therapeutic regulation of lipogenesis. Given that Golgi-to-ER transport controls SCAP expression, future studies of this other half of the SCAP cycle may also uncover new modes of biological control.
Acknowledgments
We thank Russell DeBose-Boyd (University of Texas (UT)-Southwestern Medical Center at Dallas) for sharing unpublished results, members of the Espenshade laboratory for excellent advice and discussion, Shan Zhao for outstanding technical support, and Rita Brookheart and Clara Bien Peek (Northwestern University) for help on hepatocyte isolation. We are grateful to the following investigators for sharing cell lines, antibodies, and plasmids: Michael Brown and Joseph Goldstein (UT-Southwestern), Anirban Maitra (University of Texas M. D. Anderson Cancer Center, Houston), and Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science).
This work was supported, in whole or in part, by National Institutes of Health Grant HL077588. This work was also supported by Grant 12-60-25-ESPE, the 2012 Pancreatic Cancer Action Network-AACR Innovative Grant in memory of Bonnie L. Tobin.
- SREBP
- sterol regulatory element-binding protein
- SCAP
- SREBP cleavage-activating protein
- S1P
- site-1 protease
- S2P
- site-2 protease
- ER
- endoplasmic reticulum
- LPDS
- lipoprotein-deficient serum
- ALLN
- N-acetyl-leucinyl-leucinyl-norleucinal
- CTD
- C-terminal domain
- MEF
- mouse embryonic fibroblast
- qPCR
- quantitative PCR.
REFERENCES
- 1. Cohen J. C., Horton J. D., Hobbs H. H. (2011) Human fatty liver disease: old questions and new insights. Science 332, 1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldstein J. L., Hazzard W. R., Schrott H. G., Bierman E. L., Motulsky A. G. (1973) Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J. Clin. Invest. 52, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hachinski V., Graffagnino C., Beaudry M., Bernier G., Buck C., Donner A., Spence J. D., Doig G., Wolfe B. M. (1996) Lipids and stroke: a paradox resolved. Arch. Neurol. 53, 303–308 [DOI] [PubMed] [Google Scholar]
- 4. Brown M. S., Goldstein J. L. (2009) Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J. Lipid Res. 50, (suppl.) S15–S27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rawson R. B., DeBose-Boyd R., Goldstein J. L., Brown M. S. (1999) Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 274, 28549–28556 [DOI] [PubMed] [Google Scholar]
- 6. Matsuda M., Korn B. S., Hammer R. E., Moon Y. A., Komuro R., Horton J. D., Goldstein J. L., Brown M. S., Shimomura I. (2001) SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 15, 1206–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moon Y. A., Liang G., Xie X., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Brown M. S., Goldstein J. L., Horton J. D. (2012) The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 15, 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shao W., Espenshade P. J. (2012) Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakai J., Duncan E. A., Rawson R. B., Hua X., Brown M. S., Goldstein J. L. (1996) Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 10. Nohturfft A., DeBose-Boyd R. A., Scheek S., Goldstein J. L., Brown M. S. (1999) Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. U.S.A. 96, 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hughes A. L., Todd B. L., Espenshade P. J. (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120, 831–842 [DOI] [PubMed] [Google Scholar]
- 12. Stewart E. V., Nwosu C. C., Tong Z., Roguev A., Cummins T. D., Kim D. U., Hayles J., Park H. O., Hoe K. L., Powell D. W., Krogan N. J., Espenshade P. J. (2011) Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol. Cell 42, 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughes A. L., Stewart E. V., Espenshade P. J. (2008) Identification of twenty-three mutations in fission yeast Scap that constitutively activate SREBP. J. Lipid Res. 49, 2001–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stewart E. V., Lloyd S. J., Burg J. S., Nwosu C. C., Lintner R. E., Daza R., Russ C., Ponchner K., Nusbaum C., Espenshade P. J. (2012) Yeast sterol regulatory element-binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain-containing subunit of the Golgi Dsc E3 ligase. J. Biol. Chem. 287, 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Espenshade P. J., Cheng D., Goldstein J. L., Brown M. S. (1999) Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 274, 22795–22804 [DOI] [PubMed] [Google Scholar]
- 16. Sakai J., Nohturfft A., Cheng D., Ho Y. K., Brown M. S., Goldstein J. L. (1997) Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J. Biol. Chem. 272, 20213–20221 [DOI] [PubMed] [Google Scholar]
- 17. Sato R., Yang J., Wang X., Evans M. J., Ho Y. K., Goldstein J. L., Brown M. S. (1994) Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1). J. Biol. Chem. 269, 17267–17273 [PubMed] [Google Scholar]
- 18. Yang J., Brown M. S., Ho Y. K., Goldstein J. L. (1995) Three different rearrangements in a single intron truncate sterol regulatory element-binding protein-2 and produce sterol-resistant phenotype in three cell lines. Role of introns in protein evolution. J. Biol. Chem. 270, 12152–12161 [DOI] [PubMed] [Google Scholar]
- 19. Hua X., Sakai J., Brown M. S., Goldstein J. L. (1996) Regulated cleavage of sterol regulatory element-binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 271, 10379–10384 [DOI] [PubMed] [Google Scholar]
- 20. Sakai J., Nohturfft A., Goldstein J. L., Brown M. S. (1998) Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J. Biol. Chem. 273, 5785–5793 [DOI] [PubMed] [Google Scholar]
- 21. Metherall J. E., Goldstein J. L., Luskey K. L., Brown M. S. (1989) Loss of transcriptional repression of three sterol-regulated genes in mutant hamster cells. J. Biol. Chem. 264, 15634–15641 [PubMed] [Google Scholar]
- 22. Rawson R. B., Cheng D., Brown M. S., Goldstein J. L. (1998) Isolation of cholesterol-requiring mutant Chinese hamster ovary cells with defects in cleavage of sterol regulatory element-binding proteins at site 1. J. Biol. Chem. 273, 28261–28269 [DOI] [PubMed] [Google Scholar]
- 23. Hasan M. T., Chang C. C., Chang T. Y. (1994) Somatic cell genetic and biochemical characterization of cell lines resulting from human genomic DNA transfections of Chinese hamster ovary cell mutants defective in sterol-dependent activation of sterol synthesis and LDL receptor expression. Somat. Cell Mol. Genet. 20, 183–194 [DOI] [PubMed] [Google Scholar]
- 24. Rawson R. B., Zelenski N. G., Nijhawan D., Ye J., Sakai J., Hasan M. T., Chang T. Y., Brown M. S., Goldstein J. L. (1997) Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1, 47–57 [DOI] [PubMed] [Google Scholar]
- 25. Copeland N. G., Cooper G. M. (1979) Transfection by exogenous and endogenous murine retrovirus DNAs. Cell 16, 347–356 [DOI] [PubMed] [Google Scholar]
- 26. Gluzman Y. (1981) SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23, 175–182 [DOI] [PubMed] [Google Scholar]
- 27. Graham F. L., Smiley J., Russell W. C., Nairn R. (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74 [DOI] [PubMed] [Google Scholar]
- 28. Feldmann G., Dhara S., Fendrich V., Bedja D., Beaty R., Mullendore M., Karikari C., Alvarez H., Iacobuzio-Donahue C., Jimeno A., Gabrielson K. L., Matsui W., Maitra A. (2007) Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 67, 2187–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li W., Ralphs K. L., Tosh D. (2010) Mouse Cell Culture (Ward A., Tosh D., eds) pp. 185–196, Humana Press, Totowa, NJ [Google Scholar]
- 31. Xu F., Rychnovsky S. D., Belani J. D., Hobbs H. H., Cohen J. C., Rawson R. B. (2005) Dual roles for cholesterol in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 102, 14551–14556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeBose-Boyd R. A., Brown M. S., Li W. P., Nohturfft A., Goldstein J. L., Espenshade P. J. (1999) Transport-dependent proteolysis of SREBP: relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 99, 703–712 [DOI] [PubMed] [Google Scholar]
- 33. Hawkins J. L., Robbins M. D., Warren L. C., Xia D., Petras S. F., Valentine J. J., Varghese A. H., Wang I. K., Subashi T. A., Shelly L. D., Hay B. A., Landschulz K. T., Geoghegan K. F., Harwood H. J., Jr. (2008) Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J. Pharmacol. Exp. Ther. 326, 801–808 [DOI] [PubMed] [Google Scholar]
- 34. Nohturfft A., Yabe D., Goldstein J. L., Brown M. S., Espenshade P. J. (2000) Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102, 315–323 [DOI] [PubMed] [Google Scholar]
- 35. Kamisuki S., Mao Q., Abu-Elheiga L., Gu Z., Kugimiya A., Kwon Y., Shinohara T., Kawazoe Y., Sato S., Asakura K., Choo H. Y., Sakai J., Wakil S. J., Uesugi M. (2009) A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 16, 882–892 [DOI] [PubMed] [Google Scholar]
- 36. Ciechanover A. (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87 [DOI] [PubMed] [Google Scholar]
- 37. Mellman I., Fuchs R., Helenius A. (1986) Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55, 663–700 [DOI] [PubMed] [Google Scholar]
- 38. Kisselev A. F., Goldberg A. L. (2001) Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 8, 739–758 [DOI] [PubMed] [Google Scholar]
- 39. Lee D. H., Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 40. Tanida I., Yamasaki M., Komatsu M., Ueno T. (2012) The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy 8, 88–97 [DOI] [PubMed] [Google Scholar]
- 41. Seidah N. G., Sadr M. S., Chrétien M., Mbikay M. (2013) The multifaceted proprotein convertases. Their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 288, 21473–21481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sakai J., Rawson R. B., Espenshade P. J., Cheng D., Seegmiller A. C., Goldstein J. L., Brown M. S. (1998) Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol. Cell 2, 505–514 [DOI] [PubMed] [Google Scholar]
- 43. Duncan E. A., Brown M. S., Goldstein J. L., Sakai J. (1997) Cleavage site for sterol-regulated protease localized to a Leu-Ser bond in the lumenal loop of sterol regulatory element-binding protein-2. J. Biol. Chem. 272, 12778–12785 [DOI] [PubMed] [Google Scholar]
- 44. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 45. Osborne T. F., Espenshade P. J. (2009) Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. 23, 2578–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porter J. R., Burg J. S., Espenshade P. J., Iglesias P. A. (2010) Ergosterol regulates sterol regulatory element-binding protein (SREBP) cleavage in fission yeast. J. Biol. Chem. 285, 41051–41061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bien C. M., Espenshade P. J. (2010) Sterol regulatory element-binding proteins in fungi: hypoxic transcription factors linked to pathogenesis. Eukaryot. Cell 9, 352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Espenshade P. J., Hughes A. L. (2007) Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 41, 401–427 [DOI] [PubMed] [Google Scholar]
- 49. Brown A. J., Sun L., Feramisco J. D., Brown M. S., Goldstein J. L. (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 [DOI] [PubMed] [Google Scholar]
- 50. Espenshade P. J., Li W. P., Yabe D. (2002) Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc. Natl. Acad. Sci. U.S.A. 99, 11694–11699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun L. P., Li L., Goldstein J. L., Brown M. S. (2005) Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 280, 26483–26490 [DOI] [PubMed] [Google Scholar]
- 52. Teasdale R. D., Jackson M. R. (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 12, 27–54 [DOI] [PubMed] [Google Scholar]
- 53. Radhakrishnan A., Sun L. P., Kwon H. J., Brown M. S., Goldstein J. L. (2004) Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15, 259–268 [DOI] [PubMed] [Google Scholar]
- 54. Párraga A., Bellsolell L., Ferré-D'Amaré A. R., Burley S. K. (1998) Co-crystal structure of sterol regulatory element-binding protein 1a at 2.3 Å resolution. Structure 6, 661–672 [DOI] [PubMed] [Google Scholar]
- 55. Brandizzi F., Barlowe C. (2013) Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 14, 382–392 [DOI] [PMC free article] [PubMed] [Google Scholar]