

Background: The neonatal Fc receptor (FcRn) regulates the serum half-life of human IgGs and serum albumin (SA).
Results: Structures of human FcRn bound to its two ligands shed new lights on the corresponding interactions.
Conclusion: Molecular mechanisms for IgG/SA recycling and increased IgG/FcRn binding affinity are presented.
Significance: Understanding the molecular basis of the IgG/SA/FcRn interaction is crucial to develop biologics exhibiting beneficial pharmacokinetics properties.
Keywords: Albumin, Antibodies, Crystal Structure, FC Receptors, Protein Complexes
Abstract
We report the three-dimensional structure of human neonatal Fc receptor (FcRn) bound concurrently to its two known ligands. More particularly, we solved the crystal structure of the complex between human FcRn, wild-type human serum albumin (HSA), and a human Fc engineered for improved pharmacokinetics properties (Fc-YTE). The crystal structure of human FcRn bound to wild-type HSA alone is also presented. HSA domain III exhibits an extensive interface of contact with FcRn, whereas domain I plays a lesser role. A molecular explanation for the HSA recycling mechanism is provided with the identification of FcRn His161 as the only potential direct contributor to the corresponding pH-dependent process. At last, this study also allows an accurate structural definition of residues considered for decades as important to the human IgG/FcRn interaction and reveals Fc His310 as a significant contributor to pH-dependent binding. Finally, we explain various structural mechanisms by which several Fc mutations (including YTE) result in increased human IgG binding to FcRn. Our study provides an unprecedented relevant understanding of the molecular basis of human Fc interaction with human FcRn.
Introduction
The neonatal Fc receptor (FcRn)2 plays a central role in the regulation of IgG and serum albumin (SA) levels in mammals. FcRn comprises β2-microglobulin and a membrane-anchored α-chain related to the α-chain of the class I major histocompatibility complex (1–3). It interacts in a pH-dependent fashion with SA or IgG molecules so that the strength of the corresponding interactions ranges from highest at acidic pH to lowest around neutral pH (4–8). This remarkable property is critical to recycling IgG and SA from within acidic endosomes back to the general circulation and allows their rescue from a degradative pathway (7–10), thereby prolonging their serum half-life.
Efforts to probe or optimize the corresponding interfaces have yielded several noteworthy results. For instance, a particular set of substitutions known as YTE (M252Y/S254T/T256E) resulted in an ∼10-fold pH-dependent increase in the binding of human(ized) IgGs to FcRn. Such a modified humanized IgG exhibited an ∼2–4- and 4-fold increase in its serum half-life in human and cynomolgus monkeys, respectively (11, 12). Other studies with human serum albumin (HSA) used modeling and crystallographic approaches coupled with mutagenesis to identify several important HSA residues, and pinpointed the crucial role played by its domain III (13, 14).
Here, we set out to solve the x-ray crystal structure of human FcRn bound to wild-type HSA alone, as well as to both wild-type HSA and Fc-YTE. We report diffracting crystals for each complex and have solved the three-dimensional structure of both. Of note, the only HSA·FcRn complex three-dimensional structure described so far involves a quadruple HSA mutant whose affinity for FcRn is increased by ∼300-fold (14). The crystal structures of human FcRn alone, or rat FcRn alone or in complex with rat Fc have also been determined (2, 3, 15, 16). However, structures of human FcRn bound to a human Fc or to wild-type HSA have yet to be elucidated. Our study not only provides these data for the first time, but also allows a view of FcRn bound concurrently to both of its known ligands. This work allows a more complete and relevant understanding of the molecular basis by which HSA and human IgG interact with human FcRn and are recycled in a pH-dependent manner. This fills an important knowledge gap in the field of IgG and wild-type HSA recycling in human, the understanding of which has long been limited to structural modeling, mutational approaches, and assumptions drawn from studies using rodent molecules. We also provide a detailed description of the molecular mechanisms by which YTE and other known mutations increase IgG binding affinity to FcRn. Our data should prove very useful in providing clues to further optimize IgG/FcRn and HSA/FcRn interactions and develop the next generation of IgG- or HSA-based therapeutics. These pharmacokinetically improved molecules could potentially provide many benefits, such as the ability to decrease their administration frequency or dosing requirements.
EXPERIMENTAL PROCEDURES
Conventions and Reagents
All antibody amino acid positions mentioned in the text are identified according to the EU numbering convention (17). Human embryonic kidney (HEK) 293 cells were obtained from ATCC.
Expression and Purification of Fc-YTE
A human Fc containing the M252Y/S254T/T256E (“YTE”) mutations was expressed in HEK 293 cells and purified as previously described (18). Purified Fc-YTE was further dialyzed overnight at 4 °C against 50 mm NaOAc, pH 5.2, 100 mm NaCl and concentrated to ∼4 mg/ml. This procedure allowed us to recover over 95% homogeneous Fc-YTE (Fig. 1a).
FIGURE 1.
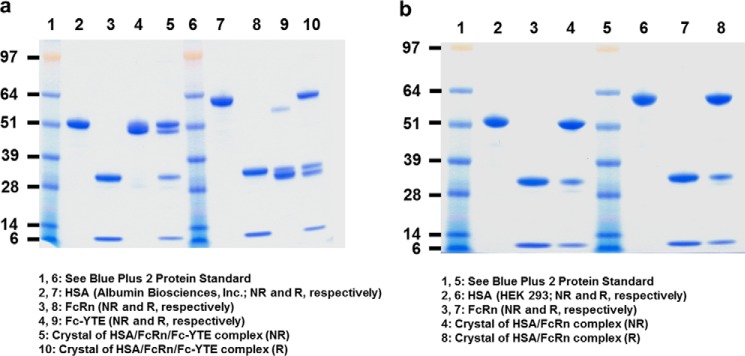
SDS-PAGE profile under reducing (R) and non-reducing (NR) conditions of (a) HSA (Albumin Biosciences, Inc.), FcRn, Fc-YTE and dissolved crystals of the HSA·FcRn·Fc-YTE complex, and (b) HSA (HEK 293-produced), FcRn and dissolved crystals of the HSA·FcRn complex. Although HSA has a predicted molecular mass of ∼65 kDa, it migrated at ∼60 and 50 kDa under R and NR conditions, respectively. This difference is likely attributable to the presence of 17 internal disulfide bonds, the differential reduction of which alters the compactness (and thus the migration) of the molecule. Fc-YTE migrated at ∼30 and 50 kDa under R and NR conditions, respectively, because of the presence of an expected interchain disulfide bond in its hinge region. Fc-YTE under R conditions seemed to run slightly slower than the expected ∼25 kDa. We attribute this difference to the presence of carbohydrates at a canonical N-glycosylation site (Asn297). Under NR conditions, this glycosylation effect is likely counterbalanced by the presence of one inter- and four intra-chain disulfide bonds that increase the compactness of the molecule, thereby accounting for an overall migration at the expected ∼50 kDa. FcRn migrated as expected as two bands corresponding to its α- and β2-microglobulin chains at ∼30 and 10 kDa, respectively (under both R and NR conditions).
Generation and Expression of Human IgG and IgG-YTE Mutants
Human IgG1 and IgG1-YTE (MedImmune) were cloned into a proprietary mammalian expression vector encoding a hybrid Cκ/pelB signal sequence, human cytomegalovirus major immediate early (hCMVie) enhancer, promoter, and 5′-untranslated region. Generation of variants at position 310 was carried out by site-directed mutagenesis using a QuikChange XL mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. HEK 293 cells were transiently transfected with the various IgG constructs using 293Fectin (Invitrogen) and standard protocols. The conditioned media was harvested at 144 h post-transfection.
Expression and Purification of FcRn
Recombinant human FcRn was expressed in HEK 293 cells as previously described (12). FcRn-containing conditioned media was collected at 72, 144, and 216 h post-transfection and pooled. After adjusting to pH 6.0 with 1 m HCl, the media was passed over a IgG-Sepharose column (GE Healthcare) previously equilibrated with 50 mm NaOAc, pH 5.2. Following washes to baseline with the same buffer, FcRn was eluted using 50 mm sodium phosphate, pH 7.8. Fractions containing FcRn were pooled and loaded onto a 5-ml HiTrap Q HP column (GE Healthcare) previously equilibrated with 50 mm Tris-HCl, pH 8.0, and eluted in a 0–0.5 m NaCl gradient. Purified FcRn was dialyzed against 50 mm NaOAc, pH 5.2, 100 mm NaCl overnight at 4 °C and concentrated to ∼4 mg/ml. This procedure allowed us to recover over 95% homogeneous FcRn (Fig. 1, a and b).
Generation, Expression, and Purification of HSA Wild-type and Variants
HSA wild-type used for preparing the binary complex was produced recombinantly in-house by cloning the corresponding DNA sequence (corresponding to residues 25–609 of entry number P02768 of the Swiss-Prot database) into a proprietary mammalian expression vector encoding a hybrid Cκ/pelB signal sequence, human cytomegalovirus major immediate early (hCMVie) enhancer, promoter, and 5′-untranslated region. HEK 293 cells were transiently transfected with this HSA construct using 293Fectin (Invitrogen) and standard protocols. The conditioned media was harvested at 144 h post-transfection. Recombinant, yeast-produced wild-type HSA used in the preparation of the ternary complex was purchased directly from Albumin Biosciences, Inc. (Huntsville, AL).
HEK 293- and yeast-produced HSA were then further purified using the same protocol. Briefly, the purchased protein (diluted to ∼5 mg/ml) or HEK 293-conditioned media was titrated into 2.5 m urea for 2 h at 37 °C in an effort to remove bound lipid molecules. The resulting protein solution was then directly applied to a 5-ml HiTrap Q HP column (GE Healthcare) previously equilibrated with 50 mm Tris-HCl, pH 8.0. After washing with the same buffer, HSA was eluted in a 0–1 m NaCl gradient. The purified protein was dialyzed against 50 mm NaOAc, pH 5.2, 100 mm NaCl overnight at 4 °C, and concentrated to ∼4 mg/ml. This procedure allowed us to obtain over 95% homogeneous HSA (Fig. 1, a and b).
HSA variants at position 510 were generated from the corresponding previously described cloned wild-type sequence. Generation of point mutations was carried out by site-directed mutagenesis using a QuikChange XL mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. HEK 293 cells were transiently transfected with the various HSA constructs using 293Fectin (Invitrogen) and standard protocols. The conditioned media was harvested at 144 h post-transfection.
Determination of HSA and IgG pH Dependence of Binding toward Human FcRn
Comparison of the interaction of soluble human FcRn with immobilized HSA wild-type and variants at acidic and neutral pH was carried out using a BIAcore 3000 (Biacore International AB, Uppsala, Sweden) in MES (50 mm MES, 0.15 m NaCl, 3 mm EDTA, 0.005% (v/v) P20, pH 6.0) or HBS-EP (10 mm HEPES, 0.15 m NaCl, 3 mm EDTA, 0.005% (v/v) P20, pH 7.4) buffer. HSA was captured directly from the corresponding conditioned media at a typical surface density of ∼800 RU using an anti-HSA antibody (Abcam, Cambridge, MA) coupled to the dextran matrix of a CM5 sensor chip (GE Healthcare) at a surface density of ∼12,000 RU. Human FcRn was used at concentrations of 1 and 10 μm at pH 6.0 and 7.4, respectively. In all cases, FcRn was also flowed over an uncoated cell, and the sensorgrams from these blank runs were subtracted from those obtained with HSA-coupled cells.
Comparison of the interaction of soluble human FcRn with immobilized human IgG, IgG-YTE, and variants thereof at acidic and neutral pH was carried out essentially as described above. In particular, IgGs were captured directly from the corresponding conditioned media at a typical surface density of ∼800–1,600 RU using a mouse anti-human κ light chain antibody (Abcam) coupled to the dextran matrix of a CM5 sensor chip (GE Healthcare) at a surface density of ∼5,200 RU.
Preparation of HSA·FcRn and HSA·FcRn·Fc-YTE Complexes
Previously purified HSA and FcRn were mixed at a, respectively, 1:1.1 molar ratio to generate the binary complex. The ternary complex was formed by mixing the previously purified HSA, FcRn, and Fc-YTE at, respectively, 1.1:1.1:0.5 molar ratio because of the presence of two potential FcRn binding sites on the Fc moiety. Further purification and buffer exchange for the binary and ternary complexes were carried out using SuperdexTM S75 10/300 GL and S200 10/300 GL columns (GE Healthcare), respectively, pre-equilibrated with 50 mm NaOAc, pH 5.2, 100 mm NaCl. Representative chromatograms of HSA, FcRn, Fc-YTE, and HSA·FcRn and HSA·FcRn·Fc-YTE complexes are shown in Fig. 2, a and b. The purified binary and ternary complexes were then concentrated to 10 and 4 mg/ml, respectively, and subjected to crystallization trials as described in the following section.
FIGURE 2.

Superimposition of the size exclusion chromatograms of (a) HSA (HEK 293-produced), FcRn and HSA/FcRn complex, and (b) HSA (Albumin Biosciences, Inc.), FcRn, Fc-YTE, and HSA·FcRn·Fc-YTE complex.
Crystallization
Sitting drop crystallization experiments were initially set up in 96-well Intelli-plates (Art Robbins Instruments, Sunnyvale, CA) using a Phoenix crystallization robot (Art Robbins Instruments). Favorable conditions were first identified using the following commercially available crystallization screens: Peg/Ion (Hampton Research, Aliso Viejo, CA) and JCSG+ Screen (Molecular Dimensions, Apopka, FL). The well and drop compartments of the 96-well plates were first filled with 50 and 0.3 μl, respectively, of the various screen solutions. 0.3 μl of HSA·FcRn or HSA·FcRn·Fc-YTE complexes at concentrations of 10 and 4 mg/ml, respectively, in 50 mm NaOAc, pH 5.2, 100 mm NaCl was then added to the drop compartment. The resulting mixture was allowed to equilibrate against the well solution at room temperature.
Based on initial screening results for the binary complex, the E9 screen solution from Peg/Ion (4% Tacsimate, pH 4.0, 12% PEG 3350) was selected for further optimization. This step included a previously described seeding procedure (19). More particularly, each well was filled with 45 and 5 μl of the E9 and various Additive solutions (Hampton Research), respectively, and thoroughly mixed. For each condition, a sitting drop was formed by mixing 0.3 μl of the corresponding well solution with 0.3 μl of the HSA·FcRn complex at 10 mg/ml in 50 mm NaOAc, pH 5.2, 100 mm NaCl. 0.05 μl of seed-containing E9 solution was then added. The best condition was found when Additive B9 (2.0 m NaSCN) was used. Further optimization in hanging drops followed using varying drop volumes (1 to 6 μl) and ratios of protein/reservoir solution in the drop (1:1 to 5:1).
Based on initial screening results for the ternary complex, the same E9 screen solution from Peg/Ion (4% Tacsimate, pH 4.0, 12% PEG 3350) was selected for further optimization. This step included the same seeding procedure as for the binary complex. More particularly, each well was filled with 45 and 5 μl of the E9 and various Additive solutions (Hampton Research), respectively, and thoroughly mixed. For each condition, a sitting drop was formed by mixing 0.3 μl of the corresponding well solution with 0.3 μl of the HSA·FcRn·Fc-YTE complex at 4 mg/ml in 50 mm NaOAc, pH 5.2, 100 mm NaCl. 0.05 μl of seed-containing E9 solution was then added. The best condition was found when Additive A9 (0.1 m YCl3) was used.
X-ray Data Collection and Processing
One diffraction dataset from a single crystal was collected at a wavelength of 1.000 Å and temperature of 100 K for the binary complex, at the IMCA-CAT 17ID Beamline of the Advanced Photon Source (APS) of the Argonne National Laboratory (University of Chicago, Chicago, IL) equipped with a PILATUS 6M detector (Paul Sherrer Institute, Villigen, Switzerland). Cryoprotection was achieved by soaking the selected crystals in 0.16 m NaSCN, 2.88% Tacsimate, pH 4.0, 8.64% PEG 3350, 20% glycerol. Crystals were then flash-cooled in liquid nitrogen. We recorded 400 frames using an oscillation range of 0.5°, a crystal to detector distance of 450 mm, and an exposure time of 1 s.
One diffraction dataset from a single crystal was collected at a wavelength of 1.000 Å and temperature of 100 K for the ternary complex, at the X06DA Beamline of the Swiss Light Source (SLS) equipped with a PILATUS 2M detector. Cryoprotection was achieved by soaking the selected crystal in 8 mm YCl3, 2.88% Tacsimate, pH 4.0, 8.64% PEG 3350, 20% glycerol. The crystal was then flash-cooled in liquid nitrogen. We recorded 444 frames using an oscillation range of 0.5°, a crystal to detector distance of 300 mm, and an exposure time of 0.3 s. All diffraction images were processed using the HKL 2000 package (20).
Determination of HSA/FcRn Three-dimensional Structure
The models of HSA and human FcRn were built from PDB entries 1AO6 (21) and 1EXU (15), respectively. The unit cell with parameters a = 136.5, b = 115.9, c = 186.2 Å, and β = 104.5° contained four nearly identical HSA·FcRn complexes. Two pairs of complexes (1–3 and 2–4) were related through pseudo-translation along the c axis (fractional 0.49). The other two pairs (1–2 and 3–4) were related through a pure non-crystallographic 2-fold rotation (179.8°). The four copies of human FcRn could be easily placed using MolRep (22). However, HSA had to be broken into separate parts to find an appropriate solution. More precisely, the four FcRn molecules were first entered as a fixed model and a solution for four copies of HSA domain I (DI, amino acids 3–203) was sought. The corresponding solution, which included four copies of each FcRn and HSA DI, was then fixed and a solution for four copies of HSA domain II (DII, amino acids 208–396) was sought and found. HSA domain III (DIII, amino acids 402–585) had to be further broken into two parts corresponding to subdomains IIIa and IIIb (DIIIa, amino acids 402–491 and DIIIb, amino acids 520–585) to find the corresponding solution. The model then underwent refinement using Refmac5 (22) with automatically generated global NCS restraints. Manual re-building was carried out with the “O” program (23). The refinement converged to a final R/Rfree of 0.23/0.26. Of note, residues between HSA DI/II and DII/III (amino acids 204–207 and 397–401, respectively) as well as the connecting peptide between HSA DIIIa and DIIIb (amino acids 492–519) were built manually during the iterative refinement/rebuilding procedure. Data and refinement statistics are presented in Table 1. The final root mean square (r.m.s.) deviations between the equivalent Cα atoms of the complexes in the asymmetric unit were 0.13, 0.13, and 0.11 Å for FcRn α-chain, HSA, and β2-microglobulin, respectively. No residues in the final model were found in the disallowed region of the Ramachandran plot (90.3% in most favored region, 9.4% in additionally allowed region, 0.3% in generously allowed region). The refined model of a representative region fitted into electron density is presented in Fig. 3a.
TABLE 1.
X-ray data collection and refinement statistics
| Complex | HSA/FcRn/Fc-YTE | HSA/FcRn |
|---|---|---|
| Light source | SLS | APS |
| Data collection | ||
| Wavelength, Å | 1.000 | 1.000 |
| Resolution, Å | 28–3.8 (4.0–3.8)a | 50–3.0 (3.05–3.00) |
| Space group | P41212 | P21 |
| Cell parameters, Å | a = 153.2, b = 153.2, | a = 136.5, b = 115.9, |
| c = 146.0 | c = 186.2, β = 104.5° | |
| Total reflections | 255,663 | 414,069 |
| Unique reflections | 17,420 | 110,017 |
| Completeness, % | 100.0 (99.9) | 99.9 (99.9) |
| Rmerge | 0.118 (0.843) | 0.250 (0.778) |
| Mean I/σ (I) | 16.1 (1.0) | 5.3 (1.0) |
| Multiplicity | 15.6 (14.2) | 3.8 (3.5) |
| Mosaicity (degree) | 0.90 | 0.55 |
| Refinement | ||
| Resolution, Å | 40–3.8 | 20–3.0 |
| Number of reflections | 15,915 | 104,460 |
| Rwork/Rfree | 0.28/0.30 | 0.23/0.26 |
| Number of atoms | 9,322 | 30,192 |
| Average B-factors, Å2 | 153.3 | 33.4 |
| Root mean square deviations | ||
| Bond length, Å | 0.009 | 0.012 |
| Bond angles (degree) | 1.4 | 1.6 |
a Values in parentheses correspond to the highest resolution shell.
FIGURE 3.
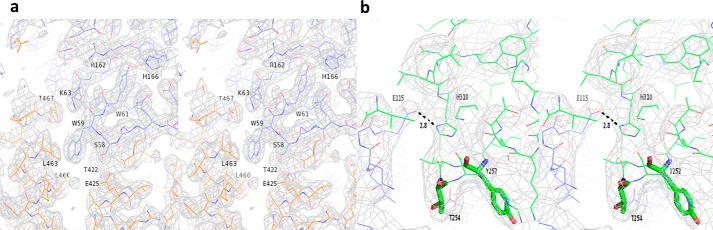
Stereographic representation of the final σA weighted electron density. Map around (a) the FcRn hydrophobic core at the HSA DIII/FcRn interface in the binary complex, and (b) the M252Y and S254T mutations (shown as green sticks) in the ternary complex. The maps are contoured at 1.0σ.
Determination of the HSA/FcRn/Fc-YTE Three-dimensional Structure
HSA/FcRn (this study) and Fc-YTE (PDB entry 3FJT) (18) were used as models. The unit cell with parameters a = b = 153.2, c = 146.0 Å contained one FcRn, one HSA, and one Fc-YTE polypeptide (half-Fc). Using Phaser (24) from the CCP4 program suite, we found one solution for each component of the complex with a final LLG of 3997.34 and R-factor of 0.48. The model then underwent rigid- and jelly-body refinement using Refmac5 (22). The refinement converged to a final R/Rfree of 0.28/0.30. Data and refinement statistics are presented in Table 1. No residues in the final model were found in the disallowed region of the Ramachandran plot (89.5% in most favored region, 10.3% in additionally allowed region, 0.2% in generously allowed region). The refined model of a representative region fitted into electron density is presented on Fig. 3b.
RESULTS AND DISCUSSION
Crystallization and Structure Determination
Diffraction quality crystals for the binary complex grew in ∼3–4 days in hanging drops where 1–5 μl of the HSA·FcRn complex at a concentration of 10 mg/ml in 50 mm NaOAc, pH 5.2, 100 mm NaCl was mixed with 1 μl of 0.2 m NaSCN, 3.6% Tacsimate, pH 4.0, 10.8% PEG 3350 and left to equilibrate against 300 μl of 0.2 m NaSCN, 3.6% Tacsimate, pH 4.0, 10.8% PEG 3350 (Fig. 4a). The symmetry of these crystals belonged to monoclinic space group P21 according to Pointless (Collaborative Computational Project 4, CCP4 suite) (25), and reached a diffraction limit down to 3.0-Å resolution. The asymmetric unit contained four HSA·FcRn complexes. This corresponds to a crystal volume per protein weight (VM) of 2.6 Å3/Da and a solvent content of 62%. SDS-PAGE analysis of these crystals confirmed that they indeed contained the expected complex formed by HSA and FcRn (Fig. 1b).
FIGURE 4.

Crystals of (a) HSA/FcRn and (b) HSA·FcRn·Fc-YTE complexes. The binary complex crystals grew to a size of up to 50 × 50 × 20 μm within 3–4 days at room temperature. The ternary complex crystals grew to a size of up to 50 × 50 × 100 μm within 4 weeks at room temperature.
Diffraction quality crystals for the ternary complex grew in ∼4 weeks in sitting drops where 0.3 μl of the HSA·FcRn·Fc-YTE complex at a concentration of 4 mg/ml in 50 mm NaOAc, pH 5.2, 100 mm NaCl was mixed with 0.3 μl of 0.01 m YCl3, 3.6% Tacsimate, pH 4.0, 10.8% PEG 3350 and left to equilibrate against 50 μl of 0.01 m YCl3, 3.6% Tacsimate, pH 4.0, 10.8% PEG 3350 (Fig. 4b). The symmetry of these crystals belonged to tetragonal space group P41212 according to Pointless (25) and reached a diffraction limit down to 3.8-Å resolution. The asymmetric unit contained one HSA·FcRn·half-Fc-YTE complex. This corresponds to a VM of 3.0 Å3/Da and a solvent content of 60%. SDS-PAGE analysis of these crystals confirmed that they indeed contained the expected complex formed by HSA, FcRn, and Fc-YTE (Fig. 1a). The three-dimensional structures of the HSA·FcRn and HSA·FcRn·Fc-YTE complexes were solved to a resolution of 3.0 and 3.8 Å, respectively, using molecular replacement (see “Experimental Procedures”).
Analysis of HSA Three-dimensional Structure
We found it necessary to break the HSA model into separate parts to find a solution during the molecular replacement procedure. Because we found a solution for each separate HSA domain, it is reasonable to assume that whereas their internal structure was preserved, their relative orientation changed upon binding to FcRn. We assessed such changes by superimposing 50 different HSA molecules from the Protein Data Bank (PDB) with our own using the LSQKAB program (26). The superimposition was carried out at full-length and individual domain levels. When full-length molecules were used, r.m.s deviations for Cα atoms varied between 2.2 and 6.6 Å (when compared with FcRn-bound HSA) and showed that DIII exhibited the largest change in its orientation relative to other domains (Fig. 5a). When superimposition was done at the domain level, deviations were considerably smaller. More precisely, HSA DII was the most conformationally conserved (r.m.s deviations for Cα atoms ranged from 0.6 to 1.3 Å when compared with FcRn-bound HSA), whereas HSA DI exhibited intermediate variability (with r.m.s deviations for Cα atoms ranging from 0.9 to 1.6 Å). HSA DIII exhibited the most variability (r.m.s deviations for Cα atoms ranged from 1.7 to 2.1 Å), which can be accounted for the most part by a significant change in the relative position of its two subdomains (Fig. 5b). This suggests that formation of the HSA·FcRn complex results in changes in the relative orientation of HSA DIII and agrees well with our finding that this domain plays a major role in binding to FcRn (see below).
FIGURE 5.
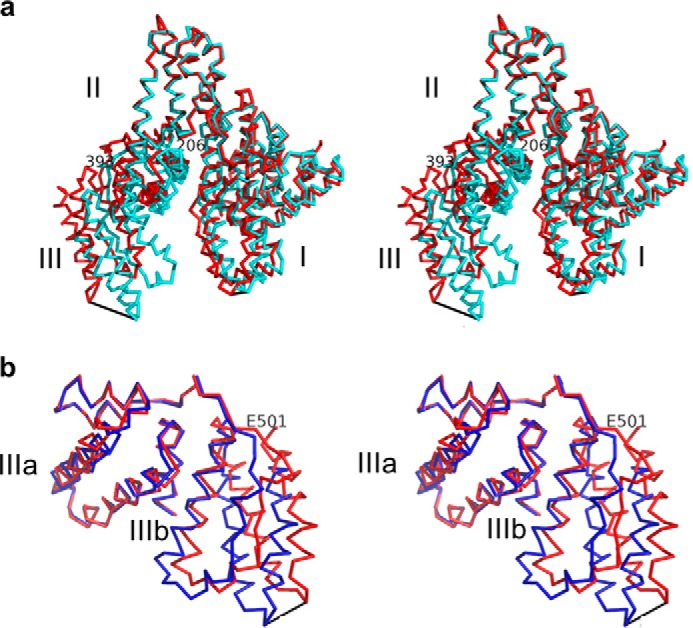
a, stereographic representation of the superimposition of full-length HSA of this study (red) with HSA corresponding to PDB entry 1AO6 (32) (blue). Superimposition was carried out through the Cα atoms of HSA DII. HSA DIII exhibits the largest change in orientation relative to other domains, with a maximum motion of 19.1 Å between the Cα atoms of Asp562. HSA DI motion was more modest with a maximum distance of 5.0 Å between the Cα atoms of Ala172. Pivot points for DI and DIII were found at Phe206 and Glu393, respectively. b, stereographic representation of the superimposition of HSA DIII of this study (red) with HSA DIII corresponding to PDB entry 1AO6 (32) (blue). The superimposition was carried out through the Cα atoms of HSA DIIIa. Significant variability in the relative orientation of HSA DIIIa and DIIIb was seen, with a maximum distance of 8.5 Å between the Cα atoms of Asp562. The pivot point was found at Glu501. This and all other figures were made using PyMOL (DeLano Scientific, Palo Alto, CA).
Analysis of HSA/FcRn Three-dimensional Structure
We present here the three-dimensional structure of wild-type HSA bound to human FcRn. A ribbon diagram of this complex is shown in Fig. 6. Overall, our complex structure is in good agreement with that involving a HSA variant (V418M/T420A/E505G/V547A) whose affinity for human FcRn was increased by ∼300-fold (14). In particular, the r.m.s deviation for all atoms is 0.8 Å. Of these four mutations, only HSA E505G seems to play a direct role in increasing FcRn/HSA binding affinity via two additional hydrogen bonds introduced between its carbonyl and FcRn Ser230 side chain. This mutation also resulted in local conformational changes between HSA Ala504 and Phe509 when compared with the wild-type molecule of this study. HSA V418M and T420A are part of the interface but do not play an obvious role in the corresponding affinity increase. HSA V547A is not part of the interface. Therefore, we postulate their impact on FcRn binding to be mediated by long-range (V547A) or indirect (V418M and T420A) effects.
FIGURE 6.
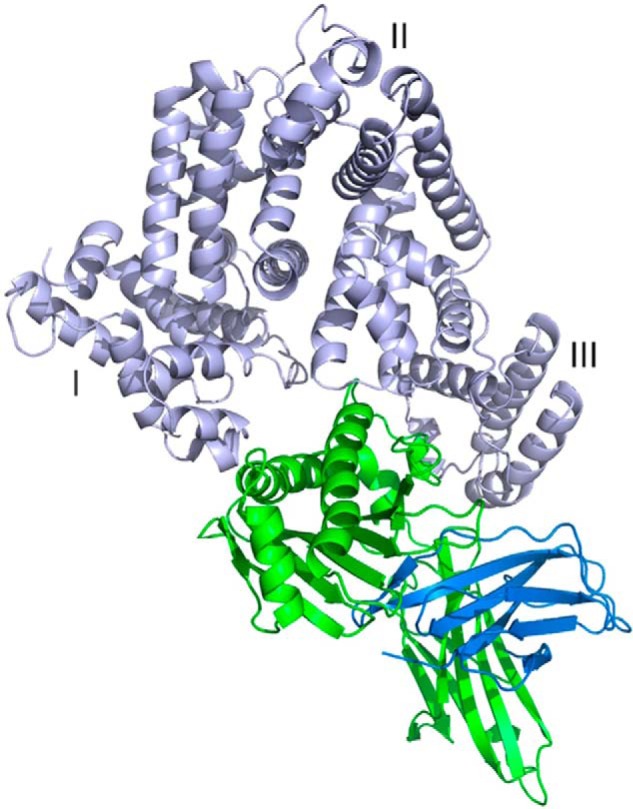
Three-dimensional view of HSA·FcRn complex. HSA (DI, DII, and DIII), FcRn α-chain and β2-microglobulin are shown in light blue, green, and dark blue, respectively. FcRn binds in a HSA crevice located between DI and DIII.
We note that no remarkable differences were detected between the conformations of human FcRn when in its unliganded (PDB entry 1EXU) (15) or bound (this study) form. FcRn binds in a crevice of the heart-shaped HSA molecule and creates direct interactions with both DI and DIII. Overall, 1800 Å2 of surface area is buried at the interface. Of that, ∼500 and ∼200 Å2 correspond to HSA DI/FcRn α-chain and HSA DIII/β2-microglobulin interfaces, respectively. The majority (∼1100 Å2) rests between HSA DIII and the FcRn α-chain. A prominent feature of FcRn is a groove made by two long α-helices (Tyr60-Leu82 and Pro132-Glu175) backed by a seven-stranded antiparallel β-sheet. A kink in the second α-helix (between Gln142 and Ala147) results in the separation of the groove into a short and long part. HSA DI is positioned above the longer portion of the groove and interacts with both α-helices. The interface mostly comprises charged or polar amino acids that are involved in direct electrostatic interactions, including two hydrogen bonds between HSA Asn111/Oδ1 and FcRn Ser58/Oγ and between HSA Arg81/Nϵ and FcRn Thr153/Oγ1 (3.6 Å distance cutoff). Other electrostatic interactions involve FcRn Ser58/Lys63/Asn149 and HSA Asp89/Asp108/Asn111 (Fig. 7). In addition, FcRn His161 contributes to this network via a weak hydrogen bond (∼4 Å) between its Nϵ2 nitrogen and the carbonyl of HSA Glu82. This histidine is one of two at the HSA/FcRn interface and likely plays a direct role in the corresponding pH dependence of binding (see below).
FIGURE 7.
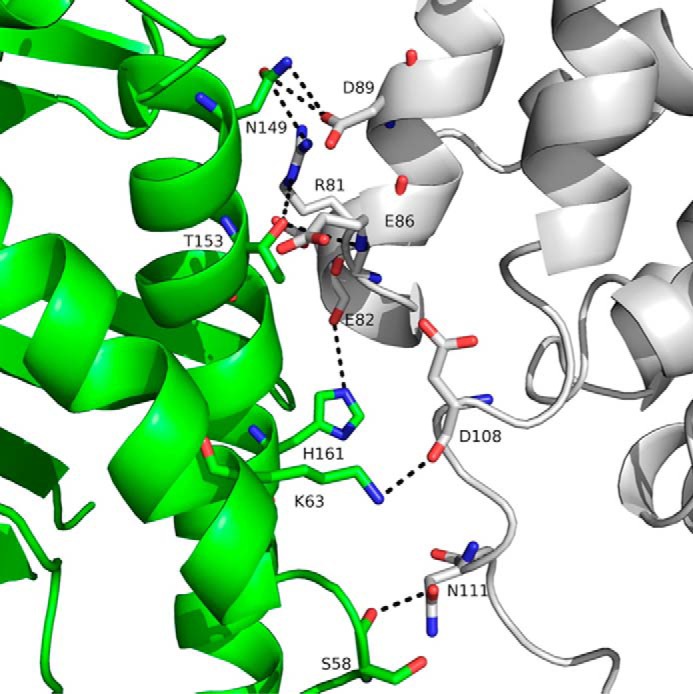
Three-dimensional representation of the interface between HSA DI (gray) and human FcRn α-chain (green). HSA DI is positioned directly above the longer portion of the FcRn groove. Although FcRn His161 is in a favorable position to create a hydrogen bond with the carbonyl of HSA Glu82, its contribution is likely minimal because of its rather large distance (∼4 Å). Possible charged interactions are indicated by black dotted lines.
No Direct Contribution from HSA DII to the HSA·FcRn Complex Was Seen
HSA DIII plays the most important role in terms of the number of amino acids it contributes to bind FcRn. The interface can be split into two parts involving subdomains DIIIa and DIIIb. We found six hydrogen bonds and salt bridges between HSA DIIIa and FcRn α-chain (3.6 Å distance cutoff). These involve FcRn Ser58/Trp59/Glu62/Arg69 and HSA Glu425/Thr422/Ser419/Pro468. Other electrostatic interactions involve FcRn Glu44/Glu62 and HSA Gln417/Val469/Ser419/Thr422 (Fig. 8a). Unlike that with HSA DI, this interface exhibits a very prominent hydrophobic core centered on FcRn Phe44/Val57/Trp59/Trp61 and HSA Gln417/Pro421/Leu460/Leu463/Val469. This is in good agreement with isothermal titration calorimetry showing that HSA binds FcRn with a large positive change in entropy (6). This area does not contain any histidine residues, and therefore does not contribute to pH dependence. The interface between HSA DIIIb (including the IIIa/IIIb connecting peptide) and FcRn is less extensive than that involving DIIIa and comprises four hydrogen bonds (3.6 Å distance cutoff). These involve FcRn Arg42/Trp53/Asn173 and HSA Tyr497/Val498/Thr506/His510. Other electrostatic interactions involve FcRn Ser230/Asp231 and HSA Glu505 (Fig. 8b). The interface is centered on FcRn Trp53, which contributes its Nϵ1 atom to hydrogen bonds with the carbonyls of HSA Thr506. It also contains His510 in a position to form a strong (2.6 Å) hydrogen bond with FcRn Asn173/Oδ1. This seems to suggest a direct role of this residue in the pH dependence of the corresponding interaction. To test this hypothesis, we substituted His510 by all possible amino acids (except cysteine), and assessed binding of the resulting variants to human FcRn at both acid and neutral pH (Fig. 9). Binding at pH 6.0 ranged from similar to significantly decreased when compared with HSA wild-type, which highlighted the overall importance of this residue to the corresponding interaction. All variants, however, maintained a pH-dependent binding profile and none bound human FcRn at pH 7.4. Therefore, we conclude that HSA His510 is not directly and majorly involved in the pH dependence of binding. We suggest that the latter likely relies on the only other interface histidine, namely FcRn His161.
FIGURE 8.

Stereographic representation of the interface between (a) HSA DIIIa and human FcRn, and (b) HSA DIIIb and human FcRn. a, the interface with HSA DIIIa is the busiest and most likely a major determinant for the affinity of the corresponding complex. It does not, however, contain any histidine residues and therefore does not contribute to the pH dependence of the interaction. Possible charged interactions are indicated by dotted lines. HSA, FcRn α-chain, and β2-microglobulin are shown in gray, green, and dark blue, respectively. b, HSA His510 in DIIIb forms a strong hydrogen bond with FcRn Asn173/Oδ1 (red dotted line). Possible charged interactions are indicated by black dotted lines. HSA and FcRn α-chain are shown in gray and green, respectively.
FIGURE 9.

BIAcore analysis of the binding of HSA wild-type and variants to human FcRn at pH 6.0 and 7.4 after correction for nonspecific binding.
β2-Microglobulin plays a relatively minor role in this complex. In particular, the guanidinium group of β2-microglobulin Arg12 is involved in a weak bidentate interaction with HSA Glu505/Oϵ1 and Oϵ2, whereas β2-microglobulin Glu50 interacts with the carbonyl of HSA Phe502 (Fig. 10).
FIGURE 10.
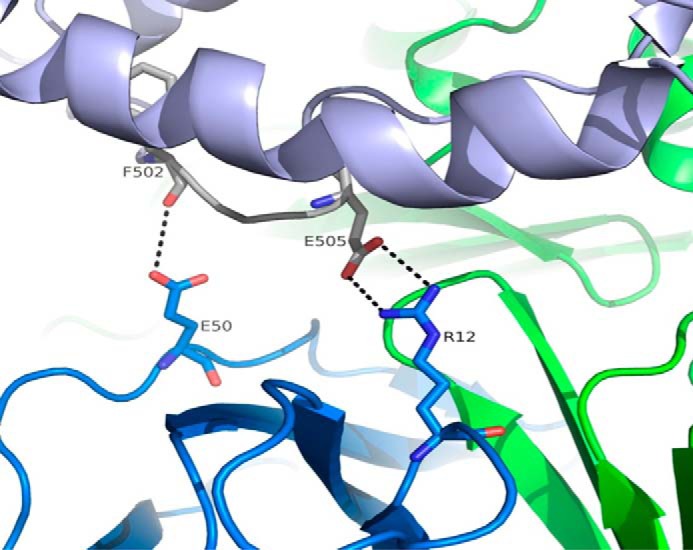
Stereographic representation of the interface between HSA IIIb and β2-microglobulin. Contribution of β2-microglobulin to HSA binding is limited to long range (∼3.7–4.0 Å) charged interactions (indicated by dotted lines). HSA, FcRn α-chain, and β2-microglobulin are shown in light blue, green, and dark blue, respectively.
HSA DIII has previously been identified as critical to the interaction with FcRn. Indeed, it was reported that DIII alone exhibits significant binding to human FcRn, whereas DI and DII individually or together show no measurable interaction (6). Andersen et al. (13) and Schmidt et al. (14) also found that DIII plays a crucial role in HSA pH-dependent binding to FcRn, although it was also suggested that DI and DII could also bring a moderate contribution (13). Our findings agree well with these data and reaffirm the crucial role played by HSA DIII, along with a significant contribution of DI. Andersen et al. (13) also built a full-length HSA/FcRn model and ran a docking procedure with bias toward solutions involving FcRn His161/His166 and HSA His464/His510/His535. We have found here, however, that FcRn His166 and HSA His464/His535 are not part of the corresponding interface and therefore do not play a direct role in this pH-dependent interaction. This led these authors to inaccurate conclusions, in particular regarding the existence of important interactions between FcRn His166 and HSA Glu505, FcRn Glu54 and HSA Glu510, FcRn Lys150/Glu151 and HSA Glu501/Lys500, and FcRn His161 and HSA Glu531. Indeed, our study shows that the corresponding residues are, respectively, ∼18, 10, 25, and 30 Å apart, and thus unable to engage in such interactions. Interestingly, the assumption that FcRn His166 is at the core of the interface seemed to be at least partially based on the inability of the H166A mutant to bind HSA (27). We propose that this mutation only acts in a indirect fashion, maybe by disrupting a network of internal interactions within FcRn (e.g. with Glu54) and leading to deleterious changes on the FcRn side of the complex.
Analysis of HSA·FcRn·Fc-YTE Three-dimensional Structure
We present here the three-dimensional structure of human FcRn bound concurrently to its two known ligands. The ribbon diagram of the HSA·FcRn·Fc-YTE ternary complex is shown in Fig. 11. The HSA·FcRn portion superimposes well with that of the binary complex, with a r.m.s deviation of Cα atoms of ∼0.2 Å. The Fc-YTE moiety also superimposes well with that of the same molecule in an unbound state (PDB entry 3FJT) (18) with a r.m.s deviation of Cα atoms of ∼0.1 Å.
FIGURE 11.

Three-dimensional view of the HSA·FcRn·Fc-YTE complex. HSA (DI, DII, and DIII), FcRn α-chain, β2-microglobulin, and Fc-YTE are shown in light blue, green, dark blue, and brown, respectively. The content of the asymmetric unit is shown in the red rectangle.
The FcRn molecule binds Fc between its CH2 and CH3 domains. The interface exhibits very strong hydrophobic properties as it involves FcRn Tyr88, Leu112, Phe117, Trp131, Pro132, and Leu135, and Fc-YTE Leu251, Tyr252, Ile253, Leu309, and Leu314. Six hydrogen bonds and salt bridges between Fc-YTE and FcRn were identified (3.6 Å distance cutoff) and included FcRn Glu133/Glu115/Glu116/Gly129 and Fc-YTE Ile253/Thr254/His310/Gln311/Asn434. In particular, the side chain of Fc-YTE His310 is positioned in such a way that its Nδ1 atom makes a strong hydrogen bond with FcRn Glu115/Oϵ1 (2.8 Å distance; Fig. 12). This is in good agreement with previous studies identifying Fc His310 as particularly important for human or murine Fc binding to murine FcRn (28–30). Interestingly, the analysis of the human(ized) IgG/human FcRn interaction had also previously revealed the critical role played by Fc Ile253 and His435 (31–33). These results can be explained by Ile253 being an important component of a large hydrophobic patch at the human IgG/FcRn interface (see above), the disruption of which likely affects formation of the complex. However, our study shows that the Fc His435 side chain does not adopt a favorable conformation to form an intermolecular hydrogen bond and is only involved in a weak π interaction involving its Nϵ2 atom and the main chain carbonyl of FcRn Asp130 (2.8 Å distance; Fig. 12). Therefore, the dramatic effects seen on human IgG·FcRn complex formation upon mutating this residue are likely not caused solely by a disruption of this interaction, but rather by other yet-to-be defined indirect effects, such as local conformational rearrangements.
FIGURE 12.
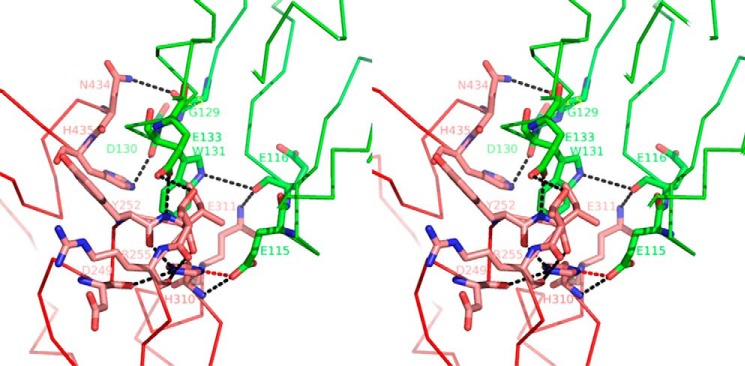
Stereographic representation of the interface between Fc-YTE (pink) and FcRn α-chain (green). Fc-YTE His310/Nδ1 forms a strong hydrogen bond with FcRn Glu115/Oϵ1 (red dotted line). Other charged interactions are marked with black dotted lines.
Finally, it has generally been assumed using rodent molecules that IgG/FcRn pH-dependent interaction relies on the titration of Fc His310/His433 and FcRn His250/His251 (5, 34). We show here that Fc His433 and FcRn His248/His249 (human equivalent of His250/His251 in mouse) are not part of the corresponding interface. Therefore, we conclude that these amino acids do not play a direct role in the corresponding pH-dependent interaction in human. Because of the central role played by His310 (see above), it is tantalizing to identify this residue as a major contributor to this process. To test this hypothesis, we substituted His310 by all possible amino acids (except cysteine), and assessed binding of the resulting IgG and IgG-YTE variants to human FcRn at both acid and neutral pH. Binding of all (non-YTE) IgG variants at both pH 6.0 and 7.4 was essentially not detectable (data not shown). Most IgG-YTE variants also did not significantly bind to FcRn at pH 6.0, with only a few exhibiting detectable, although dramatically decreased binding at pH 6.0 (H310E, H310K, H310M; Fig. 13). In conclusion, our data highlight the overall crucial importance of His310 for binding FcRn at pH 6.0, and suggest that this residue plays a significant role in the corresponding pH-dependent process.
FIGURE 13.

BIAcore analysis of the binding of IgG-YTE and variants thereof to human FcRn at pH 6.0 and 7.4 after correction for nonspecific binding.
Molecular Basis for Enhancing Human IgG Interaction with FcRn
Several Fc mutations resulting in increased human IgG binding to human FcRn have been previously described and carefully characterized at a functional level, including their effect on IgG pharmacokinetic properties and biodistribution (12, 33, 35, 36). However, the exact molecular mechanism(s) underlying the functional properties of these mutations have remained elusive. Our present study provides those for the first time.
In particular, the YTE set of substitutions (M252Y/S254T/T256E) results in an ∼10-fold increase in human IgG1 binding to human FcRn, whereas studies in human and cynomolgus monkeys indicate that the serum half-life of this mutated humanized IgG1 was increased by ∼2–4- and 4-fold, respectively, when compared with its unmutated counterpart (11, 12). We find that the YTE mutations result in the creation of two salt bridges between Fc-YTE Glu256 and β2-microglobulin Gln2 (Fig. 14). The wild-type residue at this position (Thr) would be unable to engage in such interactions because of its shorter side chain. Thr254/Oγ1 also interacts with FcRn Glu133/Oϵ1, although it is predicted that the wild-type residue at this position (Ser) would also potentially be able to engage in a similar interaction. Therefore, we propose that Glu256 plays the largest role in increasing the binding affinity between Fc-YTE and FcRn. This validates our earlier prediction on the YTE mechanism of action, where we suggested that its effects “are mediated by the gain of a few, yet-to-be accurately defined interactions with human FcRn at the substitutions sites” (18).
FIGURE 14.
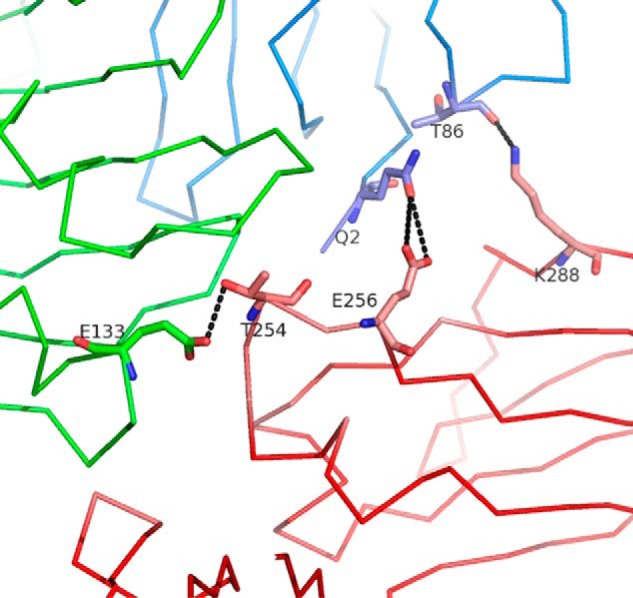
Three-dimensional view of the interface between Fc-YTE (pink), FcRn α-chain (green), and β2-microglobulin (blue) around Fc-YTE Glu256. The side chain of Glu256 allows additional interactions with β2-microglobulin Gln2 (dotted lines). Thr254/Oγ1 also interacts with FcRn Glu133/Oϵ1. These interactions likely contribute to the high affinity of Fc-YTE for FcRn.
Several other mutations resulting in increased human IgG binding to human FcRn have also been described and carefully characterized. Of those, N434A was identified by alanine scanning and resulted in an ∼3.5-fold increase in human IgG1 binding to human FcRn at pH 6.0 (32). When dosed in transgenic mice expressing human FcRn, N434A resulted in an ∼2–3-fold increase in serum half-life when compared with the same unmutated antibody (33). T250Q/M428L was also described, which, when introduced into a human IgG1, resulted in an ∼30-fold pH-dependent increase in IgG binding to human FcRn (35). The same mutant had a serum half-life ∼2.5-fold longer in rhesus monkeys than the wild-type form of the same human IgG1. Finally, M428L/N434S provided an 11-fold improvement in human IgG1 affinity to FcRn at pH 6.0 along with an ∼3–4-fold increase in serum half-life in transgenic mice expressing human FcRn (36). We find that Fc Asn434 is involved in an unfavorable interaction of polar and hydrophobic amino acids with FcRn Leu135. Upon mutation to alanine (N434A), a new hydrophobic patch is created that engages Leu135 in favorable contacts. Substitution to serine (N434S) allows the creation of additional hydrogen bonds with either or both carbonyls of FcRn Gly129 and Trp131. Finally, Fc Thr250 and Met428 are not in direct contact with FcRn and no additional direct intermolecular interaction seems to be introduced when mutated to Gln and Leu, respectively. Therefore, these two mutations likely increase human IgG binding to human FcRn in an indirect fashion, maybe through subtle favorable conformational changes at the interface or local rearrangement of the water molecules network.
Our HSA/FcRn and HSA/FcRn/Fc-YTE structures provide valuable information in the field of IgG and HSA recycling. In addition to providing new insights on these well known molecules, they are expected to guide the design and development of novel improved variants exhibiting beneficial pharmacokinetics properties. This is particularly true of the HSA/FcRn interaction where much of the interface remains to be probed. We also hope our results will provide new engineering clues and lead to a revival of studies aimed at optimizing the IgG/FcRn interaction, whose interface has been exhaustively mutated by a plethora of rational design and library-based approaches.
Acknowledgments
We thank Lena Shirinian and Srinath Kasturirangan for expert technical assistance with HSA cloning and expression. We are also grateful to Andrew Ferguson for help with data collection on the HSA·FcRn complex.
The atomic coordinates and structure factors (codes 4N0F and 4N0U) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- FcRn
- neonatal Fc receptor
- SA
- serum albumin
- HSA
- human serum albumin
- RU
- response unit
- DI
- domain I
- r.m.s.
- root mean square
- VM
- volume per protein weight.
REFERENCES
- 1. Simister N. E., Mostov K. E. (1989) An Fc receptor structurally related to MHC class I antigens. Nature 337, 184–187 [DOI] [PubMed] [Google Scholar]
- 2. Burmeister W. P., Gastinel L. N., Simister N. E., Blum M. L., Bjorkman P. J. (1994) Crystal structure at 2.2-Å resolution of the MHC-related neonatal Fc receptor. Nature 372, 336–343 [DOI] [PubMed] [Google Scholar]
- 3. Burmeister W. P., Huber A. H., Bjorkman P. J. (1994) Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 372, 379–383 [DOI] [PubMed] [Google Scholar]
- 4. Rodewald R. (1976) pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J. Cell Biol. 71, 666–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raghavan M., Bonagura V. R., Morrison S. L., Bjorkman P. J. (1995) Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 34, 14649–14657 [DOI] [PubMed] [Google Scholar]
- 6. Chaudhury C., Brooks C. L., Carter D. C., Robinson J. M., Anderson C. L. (2006) Albumin binding to FcRn. Distinct from the FcRn-IgG interaction. Biochemistry 45, 4983–4990 [DOI] [PubMed] [Google Scholar]
- 7. Chaudhury C., Mehnaz S., Robinson J. M., Hayton W. L., Pearl D. K., Roopenian D. C., Anderson C. L. (2003) The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersen J. T., Sandlie I. (2009) The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab. Pharmacokinet. 24, 318–332 [DOI] [PubMed] [Google Scholar]
- 9. Junghans R. P., Anderson C. L. (1996) The protection receptor for IgG catabolism is the β2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. U.S.A. 93, 5512–5516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghetie V., Ward E. S. (2000) Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu. Rev. Immunol. 18, 739–766 [DOI] [PubMed] [Google Scholar]
- 11. Robbie G., Criste R., Dall'Acqua W., Jensen K, Patel N., Losonsky G., Griffin P. A. (2013) Novel investigational Fc modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. A randomized study. Antimicrob. Agents Chemother. 57, 6147–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dall'Acqua W. F., Kiener P. A., Wu H. (2006) Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 281, 23514–23524 [DOI] [PubMed] [Google Scholar]
- 13. Andersen J. T., Dalhus B., Cameron J., Daba M. B., Plumridge A., Evans L., Brennan S. O., Gunnarsen K. S., Bjørås M., Sleep D., Sandlie I. (2012) Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat. Commun. 10.1038/ncomms1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt M. M., Townson S. A., Andreucci A. J., King B. M., Schirmer E. B., Murillo A. J., Dombrowski C., Tisdale A. W., Lowden P. A., Masci A. L., Kovalchin J. T., Erbe D. V., Wittrup K. D., Furfine E. S., Barnes T. M. (2013) Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure 21, 1966–1978 [DOI] [PubMed] [Google Scholar]
- 15. West A. P., Jr., Bjorkman P. J. (2000) Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry 39, 9698–9708 [DOI] [PubMed] [Google Scholar]
- 16. Martin W. L., West A. P., Jr., Gan L., Bjorkman P. J. (2001) Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex. Mechanism of pH-dependent binding. Mol. Cell. 7, 867–877 [DOI] [PubMed] [Google Scholar]
- 17. Kabat E. A., Wu T. T., Perry H. M., Gottesman K. S., Foeller C. (1991) Sequences of Proteins of Immunological Interest, 5th Ed., NIH Publications, Bethesda, MD [Google Scholar]
- 18. Oganesyan V., Damschroder M. M., Woods R. M., Cook K. E., Wu H., Dall'acqua W. F. (2009) Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol. Immunol. 46, 1750–1755 [DOI] [PubMed] [Google Scholar]
- 19. D'Arcy A., Villard F., Marsh M. (2007) An automated microseed matrix-screening method for protein crystallization. Acta Crystallogr. D 63, 550–554 [DOI] [PubMed] [Google Scholar]
- 20. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 21. Sugio S., Kashima A., Mochizuki S., Noda M. (1999) Crystal structure of human serum albumin at 2.5-Å resolution. Protein Eng. 12, 439–446 [DOI] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 23. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 25. Collaborative Computational Project, Number 4 (1994) The CCP4 Suite. Programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 26. Kabsch W. A. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 32, 922–923 [Google Scholar]
- 27. Andersen J. T., Dee Qian J., Sandlie I. (2006) The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur. J. Immunol. 36, 3044–3051 [DOI] [PubMed] [Google Scholar]
- 28. Kim J. K., Tsen M. F., Ghetie V., Ward E. S. (1994) Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur. J. Immunol. 24, 542–548 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. K., Firan M., Radu C. G., Kim C. H., Ghetie V., Ward E. S. (1999) Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur. J. Immunol. 29, 2819–2825 [DOI] [PubMed] [Google Scholar]
- 30. Medesan C., Matesoi D., Radu C., Ghetie V., Ward E. S. (1997) Delineation of the amino acid residues involved in transcytosis and catabolism of mouse IgG1. J. Immunol. 158, 2211–2217 [PubMed] [Google Scholar]
- 31. Firan M., Bawdon R., Radu C., Ober R. J., Eaken D., Antohe F., Ghetie V., Ward E.S. (2001) The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of γ-globulin in humans. Int. Immunol. 13, 993–1002 [DOI] [PubMed] [Google Scholar]
- 32. Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 33. Petkova S. B., Akilesh S., Sproule T. J., Christianson G. J., Al Khabbaz H., Brown A. C., Presta L. G., Meng Y. G., Roopenian D. C. (2006) Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model. Potential application in humorally mediated autoimmune disease. Int. Immunol. 18, 1759–1769 [DOI] [PubMed] [Google Scholar]
- 34. Vaughn D. E., Bjorkman P. J. (1998) Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 6, 63–73 [DOI] [PubMed] [Google Scholar]
- 35. Hinton P. R., Xiong J. M., Johlfs M. G., Tang M. T., Keller S., Tsurushita N. (2006) An engineered human IgG1 antibody with longer serum half-life. J. Immunol. 176, 346–356 [DOI] [PubMed] [Google Scholar]
- 36. Zalevsky J., Chamberlain A. K., Horton H. M., Karki S., Leung I. W., Sproule T. J., Lazar G. A., Roopenian D. C., Desjarlais J. R. (2010) Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 [DOI] [PMC free article] [PubMed] [Google Scholar]