

Background: Heme and [Fe-S] cluster assembly are tightly regulated processes that require mitochondrial iron.
Results: Loss of mitochondrial iron activates the [Fe-S]-dependent RNA-binding activity of IRP1 that inhibits protoporphyrin biosynthesis.
Conclusion: IRP1 forms a critical feedback mechanism, preventing protoporphyrin accumulation under limiting mitochondrial iron conditions.
Significance: This study provides evidence linking heme biogenesis to that of [Fe-S] clusters synthesis.
Keywords: Erythropoeisis, Heme, Iron, Iron-sulfur Protein, Mitochondrial Transport, Fe-S Cluster Biogenesis, Mitochondrial Solute Carrier, Protoporphyrin
Abstract
Mitochondrial iron is essential for the biosynthesis of heme and iron-sulfur ([Fe-S]) clusters in mammalian cells. In developing erythrocytes, iron is imported into the mitochondria by MFRN1 (mitoferrin-1, SLC25A37). Although loss of MFRN1 in zebrafish and mice leads to profound anemia, mutant animals showed no overt signs of porphyria, suggesting that mitochondrial iron deficiency does not result in an accumulation of protoporphyrins. Here, we developed a gene trap model to provide in vitro and in vivo evidence that iron regulatory protein-1 (IRP1) inhibits protoporphyrin accumulation. Mfrn1+/gt;Irp1−/− erythroid cells exhibit a significant increase in protoporphyrin levels. IRP1 attenuates protoporphyrin biosynthesis by binding to the 5′-iron response element (IRE) of alas2 mRNA, inhibiting its translation. Ectopic expression of alas2 harboring a mutant IRE, preventing IRP1 binding, in Mfrn1gt/gt cells mimics Irp1 deficiency. Together, our data support a model whereby impaired mitochondrial [Fe-S] cluster biogenesis in Mfrn1gt/gt cells results in elevated IRP1 RNA-binding that attenuates ALAS2 mRNA translation and protoporphyrin accumulation.
Introduction
Iron is an essential element that is incorporated into prosthetic groups that function in a multitude of biochemical processes. Two of these iron-containing prosthetic groups are heme and iron-sulfur ([Fe-S]) clusters. Heme is a moiety with diverse functions in many cell types and is an integral component of hemoglobin in erythroid cells (1). [Fe-S] clusters are requisite cofactors for many enzymes, including aconitases and succinate dehydrogenase (2, 3). Although functionally distinct, assembly of both cofactors require mitochondrial function and mitochondrial iron assimilation (4).
Iron is imported into mitochondria via two transporters: mitoferrin-1 (MFRN1, SLC25A37) and mitoferrin-2 (MFRN2, SLC25A28). MFRN2 is expressed ubiquitously, whereas MFRN1 is specifically induced in differentiating erythroid cells (5–7). Previous work has shown that loss of Mfrn1 in frascati zebrafish and mice disrupts heme and [Fe-S] cluster synthesis, owing to a severe reduction in mitochondrial iron in erythroid progenitors (7, 8). However, neither Mfrn1−/− mice nor zebrafish frascati embryos deficient in Mfrn1 develop porphyria. In fact, conditional deletion of Mfrn1 in hepatocytes only causes porphyria and hepatobiliary stasis when the animals are fed a δ-aminolevulinic acid (ALA)-rich12 diet (8). This indicates that reduction in mitochondrial iron stores may concomitantly attenuate early steps of protoporphyrin synthesis.
Recent evidence suggests that cross-talk between [Fe-S] cluster assembly and protoporphyrin synthesis pathways may explain the absence of porphyria in Mfrn1−/− animals (9). Cellular iron homeostasis is largely regulated by iron regulatory proteins-1 (IRP1) and -2 (IRP2) that can directly influence protein expression via two mechanisms (10–12). IRPs can bind to iron responsive elements (IREs) in the 5′-UTRs of mRNAs, blocking translation initiation. Alternatively, mRNAs with IRE(s) in the 3′-UTR have increased stability when bound by IRPs. As a result, this latter class of transcripts will have increased accumulation when IRPs are activated by iron deficiency. Although IRP1 and IRP2 exhibit some functional redundancy and can be regulated by common mechanisms (13–15), IRP1 is unique in that RNA-binding activity is negatively regulated by [Fe-S] cluster biogenesis. Insertion of an [Fe-S] cluster into IRP1 converts IRP1 from an IRE-RNA binding protein to cytosolic aconitase (ACO1). A decline in [Fe-S] cluster synthesis would, therefore, trigger increased IRP1 RNA-binding activity (16–19).
One mRNA harboring a 5′-IRE is erythroid-specific aminolevulinate synthase 2 (Alas2), which encodes the enzyme that catalyzes the first step in the heme biosynthetic pathway (20, 21). Wingert and colleagues (9) previously demonstrated that inhibition of glutaredoxin 5 (Glrx5), a gene required for mitochondrial [Fe-S] cluster assembly, resulted in anemia in zebrafish via increased IRP1-mediated inhibition of ALAS2 translation. Thus, a reduction in [Fe-S] cluster assembly in response to iron deficiency can feedback to attenuate the very proximal step of heme assembly (9, 22). Here, we examined whether loss of Mfrn1 can also trigger IRP1 activity in erythroid cells. We generated a model to show that in the absence of MFRN1, IRP1 blocks ALAS2 translation, preventing the accumulation of protoporphyrins in vitro and in vivo. Loss of IRP1 or the 5′-IRE in Alas2 mRNA resulted in porphyria. Our work indicates that IRP1 functions as a critical link coupling heme and [Fe-S] cluster biosynthetic pathways and that in the absence of MFRN1-mediated mitochondrial iron import, IRP1 protects erythroid cells against porphyria.
EXPERIMENTAL PROCEDURES
Cell Culture
Friend murine erythroleukemia cells were cultured and differentiated as described previously (23). Mouse embryonic stem (mES) cells containing a gene trap insertion in Mfrn1 (Mfrn1+/gt cell line, XB454, derived from strain 129P2) were obtained from the University of California, San Francisco BayGenomics (24). Insertion of the gene trap vector (containing the coding sequence for β-geo) into Mfrn1 was verified by direct sequencing of cDNA obtained by 5′-rapid amplification of cDNA ends (25). mES cells were maintained on gelatin-coated 100-mm dishes in mES cell medium (7). Null Mfrn1gt/gt mES clones were derived using G418 selection as described previously (7).
Mouse Blastocyst Injections and Knock-out Mouse Creation
ES cells were injected into C57BL/6J blastocysts and transferred to pseudopregnant C57BL/6J hosts using standard techniques (26). Male chimeras identified by agouti coat color were mated to C57BL/6J females to generate heterozygotes for interbreeding to produce homozygous knock-out progeny. Progeny were genotyped as described below. All mice were maintained at The Jackson Laboratory in a climate-controlled room with a 12-h light cycle and provided acidified water and NIH 5K52 chow ad libitum. The Jackson Laboratory Animal Care and Use Committee approved all protocols. In accordance with the International Committee on Standardized Genetic Nomenclature for Mice, the official genetic designation for this Mfrn1 targets strain is B6.129P2-Slc25a37<Gt(XB454)Byg>/Llp.
Mouse Genotyping
For analysis by Southern blotting, HindIII-digested mouse genomic or ES cell DNA was transferred onto Hybond Nylon (Amersham Biosciences), and probed with random primed labeled [α-32P]dCTP (3000 Ci/mmol; New England Nuclear) murine Mfrn1 or Escherichia coli lacZ cDNA under standard hybridization and washing conditions. Genotyping by PCR on genomic DNA was performed with the following primer pairs and resolved on a 5% native polyacrylamide gel:
The following were used in this work: for the wild-type allele (269-bp fragment), 5′-GCTTATGGAAAGGAACCCAGCC-3′ (F1 primer) and 5′-ACAAGGAAGAGCCAGGACTGTCAG-3′ (R1 primer); mutant gt allele (350-bp fragment) as shown above (F1 primer) and 5′-CGCCATACAGTCCTCTTCACAT-3′ (R2 primer); LacZ, 5′-TTATCGATGAGCGTGGTGGTTATGC-3′ (F primer) and 5′-GCGCGTACATCGGGCAAATAATATC-3′ (R primer). Primers for Irp1 mutant mice were as described previously (27).
LacZ Staining
β-Galactosidase staining of mouse embryos was performed as described previously (7).
Mixed Chimera Analysis
Mfrn1gt/gt ES cell clones were injected into C57BL/6J mouse blastocysts and transferred to C57BL/6J recipients. The degree of mosaicism in offspring was estimated by coat color. The contributions of donor (129P2) versus recipient (C57BL/6) ES cells to the red cell and leukocyte compartments were determined by cellulose acetate electrophoresis to detect strain-specific hemoglobin and glucose-6-isomerase alleles as described previously (28) in three chimeric mice (one female with moderate chimerism, two males with moderate to high chimerism) at 10 weeks of age.
o-Dianisidine Staining, CFU-E, and Cytospin Assays
Wild-type E14 and Mfrn1 null ES clones were generally split 1 day after thawing with 106 mES cells plated onto gelatinized 100-mm plates in mES media as described previously (7). The following day, mES cells were maintained in “switch media” (7). Two days after the split, the cells were collected by trypsinization and replated on untreated 100-mm dishes at a density of 3 × 103 cells/ml (total 6 × 104 cells) in embryoid body (EB) media with murine VEGF (10 ng/ml) on days 1 and 5. ALA (0.25 mm) was added to EB cultures for the last 24 h of day 6 primitive differentiation (7). Hemoglobinization of the primitive differentiated cells was assessed by o-dianisidine staining as described (29).
To achieve definitive hematopoietic differentiation, the EB medium was supplemented with mVEGF (10 ng/ml), rEPO (10 international units/ml), mSCF (5 ng/ml). Day 5 EBs were supplemented with recombinant erythropoietin (5 units/ml), mIL-3 (5 ng/ml), mIL-6 (10 ng/ml), and mSCF (100 ng/ml) and harvested on day 6. mEBs were disaggregated by trituration in 0.25% trypsin via an 18-gauge syringe. The disaggregated EBs were replated in three-dimensional methylcellulose culture for CFU-E analysis (7). Picked CFU-Es were morphologically analyzed by cytospin and Wright-Giemsa staining (7).
Semi-quantitative RT-PCR for Erythroid Markers
Total RNA was isolated from ∼105 differentiated cells from wild-type (Mfrn1+/+), heterozygous (Mfrn1+/gt), and null (Mfrn1gt/gt) embryoid bodies on day 6 using the Qiagen RNasy kit. cDNA was synthesized using random hexamers with Superscript II reverse transcriptase (Invitrogen). PCR was performed with [α-32P]dCTP (3000 Ci/mmol; New England Nuclear) and Platinum TaqDNA polymerase (Roche Applied Science) under standard conditions for 30–32 cycles. PCR products were resolved on a 5% native polyacrylamide gel, treated in 10% methanol:acetic acid fixative, dried, and exposed to autoradiography. The following murine gene-specific primers were used for semi-quantitative RT-PCR: Mfrn1, 5′-AGCTGTGTGGTGTAACTCCTGTAG-3′ (forward) and 5′-AGGACTTTAAATGACGTTTTCAGC-3′ (reverse); Mfrn2, 5′-GCCCACGCCCTCTATTTTG-3′ and 5′-CGGCGTGGCGGGAGGAC-3′; β-major globin, 5′-GGTTTAGTGGTACTTGTGAGCC-3′ and 5′-ATGGTGCACCTGACTGATGCTG-3′; βH1 globin, 5′-AGTCCCCATGGAGTCAAAGA-3′ and 5′-CTCAAGGAGACCTTTGCTCA-3′; α-globin, 5′-TGGCTAGCCAAGGTCACCAGC-3′ and 5′-CTCTCTGGGAAGACAAAAGCAAC-3′; Alas2, 5′-GATCCAAGGCATTCGCAACA-3′ and 5′-GATGGCCTGCACATAGATGC-3′; Gata-1, 5′-ACTCGTCATACCACTAAGGT-3′ and 5′-AGTGTCTGTAGGCCTCAGCT-3′; Dmt1, 5′-GGTTCTGACATGCAGGAAGT-3′ and 5′-CAAAGACATTGATGATGAAG-3′; Hprt, 5′-CACAGGACTAGAACACCTGC-3′ and 5′-GCTGGTGAAAAGGACCTCT-3′; and GAPDH, 5′-TGATGACATCAAGAAGGTGGTGAAG-3′ and 5′-TCCTTGGAGGCCATGTAGGCCAT-3′.
Iron, Heme, and Protoporphyrin Assays
Intracellular iron measurement was performed as described previously (30). Cells were washed in nitric acid, and sample iron content was determined using a Perkin-Elmer inductively coupled plasma optical emission spectrometer. Results were normalized by cell numbers. Mitochondrial iron was similarly quantitated by inductively coupled plasma spectroscopy in mitochondria that were isolated from wild-type Mfrn1+/+ or Mfrn1gt/gt EBs using the mitochondrial isolation kit according to the manufacturer's instructions (Promega, Madison, WI) and normalized by mitochondrial protein content. For heme and protoporphyrin assays, EBs or embryos were sonicated and heme and protoporphyrin were extracted in 19:1 acetone:concentrated HCl as described (31). The resulting supernatant was injected immediately into a computer-controlled Waters reverse phase high-pressure liquid chromatography system that consisted of a 996 Photodiode Array Detector, a 474 scanning fluorescence detector, an AllianceHT 2795 separations module, and a μBondapak C18 3.9 × 300 mm column with a guard column attached. Bovine Type 1 crystalline heme (Sigma, H-2250) or Protoporphyrin IX (Sigma, P-8293) was used as the standard. The absorbance values were normalized by cell number or protein concentration. EBs were supplemented with 0.25 mm ALA (Sigma) for 24 h prior to cell harvest and biochemical analysis.
Western Analysis
Immunoblotting was performed as described previously (32). Custom, affinity-purified anti-mouse MFRN1 rabbit polyclonal antibody was generated using the following peptide: C-HESHVQEVSHKTSPT (Genemed Synthesis, San Antonio, TX). Goat polyclonal anti-HSP60 (K-19) was purchased from Santa Cruz Biotechnology. Anti-ALAS2 was a generous gift of Dr. Makoto Murakami (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). Quantitative densitometry was performed using ImageJ software (NIH, Bethesda, MD) and normalized to HSP60 expression levels.
Zebrafish alas2 Expression Constructs
Wild-type zebrafish alas2 cDNA with the 5′-IRE (+IRE; GenBankTM accession no. NM_131682.2) was subcloned into the BamHI/XhoI sites of pCS2+ as described previously (33). The alas2 cDNA lacking the 5′-IRE (−IRE; 5′-GTTCGTCCTCAGTGCAGGTCAAC-3′) was amplified from this construct by PCR and subcloned into pCS2+. The alas2 cDNA lacking the 5′-IRE with a premature nonsense mutation at amino acid residue 26 (Y26X), (−IRE/Stop), was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). These clones were subsequently subcloned into MluI/XhoI sites in the pMMP-HA-IRES-eGFP retroviral vector (34) for subsequent lentivirus production.
Lentivirus Infection
Lentivirus production and infection was performed as described previously (35). Briefly, 293T cells were plated at 6 × 106 cells per 100 mm dish and incubated overnight. Cells were transfected with retroviral vectors together with psPax2 and pMD2G. Forty-eight hours after transfection, the supernatants were collected and ultra-centrifuged at 28,000 rpm for 2 h. Supernatants from three 10-cm dishes were concentrated into 100 μl of PBS. Murine ES cells in 24-well plates were transduced using various amounts of concentrated virus supplemented with 8 μg/ml polybrene. After 24 h, the cells were changed back to standard medium. Three days after transduction, the cells were dissociated by 0.05% trypsin and sorted by FACS to obtain the GFP-positive cells.
Mouse Matings between Mfrn1 and Irp1 Mice
Mfrn1+/gt mice were crossed with Irp1+/− mice (27, 36). Mfrn1+/gt;Irp1+/− offspring were intercrossed to produce Mfrn1+/gt;Irp1−/− and Mfrn1+/gt;Irp+/+ sires and dams. Embryos from mating Mfrn1+/gt;Irp1−/− sires and dams, and embryos from mating Mfrn1+/gt;Irp1+/+ sires and dams were harvested at E10.5. After removing heads for genotyping, the remaining tissue was homogenized in 20 mm HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.5, buffer for heme and protoporphyrin analysis. All use of animals met the requirements of the University of Wisconsin-Madison Institutional Animal Care and Use Committee.
RESULTS
Mfrn1gt/gt ES Cells Are Defective at Erythroid Differentiation
Mfrn1 has been studied previously in mice using tissue-specific Cre/Lox technology (8). Although global loss of Mfrn1 in this model leads to embryonic lethality due to a profound embryonic anemia, a cell-autonomous role for MFRN1 in adult hematopoiesis has never been described. Thus, to address this and to study the underlying molecular mechanisms, we developed a model where we could manipulate MFRN1 function in vitro and in vivo. Gene trap mutagenesis was used to disrupt the Mfrn1 locus between introns 1 and 2 (Fig. 1A). Proper integration in murine ES cells was confirmed using PCR and Southern blot analysis (Fig. 1, B and C). Wild-type (Mfrn1+/+), heterozygote (Mfrn1+/gt), and null (Mfrn1gt/gt) ES cells were then cultured in vitro under conditions that facilitate primitive or definitive erythropoiesis. Mfrn1gt/gt cells, lacking MFRN1 protein expression, showed defective hemoglobinization, failed to form both primitive and definitive CFU-E colonies and were morphologically distinct from wild-type differentiated erythroid cells (Fig. 2, A–E). Semi-quantitative RT-PCR analysis also revealed that expression of erythroid markers, α-globin, βH1-globin, βmajor-globin, and Alas2, were diminished in differentiating Mfrn1gt/gt cells compared with wild-type (Fig. 2F), indicating that erythroid maturation in mutant cells was blocked. In contrast, the early erythroid marker, Gata-1, and ubiquitously expressed genes such as Mfrn2, Hprt, and GAPDH were normal compared with wild-type and Mfrn1+/gt cells. Total intracellular iron, mitochondrial iron, and intracellular heme content were all decreased without protoporphyrin accumulation (Fig. 2, G–I, and data not shown). We also examined both endogenous aconitase 1 activity and IRP1 RNA-binding (data not shown). However, we were unable to detect significant changes using either assays with this embryoid culture system. We believe that this is due to a heterogeneous population of cells in our differentiation cultures. Under optimal conditions, only 30% of ES cells undergo proper erythroid maturation (Fig. 2C). RNA-binding and aconitase assays may lack sufficient sensitivity to detect moderate changes.
FIGURE 1.
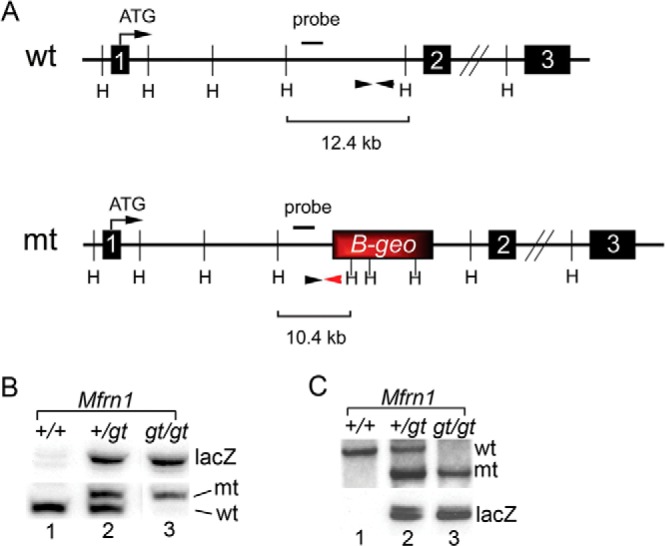
Genomic organization and genotyping of the MFRN1 gene trap mES clone. A, the wild-type (wt) and Mfrn1 mutant (mt) gene trap alleles are shown, including the site of the β-geo cassette and the primer sites used for PCR genotyping (black arrowheads). The primer-specific for the β-geo cassette is denoted by a red arrowhead. The location of the probe used for Southern blotting as well as the HindIII sites (H) are also shown. B and C, genomic DNA was isolated from Mfrn1+/+, Mfrn1+/gt, and Mfrn1gt/gt ES cells following step-up selection and subjected to PCR (B) or Southern blot analysis (C) to confirm their genotypes.
FIGURE 2.

Defect in hemoglobinization and maturation of primitive and definitive erythroblasts with Mfrn1 deficiency. A, wild-type (+/+), heterozygote (+/gt), or Mfrn1 gene-trap (gt/gt) ES cells undergoing primitive erythropoiesis were lysed and immunoblotted with anti-MFRN1 or anti-HSP60 antibodies, confirming the loss of Mfrn1 protein expression in gt/gt cells. B and C, EBs cultured under primitive (EryP) or definitive (EryD) conditions were analyzed for hemoglobinization by o-dianisidine staining using phase contrast microscopy (B) and quantified, showing a defect in hemoglobinization (C). The arrows denote cell clusters. D, quantitation of erythroid colonies (CFU-E) that formed under primitive or definitive conditions, showing defects on erythroid colony formation. E, primitive or definitive wild-type and gene trap cells were stained with Wright-Giemsa dye, showing maturation arrest of erythroblasts from Mfrn1gt/gt ES cells. Arrows denote representative erythroid cells. Mfrn1+/+ cells are morphologically normal, whereas Mfrn1gt/gt cells have a larger nuclei. F, [α-32P]dCTP-radiolabeled semi-quantitative RT-PCR analysis was performed on wild-type (+/+), heterozygote (+/gt), or two mutant (gt/gt) Mfrn1 ES cell clones undergoing primitive differentiation using the indicated gene-specific primers. G–I, total intracellular iron (Fe; G), mitochondrial iron (H), and heme (I) content of Mfrn1+/+ and Mfrn1gt/gt cells were examined, showing a consistent decrease in MFRN1-deficient embryoid bodies induced to undergo erythroid differentiation. Mean ± S.E. is shown, and all experiments were performed at least twice. IB, immunoblot.
Mfrn1 Is Required for Primitive and Definitive Erythropoiesis in Vivo
We next examined whether the primitive erythropoietic defects in ES cells can be recapitulated in vivo by injecting wild-type or Mfrn1gt/gt ES cells into early-stage embryos and generating mixed-chimeric mice. Mfrn1+/gt and Mfrn1gt/gt offspring were obtained following germ line transmission of the mutant allele. Surrogate lacZ-staining for Mfrn1 expression in heterozygote and gene-trapped embryos detected staining in the yolk sac blood islands and fetal liver at E8.5 and E11.5, respectively (Fig. 3, A and B). At E11.5, Mfrn1gt/gt embryos were severely anemic compared with wild-type littermates (Fig. 3B), and no mutant embryos survived past E11.5 (Table 1). MFRN1 is also required in vivo for definitive erythropoiesis. In mixed chimeras, only the host recipient C57BL/6J-derived hemoglobin isoform is detected by cellulose acetate electrophoresis of adult red cells (Fig. 3C). In contrast, genetic contributions from both recipient and donor (mutant, 129P2) glucose-6-isomerase isoforms were detected in leukocytes, indicating the essential role of MFRN1 in the development of the definitive erythroid lineage and its nonessential role in non-erythroid lineages in adult mice. These data confirm that our gene trap model provides an accurate representation of MFRN1 function in vitro and in vivo.
FIGURE 3.

MFRN1 is required for primitive and definitive erythropoiesis in vivo. A, LacZ staining of gene trap Mfrn1+/gt E8.5 yolk sac, showing erythroid-specific expression in sites of hematopoiesis. B, gross morphology of E11.5 wild-type (+/+) or gene trap (gt/gt) embryos. The mutant embryos were also stained for lacZ and have a profound anemia due to a defect in primitive erythropoiesis. C, erythrocytes and leukocytes from recipient, donor, or chimeric mice with medium or light penetrance (agouti coat color) were isolated and lysed, and isoforms were resolved on starch-cellulose gel electrophoresis and Ponceau-red stained for hemoglobin or glucose-6-isomerase (GPI). MFRN1-deficient donor-derived cells do not contribute to adult erythrocytes but do contribute to the myeloid lineage, indicating that MFRN1 is dispensable for myelopoiesis.
TABLE 1.
Number of Mfrn1+/+, Mfrn1+/gt, and Mfrn1gt/gt embryos during development
| Developmental stage | Total no. of embryos | Mfrn1+/+ | Mfrn1+/gt | Mfrn1gt/gt |
|---|---|---|---|---|
| E8.5 | 30 | 8 | 12 | 10 |
| E10.5 | 39 | 10 | 19 | 10 |
| E11.5 | 9 | 1 | 6 | 2 |
| E12.5 | 8 | 4 | 4 | 0 |
| E14.5 | 24 | 8 | 16 | 0 |
| E15.5 | 7 | 2 | 5 | 0 |
IRP1 Attenuates Protoporphyrin Production
Because disruption of [Fe-S] cluster synthesis has previously been linked to Alas2 mRNA translational repression by IRP1 (9), we next used our model to ask whether ALAS2 translation had any influence on protoporphyrin production in the absence of MFRN1. Mfrn1gt/gt EBs had lower expression of ALAS2 protein compared with Mfrn1+/+ EBs (Fig. 4A), suggesting that ALAS2 translation is indeed compromised in the absence of MFRN1. To examine the contribution of the 5′-IRE in MFRN1 regulation of ALAS2 function and expression, Mfrn1gt/gt ES cells were transduced with lentivirus expressing either empty vector (mock) or zebrafish alas2 cDNA constructs harboring a wild-type IRE (WT) or mutant IRE (−IRE) in the 5′-UTR. An additional mutant alas2 cDNA with a premature translational stop codon (−IRE/Stop) was also included as a negative control (Fig. 4, B and C). These cells were then differentiated into EBs. As expected, ectopic expression of WT-alas2 resulted in a modest increase in protoporphyrin levels above mock-transduced control cells (Fig. 4, C and D). However, there was almost a 2-fold increase in protoporphyrin levels in cells expressing the −IRE mutant construct due to increased ALAS2 expression (Fig. 4, C and D). The level of protoprophyrin IX (PPIX) dropped to basal levels in cells expressing the double mutant alas2 construct (−IRE/Stop) (Fig. 4, C and D). Furthermore, Mfrn1gt/gt EBs supplemented with exogenous ALA to by-pass the block in protoporphyrin synthesis resulting from diminished ALAS2 protein expression fully restored PPIX but not heme production to levels comparable with Mfrn1+/+ cells (Fig. 4E). Thus, inhibition of protoporphyrin synthesis through action of the 5′-IRE in Alas2 mRNA is substantially increased when mitochondrial [Fe-S] cluster assembly is compromised. A deficiency of MFRN1 should result in elevated cytosolic IRP1 RNA-binding activity that would attenuate translation of ALAS2 and lower PPIX production and its intracellular accumulation.
FIGURE 4.

Mitochondrial heme and Fe/S cluster biogenesis are co-regulated. A, mitochondria were isolated from undifferentiated (Undiff.) and differentiated (Diff.) Friend murine erythroleukemia (MEL) cells as well as Mfrn1+/+ and Mfrn1gt/gt embryoid bodies and subjected to Western blot analysis using the indicated antibodies. The asterisk denotes a cross-reacting band, and the arrow represents ALAS2. Densitometry analysis revealed that the intensities of the ALAS2 bands normalized to HSP60 expression levels in wild-type and gt/gt lanes are 100% and 61.5%, respectively. B and C, Mfrn1gt/gt ES cells infected with lentivirus expressing various alas2 constructs were lysed, subjected to SDS-PAGE, and immunoblotted (IB) with anti-ALAS2 and anti-HSP60 antibodies. The schematic in B highlights the features of the alas2 constructs as well as the anticipated results. D, PPIX levels were analyzed per milligram of total protein in Mfrn1gt/gt cells transduced with various lentiviruses, shown in B. E, heme and PPIX levels were measured in WT (Mfrn1+/+) and gt/gt (Mfrn1gt/gt) embryoid bodies with (+ALA) or without exogenous (−ALA), showing a dramatic increase in protoporphyrin but not heme in Mfrn1gt/gt cells because mitochondrial iron is limiting. F, murine embryos of various genotypes were examined for heme and protoporphyrin content at E10.5. Shown are mean ± S.E.; all experiments were performed at least twice. IB, immunoblot.
To address whether this was the case in vivo and to confirm whether IRP1 was responsible for negatively regulating ALAS2 expression, we examined heme and protoporphyrin concentrations in four cohorts of murine embryos, Mfrn1+/+;Irp1+/+, Mfrn1+/gt;Irp1+/+, Mfrn1+/+;Irp1−/−, and Mfrn1+/gt;Irp1−/−, at E10.5. Mfrn1 heterozygosity has little effect on heme levels in Irp1+/+ background (Fig. 4F). Protoporphyrin levels decreased, consistent with the notion that a reduction in mitochondrial iron import, while having little effect on heme biosynthesis, activates IRP1 due to a mitochondrial [Fe-S] cluster biogenesis defect (7) to inhibit ALAS2 translation. It should be noted that ferrochelatase (FECH), the terminal and rate-limiting enzyme in heme biosynthesis, is a [Fe-S] cluster protein and would also be influenced by the ability to form [Fe-S] clusters (37). However, the decreased FECH activity is unlikely to be responsible for the decline in protoporphyrin levels because diminished FECH function is predicted to exacerbate protoporphyrin accumulation. In contrast, in the absence of IRP1, loss of one copy of Mfrn1 causes a sharp rise in cellular protoporphyrin content, providing in vivo evidence that IRP1 prevents porphyria when MFRN1 is deficient. Taken together, our data strongly suggest that IRP1-mediated translational inhibition of ALAS2 in erythroid cells constitutes a feedback mechanism to attenuate protoporphyrin synthesis and accumulation when mitochondrial iron import through MFRN1 is compromised (Fig. 5).
FIGURE 5.
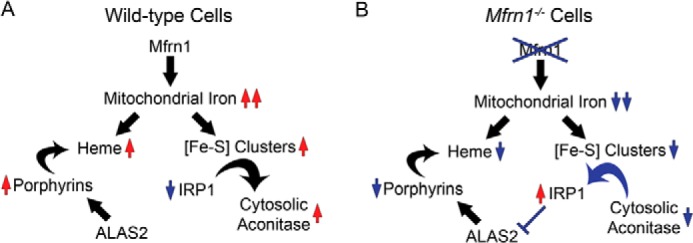
Schematic model of mitochondrial heme and Fe/S cluster co-regulation. A, MFRN1 imports iron into the mitochondria for heme and [Fe-S] cluster biosynthesis. When mitochondrial iron is readily available, sufficient [Fe-S] cluster synthesis converts IRP1 from an IRE-binding protein to cytosolic aconitase. B, however, when mitochondrial iron stores are compromised by MFRN1 mutation, both heme and [Fe-S] cluster syntheses are disrupted. Deficient [Fe-S] cluster availability results in activation of IRP1 that subsequently binds to IREs on target transcripts. IRP1 binding to the 5′-IRE of Alas2 mRNA inhibits ALAS2 translation, thereby forming a feedback loop that attenuates protoporphyrin accumulation in the absence of mitochondrial iron.
DISCUSSION
The role of MFRN1 in primitive erythropoiesis and heme synthesis has been established in zebrafish and murine models (7, 8). Although these genetic studies have been highly informative, a cell-autonomous in vivo role for MFRN1 has never been described. It was also unclear as to why porphyria was never observed with MFRN1 deficiency. In this work, we developed a genetic and biochemical model to address these questions. We provide evidence that MFRN1 is essential for primitive and definitive erythropoiesis in vivo. These defects could be recapitulated in ES cell cultures, which allowed us to study the underlying molecular mechanisms that couple mitochondrial iron homeostasis, heme metabolism, and [Fe-S] assembly. We demonstrate that IRP1 is a critical negative regulator of protoporphyrin synthesis in Mfrn1gt/gt erythroid cells via IRE-mediated repression of ALAS2 translation (Fig. 5). Mfrn1 mutant embryos only developed porphyria in the absence of IRP1 (Fig. 4).
Although our studies have focused on IRP1, both IRP1 and IRP2 function in red blood cells to regulate ALAS2 translation (9, 17, 38). Studies of translational regulation of 5′-IRE-containing mRNA in a cell culture model of erythropoiesis found that unlike ferritin mRNA, which was almost totally translationally repressed, Alas2 mRNA was partially repressed with significant amounts of the mRNA present on polyribosomes as well as in the translationally silent ribonucleoprotein pool (39). Thus, an erythropoietic phenotype tied to altered ALAS2 expression is predicted in response to either a reduction or an increase in IRP RNA-binding activity. Irp2−/− mice develop erythropoietic protoporphyria resulting from translational derepression of Alas2 mRNA and subsequent overproduction of protoporphyrin intermediates (16, 17). In contrast, impairment of [Fe-S] cluster biogenesis when ABCB7 (40–42) or GLRX5 (9, 22) are disrupted specifically activates IRP1 RNA-binding activity leading to repression of Alas2 mRNA translation. The observed increase in IRP1-mediated inhibition of ALAS2 via its 5′-IRE suggests that [Fe-S] cluster insertion into ACO1 is also compromised in the absence of MFRN1. Thus, by further supporting the concept that cytosolic [Fe-S] biogenesis is downstream of mitochondrial [Fe-S] formation that is fueled, in part, by the action of MFRN1, our findings also illustrate the unique role of IRP1 in linking mitochondrial iron metabolism with protoporphyrin synthesis in erythroid cells (9, 40, 43, 44). Taken together, these studies highlight differing roles of IRP1 and IRP2 in regulating ALAS2-IRP1 links protoporphyrin synthesis with [Fe-S] cluster synthesis and mitochondrial function, whereas IRP2 may have a more general “housekeeping” role in iron metabolism.
Our work presented here has important clinical implications. They suggest that although Mfrn1 mutations may contribute to human porphyrias, Mfrn1 inactivation alone is not likely sufficient for development of erythropoietic protoporphyria. In accord, expression of aberrant Mfrn1 and a concomitant decrease in Fech expression is associated with human erythropoietic protoporphyria (45). Interestingly, C-terminal ALAS2-activating mutations (45–47) were also found in these erythropoietic protoporphyria patients with abnormal Mfrn1 transcripts. Also, Alas2 has been shown to be a genetic modifier in patients with congenital erythropoietic porphyria (48–50). This indicates that the IRP1/ALAS2 negative feedback loop also exists in humans and that disruption of multiple regulators in the heme and [Fe-S] cluster synthesis pathways are required for protoporphyrin accumulation (45). In Mfrn1−/− conditional knock-out mice, this porphyria effect can be recapitulated by exogenous chemical supplementation of ALA in the diets (8). Given the cytotoxic effects of protoporphyrin and its biosynthetic intermediates, the IRP1/ALAS2 feedback mechanism may have evolved in mammals to tightly link protoporphyrin synthesis with iron availability to guard against their deleterious overaccumulation.
Acknowledgments
We thank Drs. Yvette Yien and Caiyong Chen for insightful discussion. We thank Prof. Makoto Murakami (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) for the anti-ALAS2 antisera. We also thank Christopher Nizzi (University of Wisconsin, Madison, WI) for preparation of the 32P-labeled RNA probe.
This work was supported by grants from the American Society of Hematology (to G. C. S.), the Canadian Institutes of Health Research (CIHR postdoctoral fellowship (to J. C.), the Federal Ministry of Education and Research (BMBF, Virtual Liver (to M. W. H.), the March of Dimes Foundation (6-FY09-289) (to B. H. P.), and the National Institutes of Health R01 HL088468 (to L. L. P.), R01 DK052380 (to J. K.), R01 DK066600 (to R. S. E.), and R01 DK070838 and P01 HL032262 (to B. H. P.).
- ALA
- δ-aminolevulinic acid
- ES
- embryonic stem
- PPIX
- protoporphyrin IX
- [Fe-S]
- iron-sulfur
- IRE
- iron-response element
- IRP1
- iron regulatory protein-1
- Alas2
- aminolevulinate synthase 2
- E1
- embryonic day 1
- EB
- embryoid body
- FECH
- ferrochelatase.
REFERENCES
- 1. Schultz I. J., Chen C., Paw B. H., Hamza I. (2010) Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 285, 26753–26759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rouault T. A., Tong W. H. (2008) Iron-sulfur cluster biogenesis and human disease. Trends Genet. 24, 398–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 4. Chung J., Chen C., Paw B. H. (2012) Heme metabolism and erythropoiesis. Curr. Opin. Hematol. 19, 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amigo J. D., Yu M., Troadec M. B., Gwynn B., Cooney J. D., Lambert A. J., Chi N. C., Weiss M. J., Peters L. L., Kaplan J., Cantor A. B., Paw B. H. (2011) Identification of distal cis-regulatory elements at mouse mitoferrin loci using zebrafish transgenesis. Mol. Cell. Biol. 31, 1344–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paradkar P. N., Zumbrennen K. B., Paw B. H., Ward D. M., Kaplan J. (2009) Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 29, 1007–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., Gwynn B., Lambert A. J., Wingert R. A., Traver D., Trede N. S., Barut B. A., Zhou Y., Minet E., Donovan A., Brownlie A., Balzan R., Weiss M. J., Peters L. L., Kaplan J., Zon L. I., Paw B. H. (2006) Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 [DOI] [PubMed] [Google Scholar]
- 8. Troadec M. B., Warner D., Wallace J., Thomas K., Spangrude G. J., Phillips J., Khalimonchuk O., Paw B. H., Ward D. M., Kaplan J. (2010) Targeted deletion of the mouse Mitoferrin1 gene: from anemia to protoporphyria. Blood 117, 5494–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wingert R. A., Galloway J. L., Barut B., Foott H., Fraenkel P., Axe J. L., Weber G. J., Dooley K., Davidson A. J., Schmid B., Schmidt B., Paw B. H., Shaw G. C., Kingsley P., Palis J., Schubert H., Chen O., Kaplan J., Zon L. I., Tübingen 2000 Screen Consortium (2005) Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 436, 1035–1039 [DOI] [PubMed] [Google Scholar]
- 10. Rouault T. A. (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 [DOI] [PubMed] [Google Scholar]
- 11. Anderson C. P., Shen M., Eisenstein R. S., Leibold E. A. (2012) Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hentze M. W., Muckenthaler M. U., Galy B., Camaschella C. (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142, 24–38 [DOI] [PubMed] [Google Scholar]
- 13. Rouault T. A. (2009) Cell biology. An ancient gauge for iron. Science 326, 676–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salahudeen A. A., Thompson J. W., Ruiz J. C., Ma H. W., Kinch L. N., Li Q., Grishin N. V., Bruick R. K. (2009) An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vashisht A. A., Zumbrennen K. B., Huang X., Powers D. N., Durazo A., Sun D., Bhaskaran N., Persson A., Uhlen M., Sangfelt O., Spruck C., Leibold E. A., Wohlschlegel J. A. (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galy B., Ferring D., Minana B., Bell O., Janser H. G., Muckenthaler M., Schümann K., Hentze M. W. (2005) Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 106, 2580–2589 [DOI] [PubMed] [Google Scholar]
- 17. Cooperman S. S., Meyron-Holtz E. G., Olivierre-Wilson H., Ghosh M. C., McConnell J. P., Rouault T. A. (2005) Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 106, 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyron-Holtz E. G., Ghosh M. C., Iwai K., LaVaute T., Brazzolotto X., Berger U. V., Land W., Ollivierre-Wilson H., Grinberg A., Love P., Rouault T. A. (2004) Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 23, 386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clarke S. L., Vasanthakumar A., Anderson S. A., Pondarré C., Koh C. M., Deck K. M., Pitula J. S., Epstein C. J., Fleming M. D., Eisenstein R. S. (2006) Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J. 25, 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamza I., Dailey H. A. (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 1823, 1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dandekar T., Stripecke R., Gray N. K., Goossen B., Constable A., Johansson H. E., Hentze M. W. (1991) Identification of a novel iron-responsive element in murine and human erythroid δ-aminolevulinic acid synthase mRNA. EMBO J. 10, 1903–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye H., Jeong S. Y., Ghosh M. C., Kovtunovych G., Silvestri L., Ortillo D., Uchida N., Tisdale J., Camaschella C., Rouault T. A. (2010) Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 120, 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen C., Garcia-Santos D., Ishikawa Y., Seguin A., Li L., Fegan K. H., Hildick-Smith G. J., Shah D. I., Cooney J. D., Chen W., King M. J., Yien Y. Y., Schultz I. J., Anderson H., Dalton A. J., Freedman M. L., Kingsley P. D., Palis J., Hattangadi S. M., Lodish H. F., Ward D. M., Kaplan J., Maeda T., Ponka P., Paw B. H. (2013) Snx3 regulates recycling of the transferrin receptor and iron assimilation. Cell Metab. 17, 343–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stryke D., Kawamoto M., Huang C. C., Johns S. J., King L. A., Harper C. A., Meng E. C., Lee R. E., Yee A., L'Italien L., Chuang P. T., Young S. G., Skarnes W. C., Babbitt P. C., Ferrin T. E. (2003) BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 31, 278–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Townley D. J., Avery B. J., Rosen B., Skarnes W. C. (1997) Rapid sequence analysis of gene trap integrations to generate a resource of insertional mutations in mice. Genome Res. 7, 293–298 [DOI] [PubMed] [Google Scholar]
- 26. Robertson E. J. (1987) Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. Oxford University Press, New York [Google Scholar]
- 27. Anderson S. A., Nizzi C. P., Chang Y. I., Deck K. M., Schmidt P. J., Galy B., Damnernsawad A., Broman A. T., Kendziorski C., Hentze M. W., Fleming M. D., Zhang J., Eisenstein R. S. (2013) The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 17, 282–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J., Astle C. M., Harrison D. E. (2000) Genetic regulation of primitive hematopoietic stem cell senescence. Exp. Hematol. 28, 442–450 [DOI] [PubMed] [Google Scholar]
- 29. Stachura D. L., Reyes J. R., Bartunek P., Paw B. H., Zon L. I., Traver D. (2009) Zebrafish kidney stromal cell lines support multilineage hematopoiesis. Blood 114, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li L., Chen O. S., McVey Ward D., Kaplan J. (2001) CCC1 is a transporter that mediates vacuolar iron storage in yeast. J. Biol. Chem. 276, 29515–29519 [DOI] [PubMed] [Google Scholar]
- 31. Cook J. D. (1980) Iron, Churchill Livingstone, New York [Google Scholar]
- 32. Shah D. I., Takahashi-Makise N., Cooney J. D., Li L., Schultz I. J., Pierce E. L., Narla A., Seguin A., Hattangadi S. M., Medlock A. E., Langer N. B., Dailey T. A., Hurst S. N., Faccenda D., Wiwczar J. M., Heggers S. K., Vogin G., Chen W., Chen C., Campagna D. R., Brugnara C., Zhou Y., Ebert B. L., Danial N. N., Fleming M. D., Ward D. M., Campanella M., Dailey H. A., Kaplan J., Paw B. H. (2012) Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature 491, 608–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brownlie A., Donovan A., Pratt S. J., Paw B. H., Oates A. C., Brugnara C., Witkowska H. E., Sassa S., Zon L. I. (1998) Positional cloning of the zebrafish sauternes gene: a model for congenital sideroblastic anaemia. Nat. Genet. 20, 244–250 [DOI] [PubMed] [Google Scholar]
- 34. Woo A. J., Moran T. B., Schindler Y. L., Choe S. K., Langer N. B., Sullivan M. R., Fujiwara Y., Paw B. H., Cantor A. B. (2008) Identification of ZBP-89 as a novel GATA-1-associated transcription factor involved in megakaryocytic and erythroid development. Mol. Cell. Biol. 28, 2675–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung J., Lau J., Cheng L. S., Grant R. I., Robinson F., Ketela T., Reis P. P., Roche O., Kamel-Reid S., Moffat J., Ohh M., Perez-Ordonez B., Kaplan D. R., Irwin M. S. (2010) SATB2 augments ΔNp63α in head and neck squamous cell carcinoma. EMBO Rep. 11, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Galy B., Ferring D., Benesova M., Benes V., Hentze M. W. (2004) Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. RNA 10, 1019–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crooks D.R., Ghosh M.C., Haller R.G., Tong W.H., Rouault T.A. (2010) Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 115, 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye H., Rouault T. A. (2010) Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 49, 4945–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schranzhofer M., Schifrer M., Cabrera J. A., Kopp S., Chiba P., Beug H., Müllner E. W. (2006) Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood 107, 4159–4167 [DOI] [PubMed] [Google Scholar]
- 40. Pondarré C., Antiochos B. B., Campagna D. R., Clarke S. L., Greer E. L., Deck K. M., McDonald A., Han A. P., Medlock A., Kutok J. L., Anderson S. A., Eisenstein R. S., Fleming M. D. (2006) The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 15, 953–964 [DOI] [PubMed] [Google Scholar]
- 41. Csere P., Lill R., Kispal G. (1998) Identification of a human mitochondrial ABC transporter, the functional orthologue of yeast Atm1p. FEBS Lett. 441, 266–270 [DOI] [PubMed] [Google Scholar]
- 42. Cavadini P., Biasiotto G., Poli M., Levi S., Verardi R., Zanella I., Derosas M., Ingrassia R., Corrado M., Arosio P. (2007) RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 109, 3552–3559 [DOI] [PubMed] [Google Scholar]
- 43. Tong W. H., Rouault T. (2000) Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 19, 5692–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kispal G., Csere P., Prohl C., Lill R. (1999) The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 18, 3981–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y., Langer N. B., Shaw G. C., Yang G., Li L., Kaplan J., Paw B. H., Bloomer J. R. (2011) Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 39, 784–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whatley S. D., Ducamp S., Gouya L., Grandchamp B., Beaumont C., Badminton M. N., Elder G. H., Holme S. A., Anstey A. V., Parker M., Corrigall A. V., Meissner P. N., Hift R. J., Marsden J. T., Ma Y., Mieli-Vergani G., Deybach J. C., Puy H. (2008) C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 83, 408–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bishop D. F., Tchaikovskii V., Hoffbrand A. V., Fraser M. E., Margolis S. (2012) X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 287, 28943–28955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. To-Figueras J., Ducamp S., Clayton J., Badenas C., Delaby C., Ged C., Lyoumi S., Gouya L., de Verneuil H., Beaumont C., Ferreira G. C., Deybach J. C., Herrero C., Puy H. (2011) ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood 118, 1443–1451 [DOI] [PubMed] [Google Scholar]
- 49. Kadirvel S., Furuyama K., Harigae H., Kaneko K., Tamai Y., Ishida Y., Shibahara S. (2012) The carboxy-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 40, 477–486 [DOI] [PubMed] [Google Scholar]
- 50. Balwani M., Doheny D., Bishop D.F., Nazarenko I., Yasuda M., Dailey H.A., Anderson K.E., Bissell D.M., Bloomer J., Bonkovsky H.L., Phillips J.D., Liu L., Desnick R.J., and Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network (2013) Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and high prevalence of X-linked protoporphyria. Mol. Med. 19, 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]