

Background: The role of caspase-1 in regulating the immunogenic properties of HMGB1 has not been previously reported.
Results: We have mapped a peptide in the A-box of HMGB1 that reverses tolerance through RAGE.
Conclusion: Inflammasome signaling regulates the immunogenic activity of HMGB1.
Significance: Immunogenic peptides within the HMGB1 A-box may be exploited to reverse immune tolerance in sepsis patients.
Keywords: Apoptosis, Caspase, Inflammation, Innate Immunity, Receptor for Advanced Glycation End Products (RAGE), Sepsis, Tolerance
Abstract
Apoptotic cells trigger immune tolerance in engulfing phagocytes. This poorly understood process is believed to contribute to the severe immunosuppression and increased susceptibility to nosocomial infections observed in critically ill sepsis patients. Extracellular high mobility group box 1 (HMGB1) is an important mediator of both sepsis lethality and the induction of immune tolerance by apoptotic cells. We have found that HMGB1 is sensitive to processing by caspase-1, resulting in the production of a fragment within its N-terminal DNA-binding domain (the A-box) that signals through the receptor for advanced glycation end products (RAGE) to reverse apoptosis-induced tolerance. In a two-hit mouse model of sepsis, we show that tolerance to a secondary infection and its associated mortality were effectively reversed by active immunization with dendritic cells treated with HMGB1 or the A-box fragment, but not a noncleavable form of HMGB1. These findings represent a novel link between caspase-1 and HMGB1, with potential therapeutic implications in infectious and inflammatory diseases.
Introduction
Sepsis is a leading cause of mortality in intensive care units. However, lack of understanding of the underlying pathological mechanisms has hindered therapeutic success in the clinic (1, 2). Sepsis is defined as the host response to overwhelming infection or injury leading to immune dysfunction and organ failure. It is now well established that following an early cytokine storm, sepsis patients become severely immunosuppressed, which renders them susceptible to secondary nosocomial infections. Human and animal studies indicate that massive leukocyte apoptosis occurs in sepsis and suppresses immunity by triggering a tolerogenic program in antigen-presenting cells (2–10). As a result, sepsis patients have impaired delayed-type hypersensitivity (DTH)7 and are often unable to mount an immune response to recall antigens (11, 12).
HMGB1 was initially identified in the early 1970s as a chromatin-associated protein, consisting of two DNA-binding domains (termed A- and B-boxes) and a highly acidic C-terminal tail (reviewed in Ref. 13). It was later recognized that HMGB1 accumulates in the circulation of patients and mice during sepsis and that its neutralization improves sepsis survival in experimental animal models (14, 15). Together these results implicated HMGB1 as a late mediator of sepsis lethality. A consensus on how HMGB1 exerts its immunomodulatory roles in sepsis remains elusive. Several studies have shown that HMGB1 functions as a cytokine by inducing the release of pro-inflammatory factors such as TNFα and IL-6, a function that has been mapped to the B-box domain (16, 17). Meanwhile other groups associated these observations with the use of HMGB1 preparations purified from bacterial systems as HMGB1 can complex with pathogen-associated molecular patterns (18–23). Interestingly, administration of recombinant A-box was reported to protect mice from experimental sepsis (15). The discovery of the regulation of HMGB1 by oxidation has exposed yet another level of biological complexity. Different configurations of oxidative states on three cysteine residues, Cys23, Cys45, and Cys106, have been correlated with modified activities of extracellular HMGB1 (reviewed in Ref. 24).
Our previous investigations of the effects of cell death modalities on the immune response have pointed to HMGB1 as a central determinant of the immunogenic versus tolerogenic effects of dying cells. We have reported that HMGB1 released from necrotic cells was immunogenic, whereas that released from apoptotic cells was tolerogenic (25). This differential effect was linked to the redox state of HMGB1, such that oxidation of Cys106 during apoptosis was sufficient to inhibit its immunogenic activity (25). In a separate study, we found that HMGB1 may be targeted for processing by caspase-1 (26), which prompted us to explore whether caspase-1 and the upstream inflammasome pathways interact with HMGB1 to modulate its immunogenic activities (25).
Here, we report that HMGB1 is sensitive to caspase-1 processing at aspartate Asp67 within the A-box. We also show that the A-box fragment contains the immunogenic determinants of HMGB1 and that it is necessary and sufficient to reverse apoptosis-induced tolerance through RAGE. In a two-hit mouse model of sepsis, we show that tolerance to a secondary Candida infection was effectively reversed by active immunization with dendritic cells (DCs)treated with HMGB1 or the A-box fragment, but not a noncleavable form of HMGB1. Collectively, these observations suggest a new link between the inflammasome and the regulation of HMGB1 activity and describe a direct functional interaction between A-box and RAGE that may shed light on the protective effects of A-box administration during experimental sepsis.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Active recombinant caspases-1, -3, and -11 were obtained from BioMol (Enzo Life Sciences, Farmingdale, NY). Active recombinant caspases-2, -5, -7, and -9 were from Merck Research Laboratories. M2 agarose, FLAG peptide, lipopolysaccharide (LPS; Escherichia coli serotype 0111:B4), ATP, 2,4,6-trinitrobenzene sulfonic acid (TNBS), puromycin dihydrochloride, anti-β-actin, anti-FLAG, and anti-HMGB1 antibodies were from Sigma-Aldrich. Anti-caspase-1 p10 was from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary immunoblot HRP-conjugated antibodies were from Amersham (GE Healthcare, Little Chalfont, UK). RNase inhibitors and protease inhibitors were from Roche Applied Science. In vitro transcription/translation kit was from Promega (Madison, WI). 35S-labeled methionine was from PerkinElmer. QuikChange site-directed mutagenesis kit was from Stratagene (La Jolla, CA). Recombinant full-length HMGB1, mutants, and fragments were generated and purified by the Biotechnology Research Institute (National Research Council of Canada, Pointe-Claire, Quebec, Canada) using a proprietary mammalian HEK293-6E cell line and Fractogel cobalt column, as described previously (27). Recombinant mammalian sRAGE was from Prospec (East Brunswick, NJ), and V-C1 and V-C1-C2 constructs were generated by Dr. Tsian Xiao at the National Institutes of Health (Bethesda, MD). HMGB1 synthetic peptides were generated at >90% purity by AnaSpec (Fremont, CA). Candida albicans antigen Candin was from Allermed Laboratories (San Diego, CA). Immortalized Hmgb1−/− and Hmgb1+/+ mouse embryonic fibroblast (MEF) cell lines were from HMGBiotech (Milan, Italy).
Animal Experiments
All knock-out mice were on a C57BL/6 background. Wild-type C57BL/6 and 129/S1 mice were from Charles River Laboratories (Wilmington, MA). Mice were housed in the McGill University Comparative Medicine Animal Resources Centre facility or the Washington University Animal facility under standard temperature and light and dark cycles. All euthanizing was done by CO2 overdose. All animal experimental protocols and clinical endpoints were approved by the McGill University Animal Care Committee or the Washington University Institutional Animal Care and Use Committee (IACUC) according to Canadian Council on Animal Care and National Institutes of Health guidelines, respectively.
Cloning and Mutagenesis of HMGB1
The open reading frame (ORF) of human HMGB1 was PCR-amplified from a pCMV-SPORT6 vector containing the HMGB1 cDNA purchased from the American Type Culture Collection (clone MGC-5223). Point mutants were generated by site-directed mutagenesis according to the manufacturer's instructions. For generation of mammalian recombinant full-length HMGB1 and fragments, inserts were PCR-amplified from the wild-type or noncleavable mutant HMGB1 cDNA in PCDNA3.1(+) and subcloned into a proprietary high expression pTT5 vector (Biotechnology Research Institute (BRI)-National Research Council Canada) to generate gene products containing 3×FLAG-His8 tags on the N terminus of the HMGB1 ORFs. To generate the D67A, D67E, D158A, and D158E single mutants, the HMGB1 wild-type construct was subsequently mutated by site-directed mutagenesis. Sequence integrity of all constructs and mutants was confirmed by DNA sequencing.
In Vitro Transcription/Translation
[35S]methionine-labeled substrates were obtained using a coupled in vitro transcription/translation TnT reticulocyte lysates system according to the manufacturer's instructions. Where indicated, in vitro transcribed/translated 35S-labeled FLAG-tagged HMGB1 was affinity-purified from the reticulocyte mixture on M2 agarose beads in maximum stringency radioimmune precipitation buffer with extensive washes and eluted from the beads by affinity competition with FLAG peptide.
Caspase Cleavage Assays
Cleavage of the in vitro transcribed/translated 35S-labeled substrates was performed as described previously (26) Proteins were resolved by Tricine-SDS-PAGE, transferred to nitrocellulose membranes, and exposed to autoradiography film for 4 h or used in Western analysis.
Isolation of DCs
CD8α+ DCs were purified from mice treated by intravenous injection of 109 PFU adenovirus-Flt3L to expand splenic DC populations. Purification was performed on spleens from treated mice using a DC enrichment kit and a phycoerythrin-positive selection kit (StemCell Technologies, Vancouver, British Columbia, Canada) with an anti-CD8α-phycoerythrin antibody (clone 53-6.7, BioLegend, San Diego, CA). Bone marrow-derived DCs were generated as described previously (25).
Delayed Type Hypersensitivity Assays
Reversal of tolerance experiments were carried out as described previously (25). For the priming assays, MEFs were first exposed to 10 mm TNBS at room temperature for 5 min, washed with PBS, made necrotic by five freeze/thaw cycles, and added to the overnight co-culture at a ratio of 1:1 DC:necrotic cell immediately after freeze/thaw. DCs exposed to necrotic cells were washed and injected subcutaneously in PBS at 106 DCs per mouse to prime immunity to TNBS. Where indicated, assays were performed by adding 250 ng/ml full-length HMGB1 and the corresponding equimolar amounts of the other recombinant proteins to the DC apoptotic or necrotic cell overnight co-cultures. Recombinant proteins were reduced prior to addition as described (25).
Generation of Stable MEFs
The neomycin cassette in the pcDNA3.1(+) vectors carrying the cDNA of wild-type (WT) HMGB1, D67A, and D158A was replaced by restriction digest and ligation with a PCR-generated puromycin cassette. For injection into MEFs to generate the stable cell lines, the plasmid constructs were linearized and purified using a PCR cleaning kit. Hmgb1−/− MEFs were seeded at 2 × 104 cells/well in a 24-well plate and transfected 24 h later with the purified linearized constructs using Lipofectamine 2000 according to the manufacturer's protocol. MEFs from each well were detached 24 h after transfection with trypsin/EDTA and resuspended in 10 ml of selection medium composed of DMEM supplemented with 10% FBS, 1% penicillin streptomycin, and 3 μg/ml puromycin. Cells were then plated immediately at 100 μl/well in 96-well plates, and selection medium changed every 2–3 days until single colonies were observed. Individual colonies were then harvested using trypsin/EDTA and reseeded into 12-well plates under selection, changing the selection medium every 2–3 days until cells became confluent. Stable cell lines were assayed for HMGB1 expression and expanded for cryopreservation.
Surface Plasmon Resonance (SPR) Binding Assays
Label-free, real-time binding assays were performed at 25 °C on a BIACORE 3000 system (GE Healthcare Life Sciences). Research-grade CM4 dextran-coated sensor chips were utilized with filtered (0.2 μm) and degassed HBS-EP running buffer (10 mm HEPES pH 7.4, 150 mm NaCl, 3.4 mm EDTA, 0.005% (v/v) Tween 20). Carrier-free, soluble RAGE was from Prospec (HEK sRAGE; PRO-601), Pierce Gentle Elution was from Thermo Scientific (21027), and detergents were from Anatrace (Tween 20 APT020 and Empigen D350); all other chemicals were reagent-grade quality. RAGE constructs (V-C1, 25 kDa; V-C1-C2, 34 kDa; sRAGE, 35 kDa; 10 μg/ml each in 10 mm sodium acetate, pH 4.0) were immobilized using the BIACORE amine coupling kit (∼250 RU each); corresponding reference surfaces were prepared in the absence of any RAGE protein. Bovine serum albumin (BSA, 66 kDa; negative control; 0–100 nm), A-box 1–67 (17 kDa; 0–100 nm), and the HMGB1 A-box synthetic peptides (∼1.3 kDa each; 0–10 μm) were titrated (2-fold serial dilutions) over reference and RAGE-immobilized at high flow rate (to minimize mass transport effects; 50 μl/min × 1-min association + 3-min dissociation). Between sample injections, sensor chip surfaces were regenerated at 50 μl/min using two 30-s pulses of solution I (running buffer containing 1 m NaCl) and II (Pierce Gentle Elution containing 0.05% (v/v) Empigen) followed by “EXTRACLEAN” and “RINSE” procedures. To cross-validate the selected SPR “ligand-analyte” orientation, similar control experiments were performed in which sRAGE (0–100 nm) was titrated (2-fold serial dilutions; 50 ml/min × 3-min association + 10-min dissociation) over amine-coupled A-box 1–67 (5 μg/ml in 10 mm sodium acetate pH 5.0; ∼150 RU final). SPR data were double-referenced (28) and are representative of duplicate injections acquired from at least two independent trials. For each replicate series, a buffer blank was injected first, the highest titrant concentration was injected second, and serial dilutions followed (from the lowest to the highest concentration repeated); comparing responses between the two highest titrant injections verified consistent immobilized surface activity throughout each assay. For the peptide titrations, apparent equilibrium dissociation constants (KD) were determined by global fitting of the data (averaged responses at the end of each association phase (Req) plotted versus concentration) to a “steady-state affinity” model in the BIAevaluation software (v 4.1). Given the inherent heterogeneity in the association phase profiles for A-box 1–67 interacting with RAGE (i.e. both ligand-analyte orientations deviated from simple 1:1 kinetics), the protein titrations were also analyzed according to the steady-state affinity model to approximate the overall KD values. In all cases, theoretical binding maxima were predicted using the following equation: Rmax = (MWA/MWL) (RL) (n); Rmax is the maximal binding response (RU) at saturating peptide/protein concentration, MWA is the molecular weight (kDa) of the peptide/protein injected in solution, MWL is the molecular weight (kDa) of the protein immobilized, RL is the amount (RU) of protein immobilized, and n is the predicted binding stoichiometry (e.g. 1:1).
Cecal Slurry and Candida Secondary Infection Assays
The cecal slurry sepsis model was adapted from Ref. 29. Five WT 8–10-week-old donor mice were euthanized, and cecal contents were extracted, pooled, weighed, and vortexed for 3 min in a sterile 5% dextrose saline solution to a final concentration of 40 mg/ml. The cecal slurry (CS) was filtered through a 100-μm cell strainer to remove fibrous debris and injected intraperitoneally at the indicated doses into recipient mice within 2 h of euthanasia of the donor mice. Abdomens of the mice were rubbed gently for 5 s immediately after injection to evenly distribute CS into the peritoneal cavity. For the Candida secondary infection model, mice were injected with 0.2 mg/g of CS and 24 h later injected intravenously with PBS or a suspension of DCs exposed to apoptotic cells and 50 μl/ml C. albicans antigen Candin with or without equimolar amounts of recombinant HMGB1 proteins (corresponding to 250 ng/ml FL WT HMGB1). Mice were injected intraperitoneally 24 h later with 5 × 105 colony-forming units (CFUs) of live C. albicans clinical isolate, and survival was monitored for 10 days.
Statistical Analysis
Data are presented as averages ± S.D. Statistical differences between groups were determined by Student's t test. A p value <0.05 was considered significant (*). For Kaplan-Meier survival curve analysis, significant differences were evaluated by both the Mantel-Cox test and the Gehan-Breslow-Wilcoxon test.
RESULTS AND DISCUSSION
In Vitro Cleavage of HMGB1 by Recombinant Caspase-1 but Not Other Inflammatory or Apoptotic Caspases at Three Aspartate Residues
Analysis of the caspase-1 digestome revealed HMGB1 as a potential caspase-1 target (26). To validate this observation and determine the specificity of this cleavage event, HMGB1 processing was evaluated in an in vitro cleavage assay. [35S]methionine-labeled in vitro transcribed and translated HMGB1 was incubated with recombinant caspases-1, -2, -3, -5, -7, -9, or -11. HMGB1 was specifically cleaved by caspase-1 but not by other apoptotic or inflammatory caspases (Fig. 1A). Caspase-1 cleaved HMGB1 in a dose-dependent manner, albeit with a slightly lower affinity as compared with poly(ADP-ribose) polymerase or its preferred substrate pro-IL-1β (Fig. 1B). This was also observed using radiolabeled FLAG-tagged HMGB1 that was stringently affinity-purified to exclude the possibility of proteolytic cofactors within the reticulocyte system contributing to the observed processing (not shown). Site-directed mutagenesis allowed the mapping of the caspase-1 cleavage sites in HMGB1 to Asp67, Asp158, and Asp169 and the generation of a noncleavable (NC) form, in which all three aspartates were replaced by alanine (Fig. 1C). Together these results suggest that HMGB1 can be specifically targeted and processed by caspase-1.
FIGURE 1.

HMGB1 is a specific caspase-1 substrate. A, HMGB1 was in vitro transcribed and translated (ITT) and [35S]methionine-labeled. ITT products (1 μl) were digested with the indicated active recombinant caspases. Radiolabeled poly(ADP-ribose) polymerase (PARP) was generated and treated with the same conditions to confirm the activity of the recombinant caspase preparations. The proteins were separated by SDS-PAGE and visualized by autoradiography. B, caspase-1 cleaves HMGB1 in a dose-dependent manner. Radiolabeled HMGB1 was generated as in A. ITT products (1 μl) were digested with increasing amounts of active recombinant caspase-1 or -3 and visualized by SDS-PAGE and autoradiography. Radiolabeled poly(ADP-ribose) polymerase and pro-IL-1β were generated and treated with the same conditions to confirm the activity of the recombinant caspase preparations. C, left, site-directed mutagenesis was used to map the caspase-1 cleavage sites in HMGB1. The aspartate to alanine mutant forms of HMGB1 were tested in a caspase-1 cleavage assay using recombinant caspase-1. Right, schematic representation of HMGB1 domain organization highlighting the three cleavage sites and the redox-sensitive cysteine residue.
The Immunogenic Function of HMGB1 in Reversing Apoptosis-induced Tolerance Resides in Its A-box
Our in vitro observations of HMGB1 cleavage by caspase-1 enticed us to map the biological activities of HMGB1. Toward this goal, we generated highly purified mammalian recombinant proteins corresponding to the observed cleavage products encompassing the A- and B-boxes as well as WT HMGB1 and the caspase-1-resistant NC form. To study the activity of these proteins, we chose to explore their effects on reversing apoptosis-induced tolerance in an established model of DTH (Fig. 2A) (25). As reported previously (25), we show that treatment of tolerogenic CD8a+ DCs (exposed to apoptotic cells) with HMGB1 was capable of reversing the tolerogenic program of these DCs, as evidenced by their ability to mediate DTH when injected in immunized mice (Fig. 2B). Surprisingly, the A-box, previously reported to antagonize full-length HMGB1 (15), reconstituted its immunogenic activity in this assay. In contrast, the B-box, which is the presumed proinflammatory domain of HMGB1 (16), did not show any activity in reversing tolerance (Fig. 2B). We verified these findings by supplementing the media of the DC-apoptotic cell co-culture system with neutralizing anti-HMGB1 antibodies specific for the A- or B-box domains. As measured by DTH and in accordance with our previous observations, neutralization of the A-box reversed the immunogenic activity of HMGB1, whereas that of the B-box did not (Fig. 2C). We next examined the activity of noncleavable HMGB1 in this assay. In contrast to WT HMGB1, the NC form failed to reverse tolerance (Fig. 2B). Consistently, full-length WT HMGB1 was nonfunctional when Casp1−/− DCs were used, whereas the A-box was fully functional in the absence of caspase-1 (Fig. 2D), suggesting that processing of HMGB1 into the A-box by caspase-1 may be necessary for biological activity. In contrast, HMGB1 was fully functional when DCs derived from 129/S1 mice, which are deficient in caspase-11 because of a premature stop codon in the Casp11 gene (30), were used (not shown), further indicating that HMGB1 activity specifically requires caspase-1 and not caspase-11. To characterize the role of the individual HMGB1 caspase-1 cleavage sites, we generated additional HMGB1 mutants in which Asp67 or Asp158 was individually substituted to Ala or Glu. Mutation of Asp67 but not Asp158 prevented HMGB1 from reversing tolerance in the DTH assay (Fig. 2E). Analysis of existing NMR and crystal structures revealed that these mutations are unlikely to change the fold of the protein, and this was supported when comparing the HPLC elution profiles of the recombinant preparations (not shown). To further validate our observations made with recombinant proteins in a more physiological system where HMGB1 would be endogenously expressed, we reconstituted Hmgb1−/− MEFs with full-length HMGB1 or the HMGB1 cleavage site mutants (Asp67 or Asp158). Because endogenous HMGB1 secreted from apoptotic cells mediates tolerance because of Cys106 oxidation (25), we used necrotic cells to compare the priming activities of cell-reconstituted HMGB1 variants. As expected, necrotic WT MEFs fed to DCs primed immunity, whereas Hmgb1−/− MEFs failed to do so. However, reconstitution of Hmgb1−/− MEFs with WT HMGB1 or the D158A mutant restored their ability to prime, to a similar extent as with the addition of exogenous recombinant HMGB1 (Fig. 2F). In contrast, reconstitution of Hmgb1−/− MEFs with HMGB1 D67A failed to restore priming. These results suggest that caspase-1 sensitivity at the Asp67 site of A-box release is required for HMGB1 immunogenicity.
FIGURE 2.
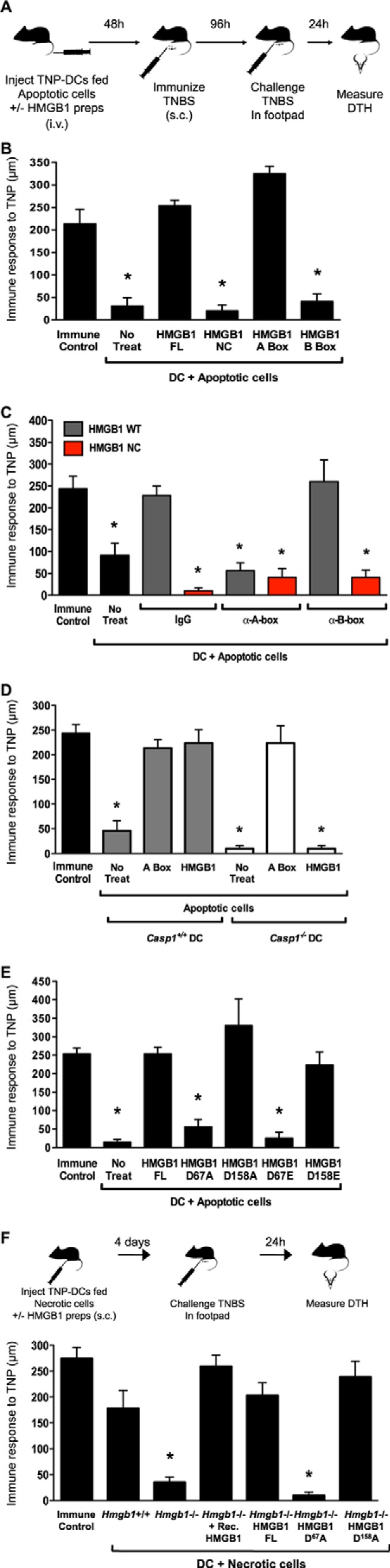
HMGB1 reverses apoptosis-induced tolerance via the A-box. A, schematic representation of the DTH assay. TNP-conjugated DCs fed apoptotic cells in the absence or presence of exogenous recombinant HMGB1 preparations were injected intravenously (i.v.). Mice were immunized 2 days later by subcutaneous (s.c.) injection of TNBS. Four days following immunization, mice were injected with TNBS in the right footpad and PBS in the left footpad. DTH was measured with a micrometer 24 h later as the difference in thickness between the right and left footpads. Immune control mice were injected with TNBS subcutaneously and challenged in the footpads. B, full-length WT (FL) HMGB1 and the A-box but not full-length NC HMGB1 or the B-box reverse tolerance in DTH. Equimolar amounts of recombinant HMGB1 proteins were incubated with TNP-conjugated WT CD8α+ DCs fed apoptotic splenocytes. The tolerogenic activity of the treated DCs was examined in the DTH assay. No treat, no treatment. C, neutralization of HMGB1 A-box reverses the effects of HMGB1 effects in blocking tolerance. DTH tolerance assay was conducted as in B with culture media supplemented with IgG control or neutralizing antibodies targeting the A-box or B-box. D, DC expression of caspase-1 is required for HMGB1 activation and reversal of tolerance in DTH. Equimolar amounts of recombinant full-length HMGB1 or the A-box were incubated with TNP-conjugated WT or Casp1−/− CD8α+ DCs fed apoptotic splenocytes. The tolerogenic activity of the treated DCs was examined in the DTH assay. E, as in B using full-length WT HMGB1 and HMGB1 mutated at Asp67 or Asp158 to Ala or Glu. F, top, schematic representation of the DTH priming assay. TNP-conjugated DCs fed necrotic cells in the absence or presence of exogenous recombinant HMGB1 preparations were injected subcutaneously in mice. Four days later, mice were injected with 10 mm TNBS in the right footpad and PBS in the left footpad. DTH was measured with a micrometer 24 h later. Immune control mice were injected with 10 mm TNBS subcutaneously. Bottom, Hmgb1+/+ MEFs, Hmgb1−/− MEFs, or Hmgb1−/− MEFs stably transfected with full-length WT HMGB1 (FL), D67A, or D158A mutants were exposed to 10 mm TNBS, made necrotic, and fed to WT CD8α+ DCs overnight in the absence or presence of recombinant HMGB1. DTH responses were measured as the DCs ability to prime immunity to TNBS.
Caspase-1 is released from cells following activation (31), and evidence suggesting that HMGB1 could be targeted by extracellular proteases upon release has previously been reported (32, 33). The commercially available antibodies we tested did not allow for robust detection of HMGB1 fragments by Western analysis in the media of DCs fed apoptotic cells (not shown), possibly due to rapid internalization and degradation of the cleavage fragments upon interaction with cell surface receptors. Alternatively, the minute amounts of HMGB1 preparations required to modulate DC responses in our assay may indicate that cleavage levels below the detection limits of available antibodies are required for signaling. Taken together, our data using caspase-1-resistant HMGB1 mutants and caspase-1-deficient DCs nevertheless suggest that the biological activity of HMGB1 depends on targeting by caspase-1. Follow-up studies using alternative detection strategies and mice engineered to express caspase-1-resistant HMGB1 will be required to further investigate these observations and validate our in vitro findings.
The Nlrp3 Inflammasome Regulates the Ability of HMGB1 to Reverse Apoptosis-induced Tolerance
To determine which inflammasome is engaged to activate caspase-1 in DCs upon sensing of apoptotic cells, we performed DTH tests using CD8α+ DCs from wild-type mice or mice deficient in inflammasome components (Fig. 3). HMGB1 was markedly ineffective in reversing tolerance in Casp1−/− DCs (Fig. 2D), Asc−/− DCs (Fig. 3A), or Nlrp3−/− DCs (Fig. 3B), whereas it was fully functional in Aim2−/− DCs (Fig. 3C) or Nlrc4−/− DCs (Fig. 3D). In contrast, the A-box was fully effective in reversing tolerance in DCs from all genotypes. These results suggest that the Nlrp3 inflammasome regulates the biological activity of HMGB1. The observation that the A-box fragment is sufficient to reproduce the immunogenic effect of full-length HMGB1 even in the absence of inflammasome signaling (Fig. 3, A--D) and caspase-1 activation (Fig. 2D) in DCs eliminates the possible contribution of other caspase-1 functions such as the processing of IL-1β or IL-18 in this immunogenic response. DCs fed apoptotic cells did not undergo pyroptosis or release their own HMGB1, as HMGB1 was not detected in the media when DCs were fed Hmgb1−/− apoptotic cells (not shown). Additionally, DC migratory ability was not modulated by dead cells (apoptotic or necrotic) and was independent of HMGB1 in Transwell migration assays (not shown). Of note, neither reduced nor oxidized mammalian recombinant HMGB1 induced IL-6 or TNFα secretion in this system (Ref. 25 and data not shown), suggesting that the observed immunomodulatory activity of HMGB1 is independent of these cytokines.
FIGURE 3.
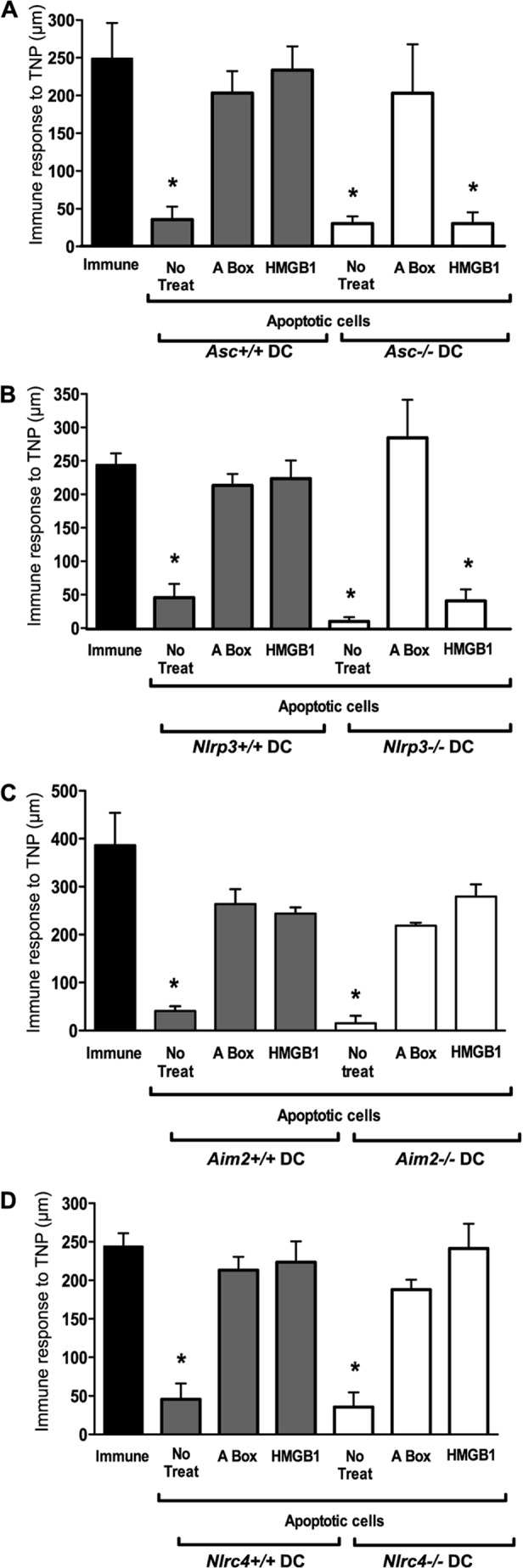
The Nlrp3 inflammasome is required in DCs for mediating the immunogenic activity of HMGB1. A–D, the DTH assay was conducted as in Fig. 2B using TNP-conjugated WT, Asc−/−, Nlrp3−/−, Aim2−/−, or Nlrc4−/− CD8α+ DCs fed apoptotic splenocytes in the absence or presence of full-length WT HMGB1 or the A-box peptide. No treat, no treatment.
The Caspase-1-generated HMGB1 A-box Reverses Tolerance through RAGE
Numerous receptors have been ascribed to HMGB1, including RAGE (34) and several Toll-like receptors (TLRs) (24, 35). The redox status of HMGB1 can affect its biological activities (reviewed in Ref. 24), and Cys106 within the B-box domain has been shown to be important for the binding of oxidized HMGB1 to TLR4 in vitro (36). A separate study proposed that HMGB1 is capable of signaling through RAGE but not TLR4 (37). Because we have previously shown that reduced, but not oxidized HMGB1 blocked tolerance (25), we addressed the receptor mediating HMGB1 action by investigating the tolerogenic response of DCs from various TLR-deficient or Rage−/− mice. We found that HMGB1 was fully functional in the absence of TLR3, -4, -6, or -9 (Fig. 4A). In contrast, full-length HMGB1 and the A-box were unable to block tolerance when Rage−/− DCs were used (Fig. 4B), indicating that RAGE is required to mediate HMGB1 activity. We next measured the affinity of A-box binding to RAGE using label-free SPR. The extracellular region of RAGE is composed of three immunoglobulin-like domains, V, C1, and C2 (Fig. 4C), each with specific affinities to RAGE ligands (38). Saturable, dose-dependent binding of recombinant A-box to three dextran-immobilized RAGE constructs (Fig. 4D, bottom) revealed similar association/dissociation kinetics and low nanomolar affinities, indicating that the A-box binds to RAGE via the V-C1 region. The A-box and sRAGE interaction was confirmed in the reverse orientation (not shown). To identify the region(s) within the caspase-1-generated A-box responsible for RAGE binding, we generated synthetic peptides of overlapping sequences spanning the A-box (Fig. 4D, top). Specific dose-dependent binding yielded equilibrium dissociation constant (KD) values in the low micromolar range in three of the peptides: one corresponding to the A-box N-terminal region (amino acids 1–22) and two overlapping medial peptides (23–45 and 40–50) (Fig. 4D, bottom). In a DTH assay, activity in reversing tolerance correlated with RAGE affinity in all except peptide 1–22. Peptides 23–45 and 40–50 both efficiently reversed tolerance (Fig. 4E). These results confirm that the HMGB1 A-box domain can bind and signal through RAGE and that the binding region between residues 23 and 50 of HMGB1 A-box constitutes the active site required for RAGE-dependent reversal of tolerance.
FIGURE 4.

A defined region in HMGB1 A-box binds to RAGE extracellular domains to reverse the tolerogenic program of DCs fed apoptotic cells. A, HMGB1 dependent reversal of tolerance does not require TLRs-3, -4, -6, and -9. The assay was carried out as in Fig. 2B using DCs from WT, Tlr3−/−, Tlr4−/−, Tlr6−/−, or Tlr9−/− mice. B, the HMGB1 A-box signals on DCs through RAGE to reverse tolerance. The assay was conducted as in Fig. 2B using DCs from Rage+/+ or Rage−/− mice. C, RAGE is composed of a short cytosolic tail (CT) involved in signal transduction, a transmembrane domain (TM), which anchors the protein to the cell membrane, two constant domains (C1 and C2), and a variable domain (V). sRAGE lacks the CT and TM domains. D, top, caspase-1-generated HMGB1 A-box fragment amino acid sequence and overlapping synthetic peptides used to map binding sites to RAGE. Bottom, representative SPR for recombinant A-box fragment (0–100 nm; 2-fold dilution series) and A-box synthetic peptides (0–10 μm; 2-fold dilution series) titrated over amine-coupled RAGE surfaces (∼250 RU each) in HBS-EP at 50 μl/min (1-min association + 3-min dissociation). Apparent equilibrium dissociation constants (KD) are indicated. N/A = no significant binding. E, A-box activity on reversal of tolerance correlates with a RAGE-binding region between amino acids 23–50. Overlapping synthetic A-box peptides (1 μg/ml) were assayed for activity in reversing tolerance as in Fig. 2B; recombinant HMGB1 was used as a positive control.
Prevention of Apoptosis-induced Tolerance in DCs by HMGB1 or A-box Rescues Septic Mice from Secondary Infection
The loss of DTH in sepsis patients has been replicated in animal models of severe sepsis such as cecal ligation and puncture. Here we show that a minimally invasive model of severe sepsis induced by administration of CS (29) effectively reproduced the loss of DTH observed in sepsis (Fig. 5A). To test whether DCs programmed by HMGB1 or the A-box can effectively modulate immunity in vivo, we utilized a DC-based immunotherapy approach. We show that immunization with DCs treated with HMGB1 rescued the DTH response in mice subjected to the CS model of severe sepsis (Fig. 5B). We then applied a clinically relevant two-hit sepsis model to address whether this DC-based therapy is effective at modulating the outcome of a secondary infection during sepsis. Mice were treated with a sublethal dose of CS 48 h before infection with live C. albicans (Fig. 5C). One day prior to Candida infection, mice were administered DCs loaded with the Candida antigen, Candin, that were also fed with apoptotic cells with or without HMGB1 proteins (FL WT, FL NC, or A-box). DCs fed apoptotic cells and Candin alone promoted tolerance to Candida such that the subsequent Candida infection was lethal (Fig. 5D). However, in the presence of caspase-1-sensitive HMGB1 (WT) or the caspase-1-generated HMGB1 A-box, the DC tolerogenic program was reversed and animals survived (Fig. 5D). DCs treated with the caspase-1-resistant HMGB1 form (NC) failed to modulate mouse survival (Fig. 5D). These findings demonstrate that the immunomodulatory functions of HMGB1-programmed DCs can be applied in vivo to reverse pathological apoptosis-induced immunological tolerance.
FIGURE 5.
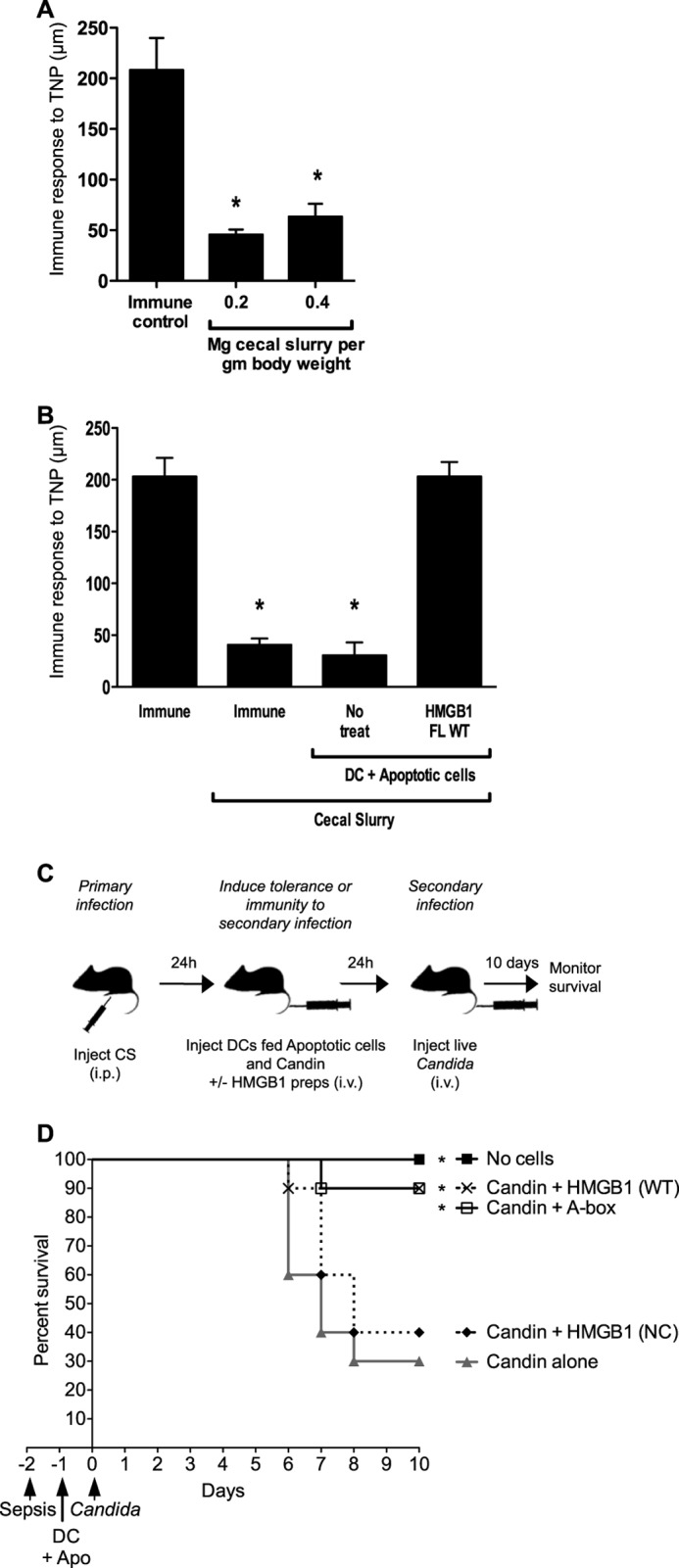
Immunization with HMGB1- or A-box-programmed DCs reverses apoptosis-induced tolerance and rescues septic mice from secondary infection. A, the CS sepsis model induces an immunosuppressed state in mice. Mice were injected with the indicated sublethal doses of CS, and DTH was assayed as a measure of immune competence. Immune control mice were injected with vehicle instead of CS. B, HMGB1 reverses tolerogenicity of DCs exposed to apoptotic cells, which restores DTH in septic immunosuppressed mice. Mice were subjected to a sublethal model of cecal ligation and puncture sepsis and then injected with TNP-conjugated CD8α+ DCs fed apoptotic cells in the absence or presence of recombinant HMGB1. DTH was assayed as in Fig. 2B. C, schematic representation of the Cecal Slurry/Candida two-hit infection model. Mice were rendered immunosuppressed by an initial sublethal intraperitoneal (i.p.) polymicrobial infection (cecal slurry, CS, 0.2 mg/g). 24 h later, mice were injected intravenously (i.v.) with PBS (No Cells) or a suspension of DCs, which were fed apoptotic cells and the C. albicans antigen Candin without (Candin alone) or with HMGB1 preparations. 24 h later, mice were exposed to a secondary infection with C. albicans (5 × 105 CFU/mouse), and survival was monitored for 10 days. Without HMGB1 treatment, the DCs were injected in this system at 24 h to promote tolerance to the subsequent Candida infection, leading to increased mortality despite the dose of Candida administered being sublethal in PBS-injected mice. D, Kaplan-Meier survival curves of mice treated with the two-hit infection model. CD8α+ DCs treated with HMGB1 full-length wild-type (WT) or A-box increase survival to Candida secondary infection, whereas those treated with caspase-1-resistant HMGB1 full-length NC do not. Data were pooled from two independent experiments with similar results. n = 10 mice per group except Candin + HMGB1 (NC) n = 5 mice. Statistically significant differences to Candin alone treatment are denoted by *.
Our findings provide novel insight into HMGB1 biology. Processing of HMGB1 by caspase-1 has to our knowledge never been previously observed and is of significant interest given the important roles these two factors exert in inflammatory settings. This observation warrants further investigation and requires the generation of specialized research tools to confirm biological relevance in physiological context. Caspase-1 appears to convert HMGB1 into an active A-box peptide that signals through RAGE to antagonize apoptosis-induced tolerance. As DCs programmed by this strategy can restore immune function in the immunosuppressed host, a DC-based immunotherapy approach to counteract the immune paralysis of sepsis patients could be explored as a future therapeutic strategy. We also define a direct functional interaction between an active site within the A-box and RAGE, potentially exposing a mechanism by which A-box administration exerts its protective effects during experimental sepsis.
Acknowledgments
We thank Sylvie Perret at BRI for the purification of recombinant HMGB1 proteins, Dr. T. Sam Xiao at the National Institutes of Health for providing the RAGE constructs, Dr. Ann-Marie Schmidt for the Rage−/− mice, Dr. Vishva Dixit for the Nlrp3−/−, mice and Dr. Denise Monack for Aim2−/− mouse femurs. The McGill SPR Facility was supported by the Canada Foundation for Innovation (CFI) and by Canadian Institutes of Health Research (CIHR) research resource grants to the Sheldon Biotechnology Centre for infrastructure support.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR-MOP 79410 and 82801), the Canada Foundation for Innovation (CFI), and the Burroughs Wellcome Fund (to M. S.). This work was also supported by the Foundation for Fighting Blindness (Owings Mills, MD) and from Research to Prevent Blindness (New York, NY).
This article was selected as a Paper of the Week.
- DTH
- delayed-type hypersensitivity
- RAGE
- receptor for advanced glycation end products
- sRAGE
- soluble RAGE
- CS
- cecal slurry
- TNBS
- 2,4,6-trinitrobenzene sulfonic acid
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- MEF
- mouse embryonic fibroblast
- DC
- dendritic cell
- RU
- resonance unit
- TLR
- Toll-like receptor
- NC
- noncleavable
- FL
- full-length
- TNP
- trinitrophenyl.
REFERENCES
- 1. Martin G. S., Mannino D. M., Eaton S., Moss M. (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 2. Hotchkiss R. S., Nicholson D. W. (2006) Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. Immunol. 6, 813–822 [DOI] [PubMed] [Google Scholar]
- 3. Ayala A., Herdon C. D., Lehman D. L., Ayala C. A., Chaudry I. H. (1996) Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood 87, 4261–4275 [PubMed] [Google Scholar]
- 4. Efron P. A., Tinsley K., Minnich D. J., Monterroso V., Wagner J., Lainée P., Lorré K., Swanson P. E., Hotchkiss R., Moldawer L. L. (2004) Increased lymphoid tissue apoptosis in baboons with bacteremic shock. Shock 21, 566–571 [DOI] [PubMed] [Google Scholar]
- 5. Lu M. C., Liu T. A., Lee M. R., Lin L., Chang W. C. (2002) Apoptosis contributes to the decrement in numbers of alveolar macrophages from rats with polymicrobial sepsis. J. Microbiol. Immunol. Infect. 35, 71–77 [PubMed] [Google Scholar]
- 6. Chung C. S., Song G. Y., Lomas J., Simms H. H., Chaudry I. H., Ayala A. (2003) Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J. Leukoc. Biol. 74, 344–351 [DOI] [PubMed] [Google Scholar]
- 7. Wesche-Soldato D. E., Chung C. S., Lomas-Neira J., Doughty L. A., Gregory S. H., Ayala A. (2005) In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood 106, 2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwulst S. J., Muenzer J. T., Peck-Palmer O. M., Chang K. C., Davis C. G., McDonough J. S., Osborne D. F., Walton A. H., Unsinger J., McDunn J. E., Hotchkiss R. S. (2008) Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock 30, 127–134 [DOI] [PubMed] [Google Scholar]
- 9. Adrie C., Bachelet M., Vayssier-Taussat M., Russo-Marie F., Bouchaert I., Adib-Conquy M., Cavaillon J. M., Pinsky M. R., Dhainaut J. F., Polla B. S. (2001) Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am. J. Respir. Crit. Care Med. 164, 389–395 [DOI] [PubMed] [Google Scholar]
- 10. Unsinger J., Kazama H., McDonough J. S., Griffith T. S., Hotchkiss R. S., Ferguson T. A. (2010) Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J. Immunol. 184, 6766–6772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meakins J. L., Christou N. V., Bohnen J., MacLean L. D. (1982) Failure of delayed hypersensitivity skin testing to predict postoperative sepsis and mortality. Br. Med. J. (Clin. Res. Ed.) 285, 1207–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christou N. V., Meakins J. L., Gordon J., Yee J., Hassan-Zahraee M., Nohr C. W., Shizgal H. M., MacLean L. D. (1995) The delayed hypersensitivity response and host resistance in surgical patients. 20 years later. Ann. Surg. 222, 534–546; discussion 546–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lotze M. T., Tracey K. J. (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342 [DOI] [PubMed] [Google Scholar]
- 14. Wang H., Bloom O., Zhang M., Vishnubhakat J. M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L., Manogue K. R., Faist E., Abraham E., Andersson J., Andersson U., Molina P. E., Abumrad N. N., Sama A., Tracey K. J. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 [DOI] [PubMed] [Google Scholar]
- 15. Yang H., Ochani M., Li J., Qiang X., Tanovic M., Harris H. E., Susarla S. M., Ulloa L., Wang H., DiRaimo R., Czura C. J., Wang H., Roth J., Warren H. S., Fink M. P., Fenton M. J., Andersson U., Tracey K. J. (2004) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. U.S.A. 101, 296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li J., Kokkola R., Tabibzadeh S., Yang R., Ochani M., Qiang X., Harris H. E., Czura C. J., Wang H., Ulloa L., Wang H., Warren H. S., Moldawer L. L., Fink M. P., Andersson U., Tracey K. J., Yang H. (2003) Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol. Med. 9, 37–45 [PMC free article] [PubMed] [Google Scholar]
- 17. Andersson U., Wang H., Palmblad K., Aveberger A. C., Bloom O., Erlandsson-Harris H., Janson A., Kokkola R., Zhang M., Yang H., Tracey K. J. (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192, 565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tian J., Avalos A. M., Mao S. Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L. L., Audoly L., La Rosa G., Bierhaus A., Naworth P., Marshak-Rothstein A., Crow M. K., Fitzgerald K. A., Latz E., Kiener P. A., Coyle A. J. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 [DOI] [PubMed] [Google Scholar]
- 19. Sha Y., Zmijewski J., Xu Z., Abraham E. (2008) HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 180, 2531–2537 [DOI] [PubMed] [Google Scholar]
- 20. Youn J. H., Oh Y. J., Kim E. S., Choi J. E., Shin J. S. (2008) High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-α production in human monocytes. J. Immunol. 180, 5067–5074 [DOI] [PubMed] [Google Scholar]
- 21. Hreggvidsdottir H. S., Ostberg T., Wähämaa H., Schierbeck H., Aveberger A. C., Klevenvall L., Palmblad K., Ottosson L., Andersson U., Harris H. E. (2009) The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol. 86, 655–662 [DOI] [PubMed] [Google Scholar]
- 22. Cassetta L., Fortunato O., Adduce L., Rizzi C., Hering J., Rovere-Querini P., Bianchi M. E., Alfano M., Poli G. (2009) Extracellular high mobility group box-1 inhibits R5 and X4 HIV-1 strains replication in mononuclear phagocytes without induction of chemokines and cytokines. AIDS 23, 567–577 [DOI] [PubMed] [Google Scholar]
- 23. Qin Y. H., Dai S. M., Tang G. S., Zhang J., Ren D., Wang Z. W., Shen Q. (2009) HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J. Immunol. 183, 6244–6250 [DOI] [PubMed] [Google Scholar]
- 24. Tang D., Kang R., Zeh H. J., 3rd, Lotze M. T. (2011) High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 14, 1315–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kazama H., Ricci J. E., Herndon J. M., Hoppe G., Green D. R., Ferguson T. A. (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shao W., Yeretssian G., Doiron K., Hussain S. N., Saleh M. (2007) The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 282, 36321–36329 [DOI] [PubMed] [Google Scholar]
- 27. Tole S., Mukovozov I. M., Huang Y. W., Magalhaes M. A., Yan M., Crow M. R., Liu G. Y., Sun C. X., Durocher Y., Glogauer M., Robinson L. A. (2009) The axonal repellent, Slit2, inhibits directional migration of circulating neutrophils. J. Leukoc. Biol. 86, 1403–1415 [DOI] [PubMed] [Google Scholar]
- 28. Myszka D. G. (1999) Improving biosensor analysis. J. Mol. Recognit. 12, 279–284 [DOI] [PubMed] [Google Scholar]
- 29. Wynn J. L., Scumpia P. O., Delano M. J., O'Malley K. A., Ungaro R., Abouhamze A., Moldawer K. L. (2007) Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock 28, 675–683 [DOI] [PubMed] [Google Scholar]
- 30. Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., Zhang J., Lee W. P., Roose-Girma M., Dixit V. M. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 [DOI] [PubMed] [Google Scholar]
- 31. Martinon F., Burns K., Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 [DOI] [PubMed] [Google Scholar]
- 32. Sparatore B., Patrone M., Passalacqua M., Pedrazzi M., Gaggero D., Pontremoli S., Melloni E. (2001) Extracellular processing of amphoterin generates a peptide active on erythroleukaemia cell differentiation. Biochem. J. 357, 569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito T., Kawahara K., Okamoto K., Yamada S., Yasuda M., Imaizumi H., Nawa Y., Meng X., Shrestha B., Hashiguchi T., Maruyama I. (2008) Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler. Thromb. Vasc. Biol. 28, 1825–1830 [DOI] [PubMed] [Google Scholar]
- 34. Hori O., Brett J., Slattery T., Cao R., Zhang J., Chen J. X., Nagashima M., Lundh E. R., Vijay S., Nitecki D., et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270, 25752–25761 [DOI] [PubMed] [Google Scholar]
- 35. Yanai H., Ban T., Wang Z., Choi M. K., Kawamura T., Negishi H., Nakasato M., Lu Y., Hangai S., Koshiba R., Savitsky D., Ronfani L., Akira S., Bianchi M. E., Honda K., Tamura T., Kodama T., Taniguchi T. (2009) HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103 [DOI] [PubMed] [Google Scholar]
- 36. Yang H., Hreggvidsdottir H. S., Palmblad K., Wang H., Ochani M., Li J., Lu B., Chavan S., Rosas-Ballina M., Al-Abed Y., Akira S., Bierhaus A., Erlandsson-Harris H., Andersson U., Tracey K. J. (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U.S.A. 107, 11942–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang D., Kang R., Cheh C. W., Livesey K. M., Liang X., Schapiro N. E., Benschop R., Sparvero L. J., Amoscato A. A., Tracey K. J., Zeh H. J., Lotze M. T. (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leclerc E., Fritz G., Vetter S. W., Heizmann C. W. (2009) Binding of S100 proteins to RAGE: an update. Biochim. Biophys. Acta 1793, 993–1007 [DOI] [PubMed] [Google Scholar]