

Background: Doc toxin, of the phd-doc toxin-antitoxin system, belongs to the Fic protein family found in all domains of life.
Results: Doc inactivates elongation factor Tu by phosphorylation of a single amino acid.
Conclusion: This phosphorylation event inhibits protein synthesis and thereby arrests cell growth.
Significance: The phosphorylation activity of Doc toxin represents a new catalytic activity for members of the Fic protein family.
Keywords: GTPase, Phosphorylation, Post-translational Modification, Protein Synthesis, Translation, Translation Elongation Factors, Fic, Adenylylation, Antitoxin
Abstract
The Doc toxin from bacteriophage P1 (of the phd-doc toxin-antitoxin system) has served as a model for the family of Doc toxins, many of which are harbored in the genomes of pathogens. We have shown previously that the mode of action of this toxin is distinct from the majority derived from toxin-antitoxin systems: it does not cleave RNA; in fact P1 Doc expression leads to mRNA stabilization. However, the molecular triggers that lead to translation arrest are not understood. The presence of a Fic domain, albeit slightly altered in length and at the catalytic site, provided a clue to the mechanism of P1 Doc action, as most proteins with this conserved domain inactivate GTPases through addition of an adenylyl group (also referred to as AMPylation). We demonstrated that P1 Doc added a single phosphate group to the essential translation elongation factor and GTPase, elongation factor (EF)-Tu. The phosphorylation site was at a highly conserved threonine, Thr-382, which was blocked when EF-Tu was treated with the antibiotic kirromycin. Therefore, we have established that Fic domain proteins can function as kinases. This distinct enzymatic activity exhibited by P1 Doc also solves the mystery of the degenerate Fic motif unique to the Doc family of toxins. Moreover, we have established that all characterized Fic domain proteins, even those that phosphorylate, target pivotal GTPases for inactivation through a post-translational modification at a single functionally critical acceptor site.
Introduction
Toxin-antitoxin (TA)4 systems are autoregulated operons composed of tandem genes encoding small (∼10 kDa) antitoxin and toxin proteins that act within the host cell. TA systems can be harbored in bacterial genomes, on extrachromosomal bacterial plasmids, or in bacteriophages. They are associated with, or implicated in, several clinically important phenomena: biofilm formation, bacterial persistence during antibiotic treatment, and bacterial pathogenesis (1–4).
On plasmids, TA systems ensure maintenance through postsegregational killing. For example, after infection the bacteriophage P1 circularizes and enters either a lysogenic or lytic program (5). The P1 phd-doc TA system comprising the Phd (prevents host death) antitoxin (8.1 kDa) and the Doc (death on curing) toxin (13.6 kDa) is involved in maintenance of the P1 prophage as a stable plasmid in response to environmental cues that direct the cell to the lysogenic state. P1 phd-doc has served as the model for the function of all phd-doc TA systems, many of which reside in the chromosomes of pathogens (6, 7). Toward that end, the regulation and general features of the toxin have been studied in detail (8–14); yet the precise mechanism underlying P1 Doc toxicity (referred to as “Doc” from here onward), and thus other family members, is not known.
Existing clues to Doc function were derived from high resolution structures (8, 15, 16) and functional studies (17). However, the activity and intracellular target(s) of Doc are still unknown. Doc exhibits structural similarity to the Fic (filimentation induced by cAMP) family of bacterial proteins (15, 16, 18). Fic proteins contain a conserved catalytic motif, HXFX(D/E)GNGRXXR (19, 20) and typically possess adenylylation (or “AMPylation”) activity (21, 22). By comparison, the consensus for the Fic motif in all Doc toxins is slightly different, HXFX(D/N)(A/G)NKR. Alterations to this motif can result in changes to the enzymatic activity of the protein. In the case of Doc, individual mutations within its Fic motif 66HIFNDANKRTAL77 (H66Y, H66R, D70N, and A76E) result in the loss of toxicity (13).
Understanding the mechanism of action of P1 Doc illuminates the function of the Doc family of toxins as a whole. Because many phd-doc TA systems are harbored in the genomes of pathogenic bacteria, they also serve as attractive antibiotic targets (23). Here we determined that Doc acts in a manner distinct from other characterized Fic domain proteins. Doc is a protein kinase that blocks translation elongation through phosphorylation of Thr-382 of the essential elongation factor EF-Tu. These studies are in agreement with those recently reported by Castro-Roa et al. (24). In addition, we also demonstrated that phosphorylation of EF-Tu alone is sufficient for translation inhibition and that the antibiotic kirromycin prevents phosphorylation by Doc. Finally, our functional data are complemented with molecular models that inform the mechanism of kirromycin-mediated phosphorylation inhibition and provide alternate mechanisms for Doc toxin binding to EF-Tu.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Reagents
The Escherichia coli strains BL21(DE3) (F- ompT hsdSβ(rβ-mβ) dcm gal (DE3) tonA) (Novagen) and BW25113 (lacIq rrnBT14 Δlac-ZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78) were used for all protein expression and toxicity studies. E. coli K-12 Mach1 T1 cells (ΔrecA1398 endA1 tonA Φ80ΔlacM15 ΔlacX74 hsdR(rk+mk+); Invitrogen) were used for all cloning experiments. The pBAD33-doc clone used in this work and our earlier publication (17) contained the doc open reading frame as a PCR-amplified 5′BamHI/HindIII3′ fragment that was cloned into the corresponding sites of pET21c to create pET21c-doc. Plasmid pET21c-doc was digested with 5′XbaI/HindIII3′ and subcloned into pBAD33 to create pBAD33-doc (17). This cloning strategy yields relatively low expression levels of wild type doc. The docH66Y mutant was created by PCR-mediated mutagenesis with 5′BamHI/HindIII3′ ends and cloned into the corresponding sites of pET21c to create pET21c-docH66Y. pBAD33-docH66Y mutant plasmid was created after digestion of pET21c-docH66Y with 5′XbaI/HindIII3′ and cloned into the corresponding sites of pBAD33. The docH66Y open reading frame was PCR-amplified as an 5′NdeI/XhoI3′ fragment and cloned into the corresponding sites of pET21c to create pET21c-docH66Y. The phd open reading frame was PCR-amplified with 5′NdeI/XhoI3′ ends and cloned into the corresponding sites of pET21c to create pET21c-phd. tufAT382A was created by PCR-mediated mutagenesis and cloned into pET21c with 5′NdeI/XhoI3′ ends to create pET21c-tufAT382A. The DNA sequences of PCR fragments used for cloning were confirmed by automated DNA sequence analysis.
Purification of Recombinant Phd-His6 and EF-Tu-His6
Phd Purification
One liter of M9 liquid medium supplemented with 0.21% glycerol, 0.2% (w/v) casamino acids, 1 mm MgSO4, 0.001% (w/v) thiamine, and 100 μg/ml ampicillin was inoculated with pET21c-phd in BL21(DE3) and grown to an A600 of 0.9. The culture was induced with 1 mm IPTG and expressed for 4 h. Cells were subsequently disrupted by sonication and the proteins purified over a nickel-nitrilotriacetic acid resin (Qiagen) as recommended.
EF-Tu Purification
EF-Tu was purified by New England Biolabs (NEB) in the absence of GDP and stored in 50 mm HEPES-KOH, pH 7.6, 100 mm KCl, 10 mm MgCl2, 7 mm β-ME, and 50% glycerol.
EF-Tu T382A Purification
pET21c-tufAT382A was transformed into chemically competent BL21(DE3) cells. Transformants were used to inoculate 500 ml of M9 liquid medium and grown to an A600 of 0.6. Expression of protein was induced by addition of 1 mm IPTG for 4 h. Cells were subsequently disrupted by sonication, and the protein extracts were applied to a nickel-nitrilotriacetic acid column to purify the proteins as recommended by Qiagen.
Purification of Recombinant Doc
BL21 Gold (DE3) pLysS cells (Agilent) containing pET21c-phd-doc were grown at 37 °C in M9 medium supplemented with 0.2% (w/v) casamino acids, 0.4% (w/v) glucose, 1 mm MgSO4, 0.001% (w/v) thiamine, and 200 μg/ml ampicillin. At A600 of 0.5–0.7, expression was induced with 0.4 mm IPTG for 3 h. Cultures were harvested by centrifugation and ruptured by sonication in a lysis buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 5 mm β-ME, 0.1 mm benzamidine, 0.1 mm phenylmethylsulfonyl fluoride (PMSF), and 0.1% Triton X-100. The sonicated mixture was subjected to centrifugation, and the supernatant containing the Phd-Doc-His6 complex was applied to a HisTrapTM FF Crude Ni2+-SepharoseTM column (GE Healthcare) equilibrated with a binding buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 10% (w/v) glycerol, 5 mm β-ME, and 20 mm imidazole. After the lysate was loaded, the column was washed for an additional 20 column volumes (CVs) with binding buffer. Denaturing buffer containing 40 mm Tris-HCl, pH 7.5, and 6 m guanidine-HCl was then applied for 15 CVs to remove Phd antitoxin. Denatured Doc-His6 was on-column refolded by gradually reducing the concentration of guanidine-HCl to 0 mm by exchanging the column solution back to the binding buffer, in 20 CVs over 8 h. Doc-His6 was eluted from the column with elution buffer (40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 10% (w/v) glycerol, 5 mm β-ME, and 300 mm imidazole). Fractions containing Doc-His6 were combined and applied to a Sephadex 75 16/60 gel filtration column (GE Healthcare) with a buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, and 5 mm β-ME.
Purification of Recombinant Doc H66Y-His6
BL21 Gold (DE3) pLysS cells containing pET21c-docH66Y were grown in Lysogeny Broth (LB) medium supplemented with 200 μg/ml ampicillin at 37 °C to an A600 of 0.5–0.7 and induced with 0.4 mm IPTG. Cultures were grown for 3 h, harvested by centrifugation, and ruptured by sonication in a lysis buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 5 mm β-ME, 0.1 mm benzamidine, 0.1 mm PMSF, and 0.1% Triton X-100. The sonicated mixture was centrifuged to obtain the supernatant containing Doc H66Y-His6. The cleared lysate was applied to a HisTrapTM FF Crude Ni2+-Sepharose column equilibrated with a binding buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 10% (w/v) glycerol, 5 mm β-ME, and 20 mm imidazole. After the lysate was loaded, the column was washed for 20 CVs with binding buffer, followed by additional washes (10 CV each) with 1 m KCl followed by 2 m KCl. Doc H66Y-His6 was eluted from the column with elution buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 10% (w/v) glycerol, 5 mm β-ME, and 300 mm imidazole. Fractions containing Doc H66Y-His6 were combined and applied to Sephadex 75 16/60 gel filtration column with a buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, and 5 mm β-ME.
Determination of Doc Target and Activity
The kinase activity of Doc in Fig. 2, A and B, was tested using the complete reaction mixture (solution A + solution B) from the PURExpress kit (NEB). Briefly, reaction mixtures were prepared as specified with or without the addition of 3.8 μm (38 pmol in 10 μl) purified Doc protein. To neutralize toxin activity, 7.5 μm (75 pmol) Phd was preincubated with 3.8 μm (38 pmol) Doc at 37 °C for 15 min. Within the 10-μl reaction volume for each sample, we used 0.5 μl of 1.7 μm γ-32P (6000 Ci/mmol) or α-32P (3000 Ci/mmol). Reactions were incubated at 37 °C for 20 min.
FIGURE 2.

Doc is a protein kinase that targets a single protein in E. coli, it is not an adenylyltransferase. A and B, PURExpress-coupled transcription/translation reactions (containing ∼90 factors required for transcription and translation) with added protein components as shown above each lane were performed with [α-32P]ATP to test for adenylylation activity (5-day exposure) (A) or [γ-32P]ATP to test for kinase activity (5-h exposure) (B). Molecular mass markers (in kDa) are on the left. C, reactions containing [γ-32P]ATP were incubated with E. coli cell lysate (Lysate only) or with lysate supplemented with purified proteins as indicated (18-h exposure). Molecular mass markers (in kDa) are on the left.
The activity of Doc against whole cell lysates was also determined. Whole cell lysates used for Fig. 2C were obtained by growing E. coli BL21(DE3) cells in M9 liquid medium to an A600 of 1.4. The cells were then pelleted and resuspended in 25 ml of 50 mm sodium phosphate monobasic, pH 8.0, 500 mm NaCl, 10 mm imidazole and lysed by sonication. The lysed cells were centrifuged and the supernatant used as cell lysate. The ability of Doc to phosphorylate components of this cell lysate was tested by adding 1.0 μm (30 pmol) wild-type or Doc H66Y to 9.5 μl of cell lysate, 3 μl of 10× reaction buffer, and 2 μl of 0.17 μm [γ-32P]ATP (6000 Ci/mmol). The total reaction volume was brought up to 30 μl, yielding 20 mm Tris-HCl, pH 7.8, 100 mm NaCl, 5 mm MgCl2, 1 mm dithiothreitol (DTT). In lysate plus Phd samples, 2.0 μm (60 pmol) Phd was added in place of Doc. To neutralize toxin activity, Phd and Doc were preincubated at 37 °C for 15 min. Reactions were incubated at 37 °C for 1, 20, and 80 min.
To all samples in experiments described above, at the appropriate time an equal volume of 2× Laemmli buffer was added to terminate the kinase reaction. Samples were heated to 95 °C for 5 min prior to separation on a 17.5% SDS-polyacrylamide gel.
Kinase Assays
Phosphorylation of pure EF-Tu (NEB) was performed in 20 mm Tris-HCl, pH 7.8, 100 mm NaCl, 5 mm MgCl2, 1 mm DTT. 4.0 μm (120 pmol) EF-Tu, 1.0 μm (30 pmol) Doc, and 2.0 μm (60 pmol) Phd were added to the reactions as indicated to a 30-μl reaction. 2 μl of 0.17 μm [γ-32P]ATP (6000 Ci/mmol) was added to all reactions. Phd neutralization of Doc was performed by preincubation of the two proteins at 37 °C for 15 min. Reactions were incubated at 37 °C for 1, 20, and 40 min. At the times indicated, an equal volume of 2× Laemmli buffer was added to terminate the kinase reaction. Samples were heated to 95 °C for 5 min prior to separation on a 17.5% SDS-polyacrylamide gel. Also at the times indicated, 1 μl of sample was removed and added to 14 μl of 4.0 n formic acid for analysis by TLC. Samples were spotted onto PEI-cellulose (EMD Millipore) and resolved using 0.4 m sodium phosphate.
Nucleotide Binding Properties and Kirromycin Treatment of EF-Tu
23 μm EF-Tu was added to a reaction containing 250 μm GDP, GTP, GTPγS, or GMP-PNP in GTP loading buffer (50 mm Tris-HCl, pH 7.8, 7.5 mm EDTA) for 15 min at 37 °C, followed by the addition of MgCl2 to a final concentration of 50 mm. A 10-fold excess of GDP, GTP, or GTP analog was used to favor formation of the desired form of EF-Tu. This preloaded EF-Tu was then used in the reactions described under “Kinase Assays” above. In the kirromycin experiments, kirromycin diluted in HPLC-grade methanol was added to the appropriate reactions to a final concentration of 10 μm. Alexander et al. established that maximal inhibition of EF-Tu phosphorylation was achieved at this concentration (38). In reactions lacking kirromycin an identical amount of methanol was added to make sure that it did not have an effect on the reaction.
Doc Translation Inhibition and Rescue by EF-Tu
To test whether addition of EF-Tu could rescue translation activity after inhibition by Doc (Fig. 5) we used the PURExpress kit following the manufacturer's instructions. Specifically, to the complete reaction mixture (solution A + solution B) Doc was added to 3.8 μm, incubated for 1.5 h, then Phd was added to 7.6 μm for 15 min to inhibit Doc activity. An equal volume of EF-Tu (to 25 μm) or water was added and incubated for 1.75 h to reconstitute translation activity. An equal volume of 2× Laemmli buffer was added to terminate the translation reaction. Samples were heated to 95 °C for 5 min prior to separation on a 17.5% SDS-polyacrylamide gel. Production of the dihydrofolate reductase protein (∼20 kDa) was assessed via [35S]methionine incorporation.
FIGURE 5.
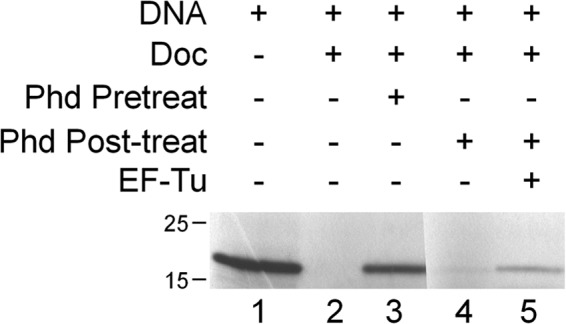
Addition of EF-Tu rescues protein synthesis in an in vitro translation system. Production of the dihydrofolate reductase protein (∼20 kDa) was assessed via [35S]methionine incorporation. Supplements were added as indicated. Doc toxin was either pretreated with Phd antitoxin for 15 min prior to the start of the translation reactions (Phd Pretreat) or Phd antitoxin was added to the reaction after 1 h 45 min of translation and allowed to incubate with reaction components (Phd Post-treat). After 15 min either water or EF-Tu was added to the mixture, and translation was allowed to continue.
Mass Spectrometry
To maximize sequence coverage, three equivalently loaded and excised gel bands of EF-Tu were digested with trypsin, chymotrypsin, or elastase, respectively. Peptide sequence analysis of each digestion mixture was performed by microcapillary reversed-phase high performance liquid chromatography coupled with nanoelectrospray tandem mass spectrometry (μLC-MS/MS) on an LTQ-Orbitrap Velos mass spectrometer (ThermoFisher Scientific). The Orbitrap repetitively surveyed an m/z range from 395 to 1600, whereas data-dependent MS/MS spectra on the 20 most abundant ions in each survey scan were acquired in the linear ion trap. MS/MS spectra were acquired with relative collision energy of 30%, 2.5-Da isolation width, and recurring ions dynamically excluded for 60 s. Preliminary sequencing of peptides was facilitated with the SEQUEST algorithm with a 30 ppm mass tolerance against the Uniprot Knowledgebase E. coli K12 reference proteome supplemented with a database of common laboratory contaminants, concatenated to a reverse decoy database. Using a custom version of Proteomics Browser Suite (PBS v.2.7, ThermoFisher Scientific) peptide-spectrum matches were accepted with mass error <2.5 ppm and score thresholds to attain an estimated false discovery rate of ∼1%. Data sets for all digest results were combined in silico, culled of minor contaminant peptide-spectrum matches, and re-searched with SEQUEST against the EF-Tu sequence without taking into account enzyme specificity and with differential modifications of phosphorylated tyrosine, serine, and threonine residues. The discovery of phosphopeptides and subsequent manual confirmation of their MS/MS spectra were facilitated by in-house versions of programs MuQuest, GraphMod, and FuzzyIons (Proteomics Browser Suite, ThermoFisher Scientific).
Doc–EF-Tu Molecular Modeling
Docking simulations between Doc and EF-Tu were performed by first implementing a Global search using the program GRAMM-X (25) between Doc protein (PDB ID 3K33, chain A) and E. coli EF-Tu from the ternary complex of EF-Tu·tRNA·GTP complex (PDB ID 1OB2, chain A) or EF-Tu·GDP complex (PDB ID 1EFC, chain A). Both EF-Tu starting models were used because there are large conformational changes that occur upon tRNA-GTP binding (26). The simulation was performed for each set independently between two proteins that were arbitrarily placed, however, with the one criterion that EF-Tu Thr-382 and His-66 of Doc protein are in proximity. The models from each major class were then fed into a local docking search program called RosettaDock (27). Independent simulations of 1000 trials were performed, and 10 best scoring structures in rank order by energy were selected.
RESULTS
Doc Is a Kinase
Fic domains (alternately referred to as “Fido” for Fic and Doc (18)) are ∼100–140 amino acids in length and share a conserved α-helical core (15, 18, 20, 28, 29). For most Fic domain proteins, this core is composed of eight α-helices; six are arranged in an up and down bundle with two lying perpendicular to the bundle (18). Doc contains a Fic domain with similar topology but with six α-helices instead of eight (15, 16, 18, 30). The Fic domain in Doc is not only important for toxicity but also for the mRNA stabilization phenotype associated with this toxin. A mutant in the conserved histidine of the Fic domain, H66Y, exhibited significantly reduced toxicity (Fig. 1A) and did not stabilize mRNA (Fig. 1B) (17). Because Doc contains a Fic domain important for its function, and proteins with Fic domains are predicted to adenylylate protein substrates (21, 22), we investigated whether Doc also possessed adenylylation activity as shown for other family members (20, 28, 31–33).
FIGURE 1.

Doc H66Y displays reduced toxicity and mRNA stabilization compared with wild type Doc. A, growth curves of E. coli BW25113 cells expressing wild type Doc (■), Doc H66Y (▴), or empty vector (♦). B, Northern blot analysis of the ompA transcript from E. coli total RNA prepared from cells expressing either empty vector (−Doc), +Doc, or +Doc H66Y for the times shown (in minutes) relative to the corresponding uninduced controls (left).
Since Doc appeared to inhibit translation elongation (17), we reasoned that we should be able to identify adenylylation targets in a coupled in vitro transcription/translation system composed of ∼90 defined components (PURExpress kit). We added either wild type Doc, wild type Doc + Phd, Doc H66Y, or Doc H66Y + Phd to this system plus [α-32P]ATP to identify potential adenylylated proteins. As a control, [γ-32P]ATP was substituted for [α-32P]ATP. No detectable signal was obtained when wild type Doc was added to the samples containing [α-32P]ATP (Fig. 2A, lane 1). However, to our surprise, control samples containing wild type Doc in the presence of [γ-32P]ATP generated a single band migrating above the 40-kDa molecular mass marker (Fig. 2B, lanes 1 and 3). This [γ-32P]ATP signal was specific to Doc toxin and not a contaminant in the recombinant protein preparation because the H66Y catalytic mutant exhibited markedly reduced incorporation (Fig. 2B, lane 3), and preincubation of wild type Doc or Doc H66Y with Phd antitoxin precluded incorporation (Fig. 2B, lanes 2 and 4). We used fresh lots of both radionucleotides and a significantly longer exposure time to compensate for the slightly lower specific activity of [α-32P]ATP (3000 Ci/mmol versus 6000 Ci/mmol for [γ-32P]ATP). Therefore, Doc did not exhibit adenylylation activity under these conditions; yet potent phosphorylation of a single product migrating just above 40 kDa was observed, indicating that Doc is in fact a kinase.
Doc Phosphorylates EF-Tu
We reviewed the molecular mass of each nucleic acid and protein component in the coupled transcription/translation system to help pinpoint the phosphorylation target of Doc. Possible targets included all proteins and nucleic acids required for translation, T7 RNA polymerase, and several accessory proteins required for energy regeneration in this in vitro system (rabbit creatine kinase, yeast myokinase, E. coli nucleotide diphosphate kinase, and E. coli pyrophosphatase). However, only six components (all of which were proteins) comprised the subset whose molecular mass was consistent with the mobility of the band we detected which migrated just above 40 kDa: histidyl-tRNA synthetase, seryl-tRNA synthetase, tyrosyl-tRNA synthetase, EF-Tu, release factor 1, release factor 2, and rabbit creatine kinase. Addition of Doc and [γ-32P]ATP to an E. coli whole cell lysate again resulted in labeling of a single protein at the same mobility just above the 40-kDa marker (Fig. 2C). Therefore, Doc phosphorylated a single E. coli protein, thus excluding rabbit creatine kinase as a possible target.
Of the remaining five potential targets with molecular masses near 40 kDa, EF-Tu was tested first because it was the only GTPase in this subset; bacterial Fic domain proteins that adenylylate mammalian proteins generally target GTPases (20, 28, 31–33). Also, the [γ-32P]ATP-labeled band in the whole cell extract was strong, i.e. visible after a short exposure (Fig. 2C, lanes 7–9), consistent with EF-Tu being one of the most abundant proteins in E. coli (constituting ∼5% of total protein) (34). Recombinant EF-Tu was incubated with wild type Doc in the presence of [γ-32P]ATP. Incorporation of radioactive phosphate into EF-Tu (43 kDa) was almost immediate (Fig. 3A, lanes 10–12). Phosphorylation of EF-Tu by Doc was also confirmed by release of [α-32P]ADP upon thin layer chromatography (Fig. 3B). This activity was not due to the presence of a contaminating kinase because preincubation of Doc with Phd prevented labeling of EF-Tu (Fig. 3A, lanes 13–15). In contrast, under the same conditions the Doc H66Y catalytic mutant was not active (Fig. 3, lanes 19–21). Therefore, because our earlier experiments (Fig. 2, B and C) demonstrated that Doc targets only one ∼ 40-kDa protein in E. coli, EF-Tu is the sole target of Doc toxin.
FIGURE 3.

Doc phosphorylates recombinant EF-Tu. A, recombinant EF-Tu was combined with wild type (WT) or mutant (H66Y) Doc in a reaction buffer containing [γ-32P]ATP as indicated. B, Doc kinase activity was confirmed by release of ADP. Reactions containing [γ-32P]ATP (ATP only) or supplemented with the indicated proteins were analyzed by thin layer chromatography. The migration of ATP and ADP is indicated on the left.
Doc Can Phosphorylate GTP- or GDP-bound EF-Tu
Next we determined whether the presence or identity of a nucleotide bound to EF-Tu influenced phosphorylation by Doc. We preincubated EF-Tu with GDP, GTP, or one of two nonhydrolyzable GTP analogs, GTPγS or GMP-PNP. Doc was able to phosphorylate EF-Tu pretreated with GDP (Fig. 4, lanes 4–6), GTPγS (Fig. 4, lanes 10–12), or GMP-PNP (Fig. 4, lanes 13–15) to approximately the same extent as it was able to phosphorylate untreated EF-Tu (Fig. 4, lanes 1–3). In contrast, pretreatment of EF-Tu with GTP markedly reduced the level of radiolabeled EF-Tu after incubation with Doc (10% relative to untreated EF-Tu; Fig. 4, lanes 7–9). Because Castro-Roa et al. demonstrated that Doc uses both ATP and GTP as a phosphate donor to modify EF-Tu (24), the presence of free GTP in the reaction most likely competes with [γ-32P]ATP and is consistent with the reduction in radiolabeled phosphate on EF-Tu. This is in agreement with studies demonstrating that the AMPylating Fic proteins VopS (28) and MtFic (35) are also able to utilize other nucleotides as substrates.
FIGURE 4.
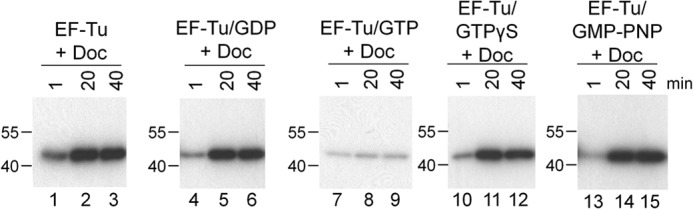
Doc can phosphorylate GTP- or GDP-bound EF-Tu. EF-Tu was incubated with [γ[-32P]ATP and Doc alone (lanes 1–3) or preincubated with GDP (lanes 4–6), GTP (lanes 7–9), GTPγS (lanes 10–12), or GMP-PNP (13–15) prior to the addition of [γ-32P]ATP and Doc. Quantification of the phosphorylated EF-Tu bands indicated that only GTP pretreatment influenced phosphorylation, reducing the signal to 10% of that of untreated EF-Tu. Molecular mass markers (in kDa) are on the left.
Phosphorylation of EF-Tu by Doc Is Sufficient for Translation Inhibition
To determine whether Doc-mediated phosphorylation of EF-Tu could inhibit protein synthesis, we examined the effect of Doc on translation using the same NEB PURExpress in vitro system with all purified components (including EF-Tu) used in Fig. 2. Addition of pure Doc completely inhibited synthesis of the ∼20-kDa dihydrofolate reductase reporter protein (Fig. 5, lane 2), and this inhibition could be prevented by preincubation of Doc with Phd antitoxin (Fig. 5, lane 3). If Phd was added to the reaction mixture after allowing time for Doc to phosphorylate EF-Tu, restoration of dihydrofolate reductase synthesis was not observed (Fig. 5, lane 4). Therefore, addition of Phd cannot reverse EF-Tu phosphorylation; it can only block the enzymatic activity of Doc. However, in a reaction where Doc is allowed to first inhibit translation, then was sequestered by treatment with Phd, addition of fresh EF-Tu partially restored translation (Fig. 5, lane 5).
Doc Inactivates EF-Tu by Phosphorylation at Thr-382
To identify the residue(s) of EF-Tu that is(are) phosphorylated, we incubated EF-Tu with Doc and unlabeled ATP, isolated EF-Tu, digested with multiple enzymes as described under “Experimental Procedures” and subjected the resulting peptides to microcapillary reverse-phase HPLC nanoelectrospray MS/MS. Sample analysis resulted in 100% protein coverage and yielded a single EF-Tu phosphorylation site in this Doc-treated sample at Thr-382 (Fig. 6, A and B). In support of our mass spectrometry data, we demonstrated that Doc was unable to phosphorylate a T382A EF-Tu mutant (Fig. 6C, lanes 10–12). Therefore, Doc is a kinase that inactivates EF-Tu at a highly conserved threonine at the C-terminal end, not at the switch I or II region of GTPases as for other Fic domain proteins (28, 31, 33, 36).
FIGURE 6.

Doc phosphorylates EF-Tu at Thr-382. A, electrospray ionization MS/MS spectra of the doubly charged precursor 640.8226 of the phosphorylated peptide EGGRTVGAGVVAK. The b and y ions are marked on the MS/MS spectra. A superscripted o (bo) indicates a b ion with a neutral loss of water. 2+ indicates a double charged b or y ion. B, scale schematic of EF-Tu highlighting the Doc phosphorylation site (red with asterisk) and flanking sequence; NH2-terminal switch regions and P-loop in blue. C, recombinant Doc combined with EF-Tu or EF-Tu T382A in reaction buffer containing [γ-32P]ATP as indicated (20-min exposure). No signal was observed with the mutant EF-Tu even after a 2-week exposure. Molecular mass markers (in kDa) are on the left.
Kirromycin Inhibits Phosphorylation of EF-Tu by Blocking Access to Doc
Twenty years ago, an enzymatic activity associated with ribosomal fractions of E. coli S15 extracts was shown to phosphorylate EF-Tu at Thr-382 (37). However, the kinase responsible for EF-Tu phosphorylation at Thr-382 had not been identified nor had its phosphorylation been linked to any phenotype in vivo (38). Nevertheless, this report revealed some mechanistic insight upon pretreatment of the S15 extract containing EF-Tu with kirromycin. This antibiotic, which inhibits the release of EF-Tu·GDP from the ribosome (39), prevented phosphorylation of EF-Tu by the S15 extract (38).
To determine whether EF-Tu phosphorylation by Doc was also inhibited, we preincubated EF-Tu with kirromycin prior to the addition of Doc. We observed complete inhibition of phosphorylation by kirromycin (Fig. 7A, lanes 7–9), analogous to the effect observed with the EF-Tu kinase activity identified in the S15 extract (38). X-ray crystal structures show that kirromycin binds EF-Tu at the cleft formed between domain 1 (also known as the G-domain) and domain 3, in proximity to Thr-382 in the EF-Tu·GNP·tRNA complex (PDB ID 1OB2). This indicates that when kirromycin is bound, EF-Tu phosphorylation by Doc is presumably inhibited by blocking access to EF-Tu Thr-382.
FIGURE 7.

Kirromycin prevents EF-Tu phosphorylation by Doc. A, EF-Tu preincubated in the presence or absence of kirromycin (Kir) in reaction buffer prior to the addition of [γ-32P]ATP alone or [γ-32P]ATP and Doc. Molecular mass markers (in kDa) are on the left. B, GDP-bound form of EF-Tu (domain 1, blue; domain 2, green; domain 3, teal; PDB ID 1EFC) with Doc (gray, PDB ID 3K33) at the proposed binding site determined using a protein-protein docking simulation with the program GRAMM-X followed by RosettaDock. GDP is shown in stick representation. Overall view is on the left, and detailed view of phosphorylation site is shown on right with the key residues (EF-Tu Thr-382 and Doc His-66) as sticks. C, GTP-bound form of EF-Tu (domain 1, blue; domain 2, green; domain 3, teal; PDB ID 1OB2) with Doc (gray, PDB ID 3K33) at the two different proposed binding sites as determined by the aforementioned programs. Overall presentation is shown in the middle, detailed views on left and right corresponding to each of the proposed binding sites. A tRNAPhe model (black/orange, PDB ID 1OB2) is also shown for a reference purpose. GNP, a GTP analog, as well as key EF-Tu and Doc residues are also shown as sticks. Kirromycin (yellow, shown as ball and sticks) binds EF-Tu near Thr-382 (PDB ID 1OB2).
Molecular Modeling of the EF-Tu/Doc Interaction
Global protein-protein docking simulations were performed with the program GRAMM-X (25) using Doc ((8) PDB ID 3K33) and EF-Tu from the ternary complex ((26) PDB ID 1OB2). This was followed by a local docking search using RosettaDock (27). The first set (Doc + EF-Tu·GDP) returned models at a structurally identical location where domain 1 and domain 2 of EF-Tu form a cleft (Fig. 7B). For the second set (Doc + EF-Tu·tRNA·GTP), 10 possible models were obtained, where 7 showed Doc placed at the surface where tRNA binds (Fig. 7C, model A, left panel) and 3 on the opposite side close to where kirromycin is located (Fig. 7C, model B, right panel). The functional implications of these models are addressed under “Discussion.”
DISCUSSION
Unlike all other proteins containing a Fic domain, the P1 Doc toxin is a kinase. Doc belongs to a Fic domain protein family whose members share an α-helical fold ranging from ∼100 to 140 amino acids in length (15, 18, 20, 28, 29). However, although the entire Doc toxin (126 amino acids) contains a topology similar to Fic domains, it is composed of six α-helices instead of eight (15, 16, 18, 30). Within this larger conserved structural fold, all Fic domains contain a short (∼10–12 amino acids) highly conserved Fic motif important for catalysis. The Fic motif HXFX(D/N)(A/G)NKR of Doc proteins differs slightly from that in other Fic proteins, HXFX(D/E)GNGRXXR (20). The differences in the sequence and structure of the P1 Doc Fic domain most likely account for its divergent enzymatic activity compared with Fic domains that confer adenylyltransferase activity. By extension, all Doc toxins with this Fic domain are predicted to function as kinases as well.
In canonical Fic proteins, the ATP substrate interacts with the conserved HXFX(D/E)GNGRXXR motif such that its α-phosphate is oriented for attack by the side chain of a target protein (19). The second conserved arginine residue at the end of the Fic motif interacts with the γ-phosphate to establish this orientation (19). This second arginine is missing in the noncanonical Fic motif in Doc, which may promote ATP reorientation in Doc such that its γ-phosphate is amenable to attack by a protein target. In agreement with the prediction of Goepfert et al. (19), the model presented by Castro-Roa et al. places the γ-phosphate of ATP in an inverted orientation relative to AMPylating Fic proteins and in proximity to Thr-382 and the catalytic His-66 of Doc (24). Furthermore, Doc proteins are missing a β-hairpin flap (18). This flap properly positions the target hydroxyl group for reaction with the α-phosphate (19). The absence of this flap may instead allow Doc to orient the target hydroxyl group of EF-Tu toward the γ-phosphate.
Interestingly, a three-dimensional structural search for proteins exhibiting structural similarity to P1 Doc uncovered the Legionella virulence protein AnkX (Dali Z-score 9.1) (40) among the other high scoring Fic family member proteins (41). Although AnkX (949 amino acids) is much larger than P1 Doc (126 amino acids), a pairwise comparison of P1 Doc and AnkX illustrates the significant structural similarity between their Fic domains (Fig. 8, B–D). As in P1 Doc, AnkX contains a degenerate catalytic Fic motif (Fig. 8A) and also does not modify proteins via adenylylation. Instead, AnkX is a type IV effector protein that inactivates a Rab GTPase through addition of phosphocholine to a single serine residue with cytidine diphosphate (CDP)-choline as the donor (36, 42). Therefore, AnkX and P1 Doc do not inactivate GTPases through adenylylation and constitute a separate branch of Fic domain proteins containing noncanonical Fic motifs (Fig. 9).
FIGURE 8.

Doc and AnkX exhibit significant structural similarity. A, schematic of the Fic domains of AnkX (yellow) and P1 Doc (gray) that are modeled in B and C. B, pairwise alignment of Doc (PDB ID 3K33) onto AnkX protein (PDB ID 4BES) by program DaliLite (Z = 9.1, root mean square deviation, 2.9 Å) colored as in A. Close up of the aligned Doc protein with Fic domain of the AnkX (CMD/Ankyrin/Insert domains are removed) is shown on the right. C, comparison of aligned Fic consensus sequence residues of Doc and AnkX. Doc residues 66–74 HIFNDANKR (labeled and underlined) and AnkX residues 229–237 HPFRDANGR (labeled, not underlined) are shown as sticks.
FIGURE 9.
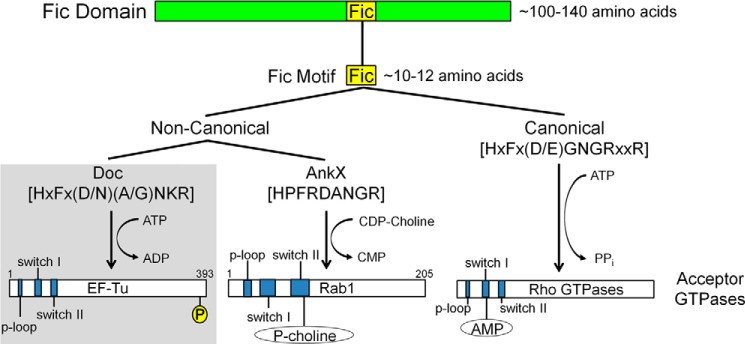
Updated overview of Fic domain proteins. Fic domain proteins are characterized by the presence of a conserved structural fold (green bar). Within this fold lies an ∼10–12-amino acid motif that is important for catalysis (yellow box). Differences in the sequence of the Fic motif allow for the designation of canonical (right branch) and noncanonical (left branch) Fic motifs. Canonical motifs are found in proteins with adenylylation activity whereas noncanonical motifs are found in proteins with other enzymatic activities. Noncanonical Fic motifs fall into two categories based on their enzymatic activities. P1 Doc (and by extension, other Doc toxins) phosphorylate GTPases at the C terminus; AnkX modifies Rab GTPase with phosphocholine. Respective Fic motif consensus sequences are bracketed. Important functional regions of the acceptor GTPases are shown in blue. P, phosphate; P-choline, phosphocholine; AMP, adenosine monophosphate. Diagrams are not drawn to scale.
Although the enzymatic activities of P1 Doc and AnkX are distinct from the Fic domain family members that adenylylate, all Fic domain proteins characterized to date are unified by three hallmark features. First, all Fic domain proteins target the same class of enzymes, GTPases. Second, all Fic domain proteins post-translationally modify a single, pivotal amino acid on the target GTPase through covalent addition of adenosine monophosphate, phosphocholine, or phosphate (as demonstrated in this work). Third, the modification event always inactivates the function of the target protein.
Despite these similarities, there are two important differences between Doc and other Fic domain proteins. First, all Doc toxins act inside the bacterial cell that synthesizes them and are activated only upon antitoxin release (11, 17, 43). In contrast, bacterial proteins containing one or more canonical Fic domains act as virulence factors that are synthesized by the bacterial pathogen and activated only after its delivery to the host cell (20). Interestingly, their activity is kept in check by an inhibitory domain that obstructs ATP binding (20). However, the molecular details that enable these virulence factors to override inhibition and stimulate ATP binding and adenylylation in the host cell are not yet known. Unlike canonical Fic domain proteins, Doc does not contain an inhibition domain. Instead, the inhibitory domain for Doc resides on the Phd antitoxin (20). The second distinguishing feature between Doc and other Fic domain proteins involves the precise site of modification as Doc targets a different domain of GTPases. All canonical Fic domain proteins inactivate GTPases through adenylylation at a single site within one of two conserved switch regions. P1 Doc inactivates EF-Tu by phosphorylation at a single essential amino acid near its C terminus, and not in a designated switch region. Therefore, P1 Doc inactivates its GTPase target in a manner that is mechanistically unique.
It is important to emphasize that not all TA system toxins containing a Fic motif phosphorylate their target. Instead, their enzymatic activity is dependent on the type of Fic motif they contain. Toxins containing a canonical Fic motif exhibit adenylyltransferase activity and are not kinases. For example, the recently characterized VbhT toxin of the VbhA/VbhT TA system from the pathogen Bartonella schoenbuchensis contains a canonical Fic motif. As with other Fic domain virulence factors, it is an adenylyltransferase that is transported out of the bacterial cell and modifies a host protein (20). Therefore, the physiological role of P1 Doc and other Doc family members is distinct from other TA system toxins containing a canonical Fic motif.
P1 Doc inhibits translation and ultimately kills bacterial cells through inactivation of EF-Tu, an essential elongation factor and the central player in tRNA selection during protein synthesis. EF-Tu is composed of three distinct domains; Thr-382 resides in domain 3 whereas three other functionally significant regions (the P-loop, switch I, and switch II) reside in domain 1 (Fig. 6B). Although the primary focus has been on the P-loop and switches I and II because they are required for GTP hydrolysis, targeting the region comprising Thr-382 for phosphorylation or with small molecule inhibitors also serves as a powerful on-off switch for growth control (37, 38, 44). Thr-382 is highly conserved among bacterial EF-Tu proteins (38) and resides in the interface between domains 1 and 3 (45–47). Therefore, EF-Tu inactivation through a single phosphorylation event would serve as a highly effective conduit for regulation of growth.
EF-Tu brings aminoacylated tRNAs to the ribosome A site as a ternary complex consisting of EF-Tu·GTP·aminoacylated tRNA. Upon anticodon-codon recognition by the ribosome, a series of conformational changes is transmitted from the 30S A site to EF-Tu via the A-site tRNA that enables EF-Tu to undergo GTP hydrolysis (48). Dissociation of EF-Tu from the ribosome then occurs, allowing the tRNA to resume a relaxed position into the A site of both the small and large subunit, a process termed “accommodation” (49, 50). Kirromycin blocks the conformational change in EF-Tu from its GTP-bound form to its GDP-bound form and thus prevents disassociation of EF-Tu from the ribosome (44). Treatment of recombinant EF-Tu with kirromycin prevented phosphorylation by Doc (Fig. 7A).
Protein-protein docking simulations between Doc and both GDP- and GTP-bound forms of EF-Tu returned three models with reasonable contact surface area between Doc and EF-Tu (800–1400 Å2) (Fig. 7, B and C). Because EF-Tu adopts different conformations depending on whether a nucleotide or tRNA is bound (26), the modeling experiments were performed with both EF-Tu forms. This is important because the accessibility of Thr-382 also changes in the different EF-Tu states. Upon modeling Doc with the GDP-bound form of EF-Tu, Doc binds at a single site at the cleft between EF-Tu domains 1 and 2 (Fig. 7B). This location is consistent with what was proposed recently by Castro-Roa et al. based upon low resolution small angle x-ray scattering and subsequent modeling experiments (24). This Doc binding site overlaps with the canonical tRNA binding site. Therefore, one interpretation indicates that when EF-Tu is in the context of the ternary complex, Thr-382 is sterically blocked from Doc access.
When we modeled Doc with EF-Tu in a GTP-bound state, we found there were two possible binding sites for Doc (Fig. 7C). In model A, the position of Doc also overlays with the normal tRNA position (Fig. 7C, left panel). If Doc were to compete with the tRNA binding pocket on EF-Tu, the loop preceding the β6 in domain 3 would block Doc His-66 access to Thr-382 (Fig. 7C, left). This model appears to be less likely given that EF-Tu is rarely found bound to GTP without tRNA. The second proposed Doc binding site is on the opposite side of EF-Tu domain 3 and the tRNA binding site (model B, Fig. 7C, right). In this orientation, Thr-382 is accessible for Doc phosphorylation (Fig. 7C, right). This is also close to where kirromycin binds, which may sterically block Doc access to Thr-382 and thus prevent EF-Tu phosphorylation.
Our biochemical data show that the nucleotide binding state of EF-Tu has no effect on the extent of Doc phosphorylation (Fig. 4). Previous experiments established that EF-Tu Thr-382 phosphorylation prevents ternary complex formation (38). Whether the ternary complex is a Doc target remains to be directly tested. Regardless, the results presented here indicate that Doc has ample opportunity to modify multiple EF-Tu states to efficiently halt protein synthesis and cease cell growth.
This work opens up many exciting avenues for exploration. First, although the noncanonical Fic motif and shorter Fic domain fold shared among all bacterial Doc family toxins suggest that all possess kinase activity, the universality of this enzymatic activity remains to be investigated. Also, because the presence of serine/threonine phosphatases has been established in many bacteria (51), it is possible that this modification is specifically removed by a phosphatase. If so, the dynamic of the phosphatase and kinase activity would add a new dimension to the regulation of toxin activity in TA systems. Second, as with all other Fic domain proteins, P1 Doc targets a GTPase. However, it is unclear whether the targeting of EF-Tu is unique to this Doc toxin or whether this highly effective mechanism of growth control is enlisted by all Doc family members. Even if other Doc toxins target EF-Tu, the switch I or II region may instead be favored over Thr-382. Finally, although kirromycin treatment of EF-Tu suggested that Thr-382 phosphorylation may mimic the effects of this antibiotic, ongoing structural studies will provide more insight into the precise mechanism by which Doc inhibits elongation through EF-Tu phosphorylation.
Acknowledgments
We thank Herbert Weissbach for numerous insightful discussions and expert guidance, Smita Patel and Anand Ramanathan for extensive feedback and expertise, and Jason Schifano for comments on the manuscript and assistance with figure preparation.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM093279 (to C. M. D.) and R01GM095693 (to N. A. W.) from the NIGMS, National Science Foundation CAREER Award MCB 0953714 (to C. M. D.), and National Institutes of Health Training Grant T32AI007403, Virus-Host Interactions in Eukaryotic Cells (to J. W. C. and F. P. R., awarded to G. Brewer), from the NIAID.
- TA
- toxin-antitoxin
- β-ME
- β-mercaptoethanol
- CV
- column volume
- GMP-PNP
- guanosine 5′-[β,γ-imido]triphosphate
- GNP
- phosphoaminophosphonic acid-guanylate ester
- GTPγS
- guanosine 5′-[γ-thio]triphosphate
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- MS/MS
- tandem mass spectrometry
- PDB
- Protein Data Bank.
REFERENCES
- 1. Dawson C. C., Intapa C., Jabra-Rizk M. A. (2011) “Persisters”: survival at the cellular level. PLoS Pathogens 7, e1002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerdes K., Maisonneuve E. (2012) Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66, 103–123 [DOI] [PubMed] [Google Scholar]
- 3. Kint C. I., Verstraeten N., Fauvart M., Michiels J. (2012) New-found fundamentals of bacterial persistence. Trends Microbiol. 20, 577–585 [DOI] [PubMed] [Google Scholar]
- 4. Wang X., Wood T. K. (2011) Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Łobocka M. B., Rose D. J., Plunkett G., 3rd, Rusin M., Samojedny A., Lehnherr H., Yarmolinsky M. B., Blattner F. R. (2004) Genome of bacteriophage P1. J. Bacteriol. 186, 7032–7068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makarova K. S., Wolf Y. I., Koonin E. V. (2009) Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pandey D. P., Gerdes K. (2005) Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R. D., Charlier D., van Nuland N. A., Loris R. (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142, 101–111 [DOI] [PubMed] [Google Scholar]
- 9. Gazit E., Sauer R. T. (1999) The Doc toxin and Phd antidote proteins of the bacteriophage P1 plasmid addiction system form a heterotrimeric complex. J. Biol. Chem. 274, 16813–16818 [DOI] [PubMed] [Google Scholar]
- 10. Gazit E., Sauer R. T. (1999) Stability and DNA binding of the Phd protein of the phage P1 plasmid addiction system. J. Biol. Chem. 274, 2652–2657 [DOI] [PubMed] [Google Scholar]
- 11. Lehnherr H., Yarmolinsky M. B. (1995) Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 3274–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Magnuson R., Lehnherr H., Mukhopadhyay G., Yarmolinsky M. B. (1996) Autoregulation of the plasmid addiction operon of bacteriophage P1. J. Biol. Chem. 271, 18705–18710 [DOI] [PubMed] [Google Scholar]
- 13. Magnuson R., Yarmolinsky M. B. (1998) Corepression of the P1 addiction operon by Phd and Doc. J. Bacteriol. 180, 6342–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McKinley J. E., Magnuson R. D. (2005) Characterization of the Phd repressor-antitoxin boundary. J. Bacteriol. 187, 765–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arbing M. A., Handelman S. K., Kuzin A. P., Verdon G., Wang C., Su M., Rothenbacher F. P., Abashidze M., Liu M., Hurley J. M., Xiao R., Acton T., Inouye M., Montelione G. T., Woychik N. A., Hunt J. F. (2010) Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 18, 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia-Pino A., Christensen-Dalsgaard M., Wyns L., Yarmolinsky M., Magnuson R. D., Gerdes K., Loris R. (2008) Doc of prophage P1 is inhibited by its antitoxin partner Phd through fold complementation. J. Biol. Chem. 283, 30821–30827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu M., Zhang Y., Inouye M., Woychik N. A. (2008) Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc. Natl. Acad. Sci. U.S.A. 105, 5885–5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kinch L. N., Yarbrough M. L., Orth K., Grishin N. V. (2009) Fido, a novel AMPylation domain common to Fic, Doc, and AvrB. PloS One 4, e5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goepfert A., Stanger F. V., Dehio C., Schirmer T. (2013) Conserved inhibitory mechanism and competent ATP binding mode for adenylyltransferases with Fic fold. PloS One 8, e64901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engel P., Goepfert A., Stanger F. V., Harms A., Schmidt A., Schirmer T., Dehio C. (2012) Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110 [DOI] [PubMed] [Google Scholar]
- 21. Woolery A. R., Luong P., Broberg C. A., Orth K. (2010) AMPylation: something old is new again. Front. Microbiol. 1, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itzen A., Blankenfeldt W., Goody R. S. (2011) Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem. Sci. 36, 221–228 [DOI] [PubMed] [Google Scholar]
- 23. Williams J. J., Hergenrother P. J. (2012) Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 20, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castro-Roa D., Garcia-Pino A., De Gieter S., van Nuland N. A., Loris R., Zenkin N. (2013) The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat. Chem. Biol. 9, 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tovchigrechko A., Vakser I. A. (2006) GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, W310–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nissen P., Kjeldgaard M., Thirup S., Polekhina G., Reshetnikova L., Clark B. F., Nyborg J. (1995) Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science 270, 1464–1472 [DOI] [PubMed] [Google Scholar]
- 27. Lyskov S., Gray J. J. (2008) The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 36, W233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mattoo S., Durrant E., Chen M. J., Xiao J., Lazar C. S., Manning G., Dixon J. E., Worby C. A. (2011) Comparative analysis of Histophilus somni immunoglobulin-binding protein A (IbpA) with other Fic domain-containing enzymes reveals differences in substrate and nucleotide specificities. J. Biol. Chem. 286, 32834–32842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao J., Worby C. A., Mattoo S., Sankaran B., Dixon J. E. (2010) Structural basis of Fic-mediated adenylylation. Nat. Struct. Mol. Biol. 17, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goepfert A., Harms A., Schirmer T., Dehio C. (2013) in Prokaryotic Toxin-Antitoxins (Gerdes K., ed) pp. 177–187, Sorubger-Verlag, Heidelberg [Google Scholar]
- 31. Yarbrough M. L., Li Y., Kinch L. N., Grishin N. V., Ball H. L., Orth K. (2009) AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272 [DOI] [PubMed] [Google Scholar]
- 32. Palanivelu D. V., Goepfert A., Meury M., Guye P., Dehio C., Schirmer T. (2011) Fic domain-catalyzed adenylylation: insight provided by the structural analysis of the type IV secretion system effector BepA. Protein Sci. 20, 492–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Worby C. A., Mattoo S., Kruger R. P., Corbeil L. B., Koller A., Mendez J. C., Zekarias B., Lazar C., Dixon J. E. (2009) The Fic domain: regulation of cell signaling by adenylylation. Mol. Cell 34, 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weijland A., Harmark K., Cool R. H., Anborgh P. H., Parmeggiani A. (1992) Elongation factor Tu: a molecular switch in protein biosynthesis. Mol. Microbiol. 6, 683–688 [DOI] [PubMed] [Google Scholar]
- 35. Mishra S., Bhagavat R., Chandra N., Vijayarangan N., Rajeswari H., Ajitkumar P. (2012) Cloning, expression, purification, and biochemical characterisation of the FIC motif containing protein of Mycobacterium tuberculosis. Protein Expression Purification 86, 58–67 [DOI] [PubMed] [Google Scholar]
- 36. Mukherjee S., Liu X., Arasaki K., McDonough J., Galán J. E., Roy C. R. (2011) Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lippmann C., Lindschau C., Vijgenboom E., Schröder W., Bosch L., Erdmann V. A. (1993) Prokaryotic elongation factor Tu is phosphorylated in vivo. J. Biol. Chem. 268, 601–607 [PubMed] [Google Scholar]
- 38. Alexander C., Bilgin N., Lindschau C., Mesters J. R., Kraal B., Hilgenfeld R., Erdmann V. A., Lippmann C. (1995) Phosphorylation of elongation factor Tu prevents ternary complex formation. J. Biol. Chem. 270, 14541–14547 [DOI] [PubMed] [Google Scholar]
- 39. Wolf H., Chinali G., Parmeggiani A. (1974) Kirromycin, an inhibitor of protein biosynthesis that acts on elongation factor Tu. Proc. Natl. Acad. Sci. U.S.A. 71, 4910–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 42. Tan Y., Arnold R. J., Luo Z. Q. (2011) Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc. Natl. Acad. Sci. U.S.A. 108, 21212–21217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gerdes K., Rasmussen P. B., Molin S. (1986) Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. U.S.A. 83, 3116–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parmeggiani A., Swart G. W. (1985) Mechanism of action of kirromycin-like antibiotics. Annu. Rev. Microbiol. 39, 557–577 [DOI] [PubMed] [Google Scholar]
- 45. Berchtold H., Reshetnikova L., Reiser C. O., Schirmer N. K., Sprinzl M., Hilgenfeld R. (1993) Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature 365, 126–132 [DOI] [PubMed] [Google Scholar]
- 46. Kjeldgaard M., Nissen P., Thirup S., Nyborg J. (1993) The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure 1, 35–50 [DOI] [PubMed] [Google Scholar]
- 47. Kjeldgaard M., Nyborg J. (1992) Refined structure of elongation factor EF-Tu from Escherichia coli. J. Mol. Biol. 223, 721–742 [DOI] [PubMed] [Google Scholar]
- 48. Voorhees R. M., Schmeing T. M., Kelley A. C., Ramakrishnan V. (2010) The mechanism for activation of GTP hydrolysis on the ribosome. Science 330, 835–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hopfield J. J. (1974) Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. U.S.A. 71, 4135–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ninio J. (1975) Kinetic amplification of enzyme discrimination. Biochimie 57, 587–595 [DOI] [PubMed] [Google Scholar]
- 51. Pereira S. F., Goss L., Dworkin J. (2011) Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75, 192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]