

Background: The glycoproteins contactin and neurofascin-155 are implicated in axo-glial junctions insulating the node of Ranvier.
Results: N-Glycosylated contactin is targeted in inflammatory neuropathy.
Conclusion: Autoantibodies reveal specific N-glycans that are implicated in adhesive interaction between contactin and neurofascin-155.
Significance: Autoantibodies against N-glycosylated contactin may be pathogenic via functional blocking.
Keywords: Cell Adhesion, Cell Surface Protein, Glycoprotein, Myelin, Neuroinflammation
Abstract
Cell adhesion molecules (CAMs) play a crucial role in the formation of the nodes of Ranvier and in the rapid propagation of the nerve impulses along myelinated axons. These CAMs are the targets of autoimmunity in inflammatory neuropathies. We recently showed that a subgroup of patients with aggressive chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) shows autoantibodies to contactin (1). The complex of contactin·Caspr·neurofascin-155 (NF155) enables the formation of paranodal junctions, suggesting that antibody attack against paranodes may participate in the severity of CIDP. In the present study, we mapped the molecular determinants of contactin targeted by the autoantibodies. In three patients, immunoreactivity was directed against the Ig domains of contactin and was dependent on N-glycans. The serum of one patient was selectively directed against contactin bearing mannose-rich N-glycans. Strikingly, the oligomannose type sugars of contactin are required for association with its glial partner NF155 (2). To investigate precisely the role of contactin N-glycans, we have mutated each of the nine consensus N-glycosylation sites independently. We found that the mutation of three sites (N467Q/N473Q/N494Q) in Ig domain 5 of contactin prevented soluble NF155-Fc binding. In contrast, these mutations did not abolish cis-association with Caspr. Next, we showed that the cluster of N-glycosylation sites (Asn-467, Asn-473, and Asn-494) was required for immunoreactivity in one patient. Using cell aggregation assays, we showed that the IgGs from the four CIDP patients prevented adhesive interaction between contactin·Caspr and NF155. Importantly, we showed that the anti-contactin autoantibodies induced alteration of paranodal junctions in myelinated neuronal culture. These results strongly suggest that antibodies to CAMs may be pathogenic and induce demyelination via functional blocking activity.
Introduction
Cell adhesion molecules (CAMs)3 are implicated in the insulation of myelin and in the formation of the nodes of Ranvier at the basis of the rapid propagation of the nerve influx. In the peripheral nervous system, the domain organization of myelinated axons depends on specific axo-glial contacts between the axonal membrane and Schwann cells at node, paranode, and juxtaparanode. At the nodal gap, gliomedin and NrCAM secreted by the Schwann cell microvilli interact with the axonal neurofascin-186 and mediate the clustering of voltage-gated sodium channels (3–5). At paranodes, neurofascin-155 (NF155) is expressed by the terminal loops of myelin and associates with the axonal CAMs contactin and Caspr (6). This ternary complex of glycoproteins is required for the formation and stability of the septate-like junctions (7, 8). At juxtaparanodes, Caspr2 associated with TAG-1 dictates the clustering of voltage-gated Kv1 channels (9, 10). The paranodal junctions act as a fence to separate nodal sodium channels and juxtaparanodal potassium channels and regulate axonal conduction by precluding current leakage across the paranode.
N-Glycosylations play a crucial role in the cell surface targeting and in the assembly of the paranodal complex. These post-translational modifications are of general importance for the quality control and folding assistance of glycoproteins along the secretory pathway and are also involved in a large number of recognition events (11). As a matter of fact, the export of Caspr and contactin is a tightly controlled process. Caspr associates in heterodimers with contactin to adopt a transport-permissive conformation that is checked through the cycle of the lectin chaperones calnexin·calreticulin (12). The complex of Caspr·contactin bearing ER-type mannose-rich N-glycans is addressed to the cell surface in an unconventional way (13). Contactin expressed without Caspr is usually modified through the Golgi apparatus with complex N-glycans. Strikingly, N-glycosylation is required for the binding of NF155 on contactin, and NF155 preferentially binds the glycoform of contactin with mannose-rich oligosaccharides (2). However, how this may determine the assembly or stability of the paranodal complex is still elusive.
The CAMs at the node of Ranvier have been recently identified as targets of autoimmunity in a small proportion of patients with autoimmune neuropathies. Autoantibodies against gliomedin, neurofascins, NrCAM, and contactin have been identified in subgroups of patients with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) or Guillain-Barré syndrome (1, 14–17). These autoantibodies to nodal and paranodal CAMs may contribute to the pathology by disrupting the proper axonal domain organization, leading to conduction defects as reported in rat models of Guillain-Barré syndrome (18, 19).
We recently reported that a subgroup of patients with aggressive CIDP display autoantibodies to contactin also named CNTN1. These antibodies react against conformational epitopes of contactin or contactin·Caspr and bind the paranodal junctions in teased sciatic nerves of rats (1). Here, we showed that these autoantibodies abrogated cell-cell adhesion based on Caspr·contactin and NF155 trans-interaction and perturbed paranodal junctions in myelinating culture of dorsal root ganglia (DRG) neurons. Next, we used these functional blocking antibodies combined with mutagenesis of N-glycosylation sites to dissect the role of the N-linked carbohydrates of contactin. Our data demonstrated the critical role of N-glycans in the assembly of the paranodal complex. Worth noting, they underscore that CIDP antibodies may participate in the demyelination process by interfering with the paranodal antigens.
EXPERIMENTAL PROCEDURES
Constructs
The mouse contactin, contactin-ΔIg, and contactin-ΔFnIII in pRC-CMV, mouse contactin-GFP, rat Caspr in pBKCMV, and Caspr-GFP constructs have been described previously (2, 12). GFP has been inserted at the C terminus both for Caspr and contactin. The rat NF155 coding sequence (GenBankTM number AY061639.1) was amplified by PCR and inserted into the XhoI-HindIII sites of pmCherry-N1. Mutations of the N-glycosylation sites of contactin-GFP were generated by QuikChange mutagenesis (Agilent Technologies). The nine consensus N-glycosylation sites (residues 208, 258, 338, 457, 473, 494, 521, 593, and 935) were independently mutated from NX(S/T) to QX(S/T). Double N521Q/N593Q and triple N457Q/N473Q/N494Q mutations were sequentially generated. PCR-amplified products were verified by sequencing (Beckman Coulter Genomics). The constructs were analyzed after transfection in N2a cells. Cells were lysed for 30 min on ice with 50 mm Tris, pH 7.5, 1% Nonidet P-40, 10 mm MgCl2, and protease inhibitors and centrifuged at 4 °C for 15 min at 15,000 rpm. The lysates were analyzed by SDS-PAGE and immunoblotting with anti-GFP mab.
Antibodies and Chemicals
The IgGs from patients' sera were purified using the Melon gel kit (Pierce). We used a rabbit antiserum reacting against the intracellular region of Caspr (20). Mouse anti-panNav mAb was purchased from Sigma, rat anti-MBP mAb was from Abcam, mouse anti-GFP mAb was from Roche Applied Science, and Alexa-488, -568, and -647-conjugated secondary antibodies were from Molecular Probes.
ELISA
96-well ELISA plates (MaxiSorp®, Nunc) were coated with 1 μg/ml human recombinant CNTN1 protein (Sino Biological Inc.) overnight at 4 °C. Wells were blocked with 5% nonfat milk in PBS for 1 h, incubated with sera diluted in blocking buffer for 1 h, and then incubated with peroxidase-conjugated mouse anti-human IgG1–4 secondary antibodies (Invitrogen) for 1 h at room temperature. ELISA was developed with tetramethylbenzidine solution (Biolegend), and the reaction was stopped with 1 m sulfuric acid. Optical density was measured at 450 nm in a Beckman AD340 plate reader (Beckman-Coulter). All samples were tested in duplicate. For cell ELISAs, CHO Lec1 cells grown in 96-well plates were transfected with pEGFP-N1 or contactin-GFP native or mutated for the different N-glycosylation sites. Cells were incubated 48 h after the transfection with mouse anti-GFP mAb (1:200) at 37 °C for 30 min, fixed with 4% paraformaldehyde, washed, and incubated with peroxidase-conjugated anti-mouse antibody in 3% goat serum for 1 h. After five washes, the chromogenic substrate (SuperSignal ELISA Femto, Pierce) was added, and immunoreactivity was detected at 492 nm in a Mithras LB940 reader (Berthold Technologies). Values in triplicate were normalized with respect to the total protein amounts.
Cell Cultures
The N-glycosylation mutant CHO cell line Lec1 (21), neuroblastoma N2a cells, and HEK-293 cells were grown in DMEM containing 10% FCS and were transiently transfected using jet PEI (Polyplus Transfection, Ozyme). Treatment with tunicamycin (Sigma) (2 μg/ml) was performed 4 h after transfection for 16 h before immunostaining. NF155-Fc was produced in the supernatant of transfected HEK-293 cells and affinity-purified using protein A-Sepharose. Lec1 cells transfected with control or mutant contactin-GFP were incubated with NF155-Fc (10 μg/ml) preclustered with Alexa-568-conjugated anti-human Fc (50 μg/ml) for 30 min at 37 °C.
Immunofluorescence and Confocal Microscopy
Immunostaining with IgGs from CIDP patients was performed on live cells or on cells fixed and permeabilized. Cells were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.1% Triton X-100 for 10 min or fixed with methanol for 10 min at −20 °C. Immunofluorescence staining was performed using rabbit anti-Caspr (1:2000), mouse anti-GFP mAb (1:500), and IgGs from patients (1:500 to 1:2000) diluted in DMEM with 10% FCS for 30–60 min and with secondary antibodies diluted in DMEM with 10% FCS for 30 min. Diluted IgGs from patients were incubated with 2–20 μm oligomannose-5 (Dextra) for 15 min before immunostaining. Cells were mounted in Mowiol (Calbiochem). Confocal image acquisition was performed on a Zeiss LSM780 microscope equipped with a ×63/1.32 numerical aperture oil immersion objective. Fluorescence images were acquired using an ApoTome AxioObserver Z1 inverted microscope under the control of Axiovision software (Carl Zeiss MicroImaging GmbH).
Cell Aggregation Assays
N2a cells transfected with NF155-mcherry, Caspr-GFP, and contactin or Caspr2-GFP were harvested and preincubated for 20 min with purified IgGs (1:50 and 1:200) from control or CIDP patients. Cells were mixed at a density of 2 × 105 cells/ml and incubated for 2 h at 37 °C on a rotary shaker at 80 rpm. Cell suspension (30 μl) was placed on coverslips and observed on inverted microscope. The number of cells in mixed aggregates/field was evaluated in 10 fields at the ×20 objective. A mixed aggregate was defined as formed by at least n = 5 cells, with a ratio of green to red cells between 1:2 and 2 and with the number of contacts between red and green cells >n/2. Experiments were performed in triplicate.
Perturbation of Nodes of Ranvier
Myelinating cultures of DRG neurons and Schwann cells were prepared from embryonic day 16 Wistar rats as described previously (22). Purified IgGs from control and patients 1 and 4 were added at a 1:200 dilution to DRG/Schwann cells cultured for 7 weeks. After 36 h, cells were fixed with methanol, and immunofluorescence staining was performed with rat anti-MBP mAb (1:200), mouse anti-pan-Nav mAb (1:1000), and rabbit anti-Caspr antibody (1:2000). Experiments were performed in triplicate. Parameters of node and paranode morphology were evaluated on confocal sections acquired with identical settings. The lengths of paranodes and nodal gap were estimated using immunostaining for Caspr, and the ratio of integrated fluorescence intensity for Caspr and Nav was determined using the Axiovision software. More than 70 nodes of Ranvier were analyzed for each condition.
RESULTS
In the present study, we analyzed the reactivity of autoantibodies to contactin from four patients with CIDP. Three patients shared common clinical features with aggressive symptom onset and early axonal and predominantly motor involvement and were described by Querol et al. (1). In this previous study, we showed that these patients' sera reacted strongly against paranodal structures on rat teased peripheral nerves. Contactin was identified as a target antigen in patients 1 and 2, and contactin·Caspr was identified as a target in patient 3 using immunoprecipitation from lysate of rat hippocampal neurons and mass spectrometry. A fourth patient with CIDP has recently been identified who reacted against contactin and showed exactly the same clinical features as patient 1 (Table 1). Moreover, the antibodies in patients 1, 2, and 4 are mainly IgG4, as assayed using ELISA with coated recombinant human contactin (Fig. 1). The autoantibodies in patient 3 do not react in ELISA and are also mainly IgG4, as determined by isotyping using immunofluorescence staining on HEK cells transfected with contactin·Caspr (Table 1). In addition, the sera from these four patients are negative for antiganglioside antibodies. This is an indication that these antibodies may not mediate complement activation or bind Fc receptors on effector cells and is consistent with the poor response to treatment with intravenous immunoglobulin. Therefore, we asked whether these antibodies may induce paranodal demyelination via an antigen-blocking function.
TABLE 1.
Basic epidemiological, immunological, and clinical features of CIDP patients with antibodies against contactin
Detailed features of patients 1, 2, and 3 were reported previously (1). CNTN1, contactin; CASPR1, Caspr. Titers were determined using ELISA. ND, not determined.
| Patient | Age at onset (gender) | Reactivity | Titers | Anti-contactin isotypes | Clinical at onset |
|---|---|---|---|---|---|
| 1 | 77 (female) | CNTN1 | 1:8100 | IgG4 ⋙ IgG2 ELISA | Rapidly progressive, aggressive weakness. Bed-bound |
| 2 | 60 (male) | CNTN1 | 1:900 | IgG4 ⋙ IgG2 ELISA | Progressive weakness, ataxia |
| 3 | 76 (female) | CNTN1/CASPR1 | ND | IgG4 ⋙ IgG2 cell-binding assay | Progressive weakness, sensory disturbances. Wheelchair-bound |
| 4 | 71 (male) | CNTN1 | 1:8100 | IgG4 ⋙ IgG3 > IgG2 ELISA | Rapidly progressive, aggressive weakness. Bed-bound |
FIGURE 1.
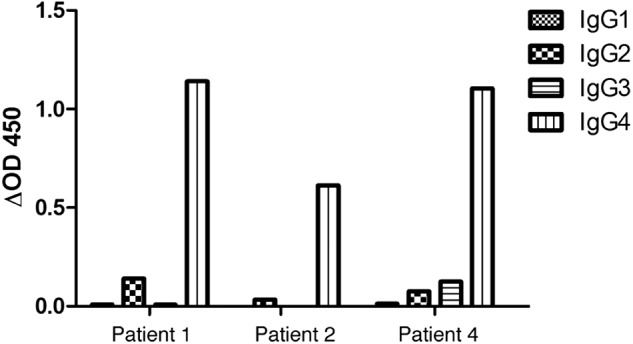
CIDP autoantibodies against contactin are of the IgG4 subclass. Shown is isotype analysis of the sera from patients 1, 2, and 4. ELISA plates were coated with human recombinant contactin (CNTN1). Optical density was measured at 450 nm after incubation with serial dilutions of CIDP sera and peroxidase-conjugated anti-human IgG1–4 secondary antibodies and reaction with tetramethylbenzidine solution.
The CIDP Autoantibodies Prevent Cell Aggregation Mediated by Caspr·Contactin and NF155
To evaluate whether the CIDP autoantibodies display functional blocking activity, we set up a cell aggregation assay based on the trans-interaction between Caspr·contactin and NF155. Neuroblastoma N2a cells co-transfected with Caspr-GFP and contactin or transfected with NF155-mcherry (Fig. 2, A and B) were mixed together and incubated on a rotating shaker. After 2 h, small mixed aggregates of 5–20 cells were observed, resulting from the preferential heterotypic adhesion between Caspr-GFP/contactin- and NF155-mcherry-expressing cells, as shown in Fig. 2D. As a negative control, N2a cells transfected with Caspr2-GFP did not co-aggregate with NF155-mcherry-expressing cells (Fig. 2C). Next, N2a cells transfected with Caspr-GFP/contactin were preincubated for 20 min with IgGs from control or CIDP patients diluted at 1:50 or 1:200 prior to cell aggregation assays. The formation of heterotypic aggregates was prevented in the presence of IgGs from CIDP patients 1, 3, and 4 and diluted 1:200, and small aggregates were formed comprising untransfected or transfected cells, as shown in Fig. 2, F, H, and I. The IgGs from patient 2 blocked the formation of mixed aggregates at 1:50 but not at 1:200 dilution (Fig. 2, E and G). This is in good agreement with the lower titer of IgGs from patient 2 (1:900) by comparison with patients 1 and 4 (1:8100) (Table 1). The quantitative analysis of mixed aggregates indicated that the CIDP autoantibodies directed against contactin are able to significantly prevent cell-cell adhesion mediated by the Caspr, contactin, and NF155 ternary complex (Fig. 2J). This result points out that anti-contactin IgG4 in this subgroup of CIDP patients may display functional blocking activity.
FIGURE 2.

CIDP autoantibodies against contactin display functional blocking activity. Shown are cell aggregation assays using N2a cells co-transfected with Caspr-GFP and contactin (A) or transfected with NF155-mcherry (B). Cells were harvested and incubated on a rotary shaker for 2 h at 37 °C. As a negative control, cells transfected with Caspr2-GFP and NF155-mcherry did not form mixed aggregates (C). Caspr-GFP/contactin- and NF155-mcherry-expressing cells formed mixed aggregates in the presence of control IgGs diluted to 1:50 (D). IgGs from patients 1, 3, and 4 prevented the formation of mixed aggregates at a 1:200 dilution (F, H, and I). Incubation with IgGs from patient 2 blocked the formation of mixed aggregates at a 1:50 (E) but not at a 1:200 dilution (G). C–I, phase-contrast and fluorescence overlay (left) and fluorescence overlay (right). Bar, 40 μm. J, mixed aggregates were defined as described under “Experimental Procedures” after counting the green and red cells and the number of heterotypic contacts (white bars), as illustrated. 10 fields at the ×20 objective were analyzed for each condition. Shown is the mean number ± S.E. (error bars) of cells recruited in mixed aggregates per field. *, significant difference (p < 0.01) by comparison with control IgGs using analysis of variance and Fisher's test.
The CIDP Autoantibodies Induce Paranodal Alterations in Myelinated DRG/Schwann Cell Culture
Next, we used myelinated DRG/Schwann cell culture from rats to determine whether the anti-contactin autoantibodies may have a perturbating effect on paranodal junctions. DRG neurons cultured for 7 weeks with ascorbic acid displayed a high density of myelinated segments, numerous heminodes, and nodes of Ranvier (Fig. 3D). The paranodal junctions were strongly labeled with IgGs from CIDP patients (Fig. 3, A–C). Immunostaining was performed after cell fixation and permeabilization using patients' IgGs diluted at 1:500. In perturbating assays, myelinating cultures were incubated for 36 h with IgGs from control or patients 1 and 4 diluted at 1:200. We estimated the number of MBP-positive myelinated segments and Caspr/Nav-positive nodes observed after treatment with control or CIDP IgGs (Fig. 3, D and E). The number of myelinated segments/field was unchanged (10.3 for control versus 12.1 for patient 1 and 9.9 for patient 4 IgGs). The number of node/myelinated segment was not significantly modified (0.27 for control versus 0.29 for patient 1 and 0.23 for patient 4 IgGs).
FIGURE 3.
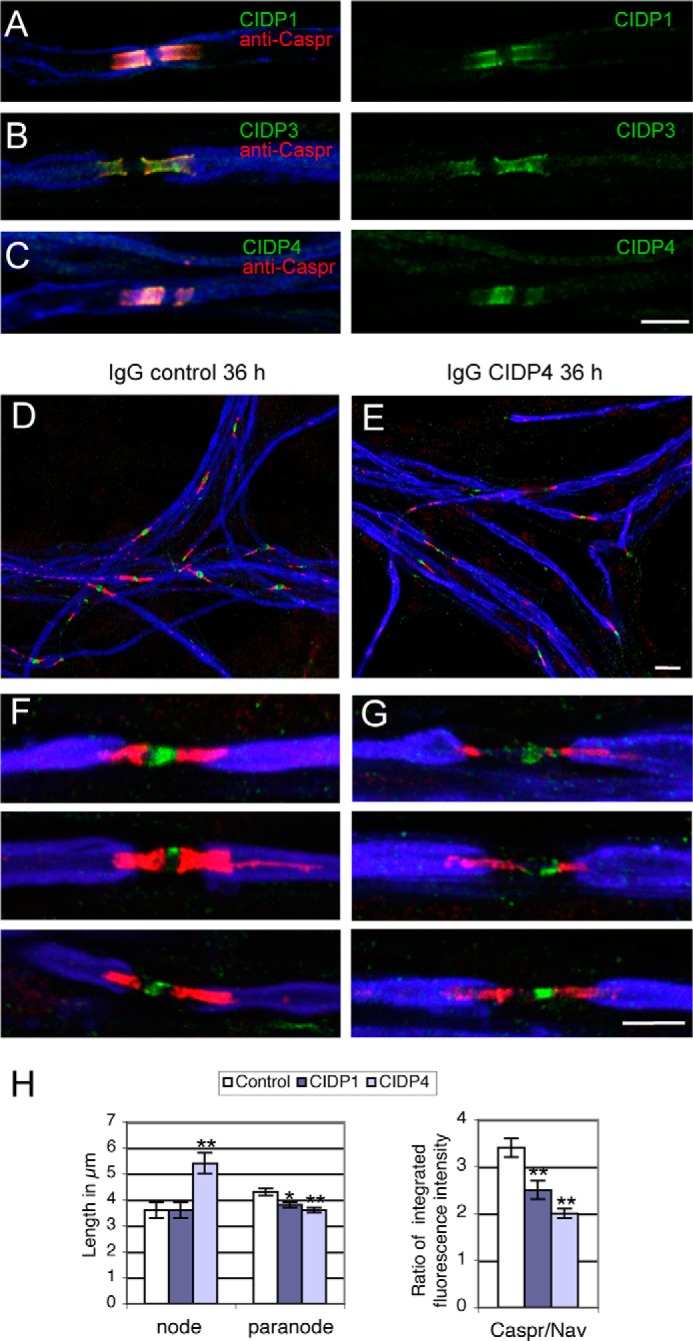
CIDP autoantibodies against contactin induce paranodal alteration in myelinated DRG neurons in culture. DRG neuron/Schwann cell mixed cultures were induced for myelination for 7 weeks in vitro. A–C, serum IgGs from CIDP patients 1, 3, and 4 binds the paranodes (green). Cells were fixed with 1% paraformaldehyde and permeabilized with 0.1% Triton X-100 before immunostaining with IgGs from CIDP patients diluted 1:500 (green), rabbit anti-Caspr (red), and rat anti-MBP (blue) as a myelin marker. In functional assays, cells were incubated with control (D and F) or patient 4 (E and G) IgGs diluted 1:200 for 36 h. Cells were fixed with methanol and immunostained with rabbit anti-Caspr (red), mouse anti-pan Nav (green), and rat anti-MBP (blue). Note that IgGs from CIDP patient 4 induced alteration of paranodal immunostaining and enlargement of the nodal region. H, quantitative analysis of nodal and paranodal lengths and of the ratio between Caspr and Nav integrated fluorescence intensity after incubation with IgGs of control and CIDP patients 1 and 4. More than 70 nodes were analyzed under each condition. * and **, significant difference (p < 0.05 and p < 0.01, respectively) in comparison with control IgGs using analysis of variance and Fisher's test. Bar in A–C, F, and G, 5 μm; bar in D and E, 10 μm. Error bars, S.E.
After a 36-h incubation with control IgGs, numerous nodes of Ranvier displayed compacted paranodal junctions labeled for Caspr and short nodal gap enriched in Nav channels (Fig. 3F), as observed in untreated cultures. When incubated with patients' IgGs, a striking alteration in Caspr immunostaining was observed (Fig. 3G). Quantitative analysis after incubation with control IgGs indicated a mean nodal gap length of 3.6 ± 0.3 μm and paranodal length of 4.3 ± 0.13 μm. The nodal gap was significantly enlarged (5.4 ± 0.4 μm) after treatment with IgGs from patient 4 (Fig. 3, G and H) but not from patient 1. The mean paranodal length was slightly reduced after treatment with IgGs from both patients (Fig. 3H). The most significant effect consisted in a decrease in Caspr immunostaining at paranodes with respect to nodal Nav labeling (Fig. 3H), as illustrated in Fig. 3G for patient 4. These data indicate that anti-contactin antibodies from these CIDP patients may contribute to the pathogenesis through alteration of the paranodal adhesive complex.
The Ig Domains of Contactin Are Selectively Targeted by CIDP Autoantibodies
Because the anti-contactin IgGs from patients display functional blocking activity, we decided to further characterize their reactivity. Contactin is a multimodular glycoprotein that contains six IgC2 domains and four fibronectin-like type III (Fn) repeats. Contactin interacts through its Ig domains with multiple ligands, including neurofascins, NrCAM, the receptor protein tyrosine phosphatase RPTP-β, or tenascin-C (23–25), which are implicated in the organization of the nodes of Ranvier and myelination (26). The serum of patients immunoreacted at the same dilution against human contactin (CNTN1) transfected in HEK cells (Fig. 4, M and N) and mouse contactin transfected in N2a cells (Fig. 4, A and C), as illustrated for patients 1 and 2. It should be noted that contactin exhibits 97% amino acid identity between human and mouse or rat sequences. Therefore, the anti-contactin autoantibodies display a very high degree of species cross-reactivity.
FIGURE 4.

Autoantibodies from CIDP patients 1, 2, and 4 are targeted against the N-glycosylated Ig domains of contactin. N2a cells were transfected with contactin (A and C), contactin-Ig showing a deletion of the four Fn domains (B and D), or HA-tagged contactin-Fn showing a deletion of the six Ig domains (E and F). Surface labeling indicated that IgGs from CIDP patients 1 and 2 recognized contactin and contactin-Ig but not contactin-Fn. Double staining of contactin-Fn using anti-HA mAb (green) in E and F. G–L, N2a cells transfected with contactin-GFP were untreated (G–I) or treated (J–L) with tunicamycin (2 μg/ml). Cells were fixed and permeabilized before immunostaining with IgGs from CIDP patients 1, 2, and 4 (red). Unglycosylated contactin-GFP was retained in the ER and was not recognized by the autoantibodies. M and N, the sera from patients 1 and 2 recognized human CNTN1 transfected in HEK cells. Bars in A–F, M, and N, 10 μm; bars in G–L, 20 μm.
We analyzed whether anti-contactin autoantibodies may be directed against the Ig or Fn domains. Full-length contactin or contactin mutants deleted from its Fn or Ig domains were transfected in N2a cells (Fig. 4). Using immunofluorescence staining on live cells, we showed that contactin-Ig was targeted by autoantibodies in CIDP patients 1 and 2 (Fig. 4, B and D), whereas contactin-Fn labeled with anti-HA mAb was not (Fig. 4, E and F), as also observed in patient 4 (not shown). In contrast, contactin-Ig (Fig. 5D) and contactin-Fn (not shown) were not immunolabeled by the serum from patient 3, indicating that only the full-length structure of contactin was recognized.
FIGURE 5.

Autoantibodies from CIDP patient 3 specifically recognize contactin bearing mannose-rich N-glycans. N2a cells were co-transfected with contactin-GFP and Caspr (A) or transfected with contactin-GFP alone (B) and surface-labeled with purified IgGs from CIDP patient 3 (red). CHO Lec1 mutant cells transfected with contactin (C) or contactin-Ig (D) were surface-labeled with IgGs from CIDP patient 3 (C and D, red) and rabbit anti-contactin antibody (green). CHO Lec1 cells were transfected with contactin and surface-labeled with IgGs from patient 1 (E) and patient 2 (F). As schematized, contactin co-expressed with Caspr in N2a cells or contactin expressed in Lec1 cells bears mannose-rich N-glycans. In contrast, contactin expressed in N2a cells bears complex N-glycans. Only mannose-rich contactin was targeted by autoantibodies from CIDP patient 3. G and H, N2a transfected with Caspr and contactin (G) and Lec1 cells transfected with contactin alone (H). Preincubation of IgGs from patient 3 with 20 μm oligomannose-5 did not prevent surface labeling. Bar, 10 μm.
Contactin N-Glycosylation Is Implicated in Recognition by CIDP Autoantibodies
We showed previously that contactin associated with Caspr bears mannose-rich N-glycans, whereas contactin expressed alone is processed with complex N-glycans, as illustrated in Fig. 5 (2). The mannose-rich glycoform of contactin preferentially binds NF155, its glial partner at paranodes, as demonstrated using the glycosyltransferase mutant Lec1 cells (2). In the same manner, Lec1 cells were used here for expressing N-glycosylated proteins of the mannose-rich subtype, as schematized in Fig. 5 (21).
The autoantibodies from patients 1 and 2 recognized both the complex and mannose-rich glycoforms of contactin expressed in N2a and Lec1 cells, respectively (Figs. 4 (A and C) and 5 (E and F)). Next, N2a cells were treated with 2 μg/ml tunicamycin, which was added 4 h after transfection to completely block the linkage of N-glycans. Because N-glycosylation is tightly implicated in the quality control and export of glycoproteins to the cell surface, tunicamycin treatment prevented cell surface expression of contactin in N2a cells. Therefore, immunofluorescence labeling after cell fixation and permeabilization was required to analyze whether CIDP autoantibodies reacted against unglycosylated contactin-GFP retained in the ER. Under this condition, immunoreactivity for contactin was completely prevented, indicating that the autoantibodies from patients 1, 2, and 4 only recognized N-glycosylated contactin (Fig. 4, G–L).
Next, we found out that autoantibodies of patient 3 were specifically directed against contactin bearing mannose-rich N-glycans (Fig. 5). Indeed, contactin-GFP expressed alone in N2a cells was not recognized (Fig. 5B), whereas a strong labeling was observed when contactin-GFP was associated with Caspr (Fig. 5A). Contactin transfected alone in Lec1 cells and double-stained with rabbit anti-contactin (green) was recognized by the IgGs of patient 3 (Fig. 5C). Moreover, the serum of patient 3 contained antibodies directed against Caspr, as shown in Fig. 6A. Mutants of Caspr with complementary deletions of the N- or C-region of the ectodomain were expressed in N2a cells and double-stained using rabbit anti-Caspr antibodies (Fig. 6, A–C, right). These deletion mutants were not recognized by the autoantibodies (Fig. 6, B and C). Finally, treatment with tunicamycin indicated that autoantibodies of patient 3 did not recognize the unglycosylated protein core of Caspr, which was double-labeled using rabbit anti-Caspr antibodies (Fig. 6, D and E). Thus, the serum of CIDP patient 3 contains antibodies directed against the complex of Caspr and contactin bearing mannose-rich N-glycans. In order to test whether the mannose residues could be targeted alone, IgGs of CIDP patients were preincubated with 2 or 20 μm oligomannose-5 before the immunofluorescence assay. As shown in Fig. 5, G and H, the presence of oligomannose-5 did not block the binding of IgGs of patient 3 on contactin-GFP·Caspr co-expressed in N2a cells or on contactin-GFP transfected alone in Lec1 cells. We also determined that the immunoreactivity of IgGs from patients 1 and 2 was not prevented by oligomannose-5, suggesting that oligomannose-5 alone is not the target of those antibodies.
FIGURE 6.
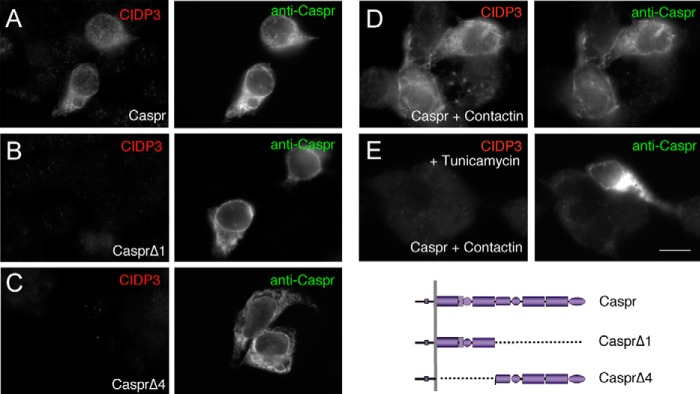
Antibodies from CIDP patient 3 are directed against N-glycosylated Caspr. N2a cells were transfected with Caspr (A), CasprΔ1 (B), or CasprΔ4 (C), which are deleted from the N- or C-region of the ectodomain, respectively. Cells were fixed with methanol and double-stained with IgGs from patient 3 (left) and rabbit anti-Caspr antibodies (right). N2a cells co-transfected for Caspr and contactin were untreated (D) or treated (E) with tunicamycin and fixed with methanol. Note that Caspr was expressed at the plasma membrane only when co-expressed with contactin (compared D with A). The autoantibodies from patient 3 reacted against full-length N-glycosylated Caspr. Bar, 10 μm.
The N-Linked Carbohydrates on the Ig5 Domain of Contactin Are Required for Neurofascin Binding
Because contactin N-glycans are crucial for NF155 binding and may determine the assembly or stability of the paranodal complex, we started a functional analysis of the N-glycosylation sites of contactin using mutagenesis. Contactin contains nine consensus sites of N-glycosylation (NX(S/T)) mainly present in the Ig domains. All of these sites have been conserved during evolution from Xenopus to humans. Each of these sites was mutated independently, with asparagine changed to glutamic acid, in contactin fused with GFP. Notably, we performed a triple mutation of the three sites in Ig5 and double mutation of the two sites in Ig6 domains as depicted in Fig. 7. The mutants were expressed in N2a cells and analyzed by SDS-PAGE and immunoblotting with anti-GFP mAb (Fig. 7F). The mutant with three mutations on the Ig5 (N457Q/N473Q/N494Q) was detected with a shift to a lower apparent molecular weight by comparison with control contactin-GFP. This indicates that the predicted N-glycosylation sites on Ig5 were occupied.
FIGURE 7.
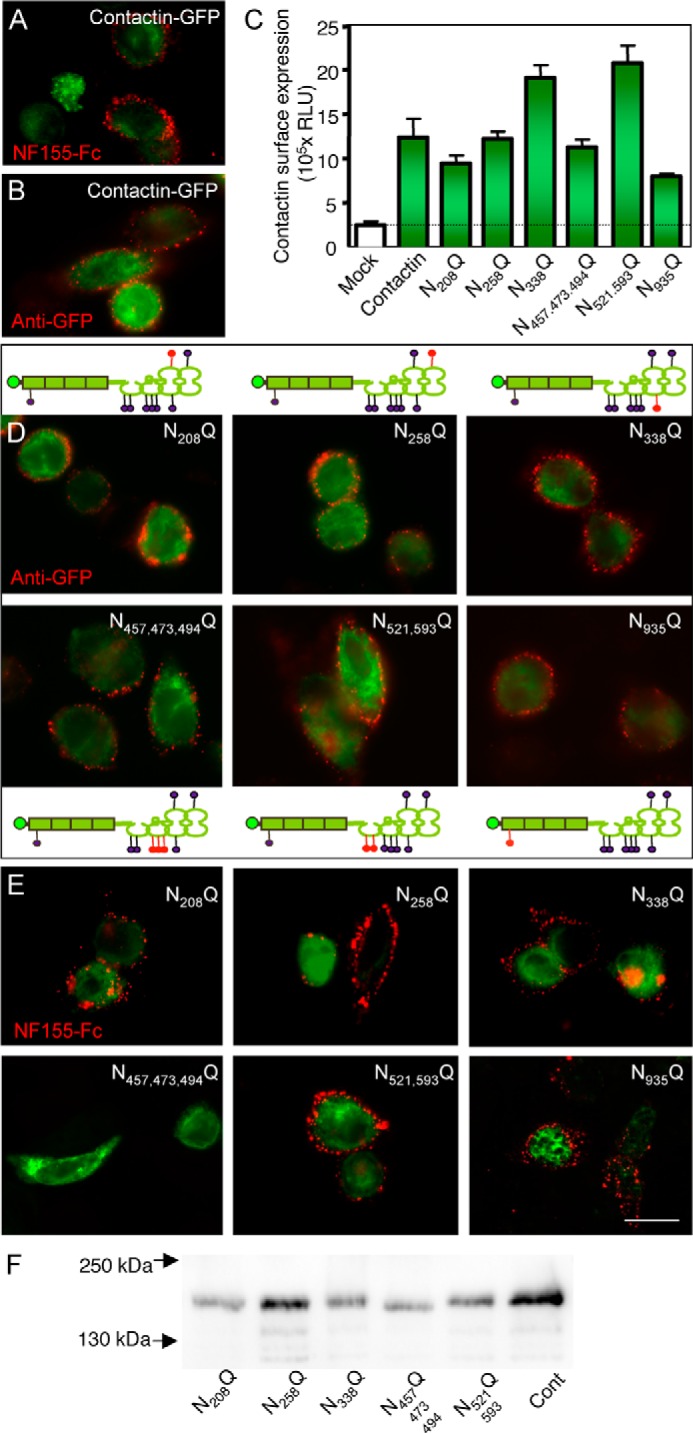
Selective mutations of N-glycosylation sites of contactin prevent its trans-interaction with NF155. Lec1 cells were transfected with contactin-GFP (A and B) or contactin-GFP mutated for its N-glycosylation sites (D and E) as schematized, with the mutated sites indicated in red. All of the mutated constructs were detected at the cell surface using mouse anti-GFP and Alexa-568-conjugated secondary antibodies (A and D). C, quantitative analysis of the cell surface expression of contactin-GFP mutants using cell ELISA. B and E, cells were incubated with 50 nm NF155-Fc chimera and Alexa-568-conjugated anti-human antibodies (red). Note that mutation of the three sites Asn-457, Asn-473, and Asn-494 in the Ig5 domain of contactin prevented NF155-Fc binding. F, Western blot analysis of contactin-GFP mutants expressed in N2a cells and detected using mouse anti-GFP mAb. Note the lower apparent molecular weight of the N457Q/N473Q/N494Q mutant by comparison with the other mutants and control contactin-GFP (Cont). Bar, 10 μm. Error bars, S.E. N521,593Q, N521Q/N593Q; N457,473,494Q, N457Q/N473Q/N494Q.
We checked that all of the contactin mutants were properly targeted at the plasma membrane using surface labeling with anti-GFP antibody (Fig. 7D). In addition, using cell ELISA, we showed no significant difference in the cell surface expression of the contactin mutants, indicating that the mutations did not alter their export (Fig. 7C). Binding experiments were performed on Lec1 cells transfected with contactin-GFP mutants and incubated with soluble NF155-Fc chimera. Strikingly, we observed that NF155 binding was completely abolished on contactin N457Q/N473Q/N494Q mutant (Fig. 7E) but not on the other mutants. Independent mutations of each of these sites of Ig5 had no effect on NF155-Fc binding (not shown). Next, we tested whether mutations of N-glycosylation sites may impair the interaction between contactin and Caspr. Contactin associates in cis with Caspr and induces its release from the ER and its export at the cell surface, as shown in Fig. 8. We observed that all of the contactin mutants induced the cell surface targeting of Caspr when co-transfected in N2a cells, as did the wild-type contactin (Fig. 8). This is a strong indication that the overall conformation of contactin is not strongly altered in the mutants. These data suggest that NF155 may bind a domain of contactin that comprises a cluster of carbohydrates of Ig5.
FIGURE 8.
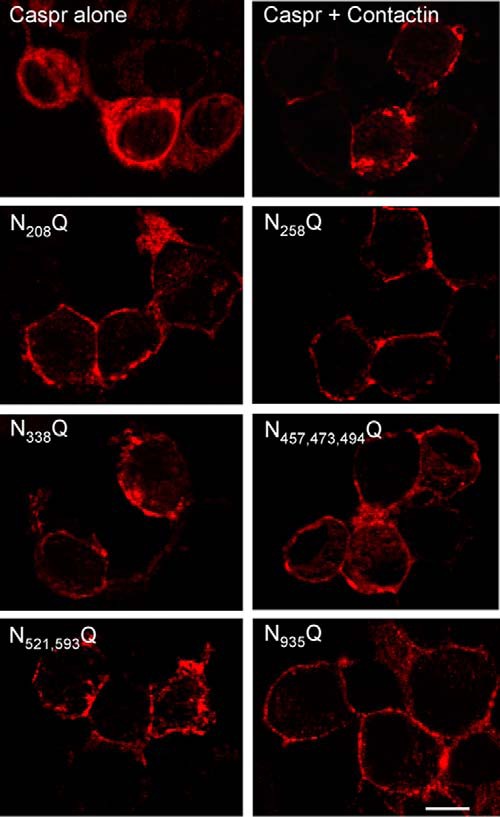
Mutations of N-glycosylation sites of contactin do not prevent its cis-association with Caspr. N2a cells were transfected with Caspr alone or co-transfected with Caspr and contactin-GFP or with Caspr and contactin-GFP mutated for its N-glycosylation sites. Cells were fixed with methanol and immunostained with rabbit anti-Caspr antibodies and Alexa-568-conjugated secondary antibodies. The co-expression of wild-type or mutated contactin-GFP induces ER exit and cell surface targeting of Caspr. Bar, 5 μm. N521,593Q, N521Q/N593Q; N457,473,494Q, N457Q/N473Q/N494Q.
The N-Glycans on the Ig5 Domain of Contactin Are Required for Recognition by CIDP Autoantibodies in Patient 2
To determine whether specific N-glycosylated residues of contactin may be required for the binding of CIDP autoantibodies, the N-glycosylation mutants were transfected in N2a cells and assayed for immunoreactivity. Strikingly, we observed that the serum of patient 2 did not recognize contactin N457Q/N473Q/N494Q but recognized the other mutants (Fig. 9B). In contrast, immunoreactivity of the serum of patient 1 was not prevented by any mutation of the N-glycosylation sites (Fig. 9A), as also observed for patient 4 serum (not shown). Immunoreactivity of the serum of patient 3, which is specific for the mannose-rich glycoform of contactin, was tested using transfected Lec1 cells and was not reduced by any of the mutations (Fig. 9C). Therefore, the cluster of N-glycans on contactin Ig5 that determines NF155 binding is selectively targeted by autoantibodies in CIDP patient 2.
FIGURE 9.
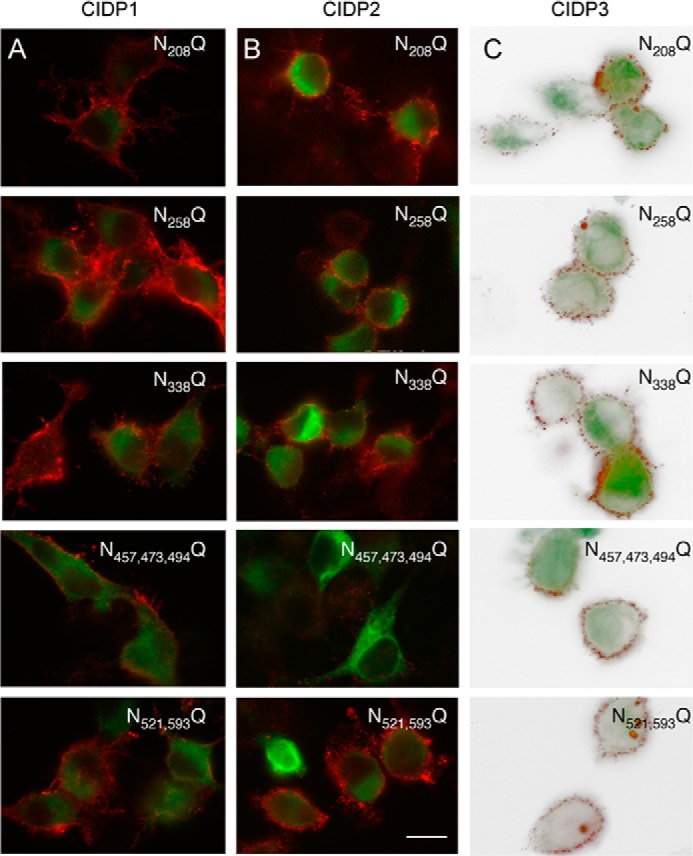
The N-glycosylation sites of the Ig5 domain of contactin are selectively required for immune reactivity of CIDP patient 2. N2a cells (A and B) or Lec1 cells (C) were transfected with contactin-GFP or contactin-GFP mutated for its N-glycosylation sites. Cells were fixed with paraformaldehyde and immunostained with IgGs from CIDP patient 1 (A), patient 2 (B), and patient 3 (C) and Alexa-568-conjugated anti-human antibodies (red). Note that mutation of the three sites Asn-457, Asn-473, and Asn-494 in contactin Ig5 prevented immunoreactivity with IgGs from patient 2 but not from patients 1 and 3. Bar, 10 μm. N521,593Q, N521Q/N593Q; N457,473,494Q, N457Q/N473Q/N494Q.
DISCUSSION
We have developed cell-binding assays to characterize the selectivity of anti-contactin autoantibodies of four patients with an aggressive form of CIDP (Table 2). Importantly, we showed that in these patients, anti-contactin IgGs block cell aggregation mediated by the Caspr·contactin·NF155 complex. In addition, these autoantibodies induce alteration of paranodes, as shown using an in vitro model of myelinated DRG neurons. These anti-contactin antibodies are of the IgG4 isotype and are expected to be pathogenic in a complement-independent way. Our data suggest that they may be pathogenic by disrupting paranodal junctions, thereby inducing a severe reduction of conduction velocity. The different selectivity of these autoantibodies highlights the complex role of N-linked carbohydrate residues of contactin in paranode formation: 1) in patient 2, immune reactivity relies on three N-glycosylation sites of the Ig5 domain of contactin that are implicated in NF155 binding; 2) in patient 3, anti-contactin antibodies specifically react with the high mannose glycoform of contactin, which preferentially binds NF155.
TABLE 2.
Specificity of anti-contactin antibodies in CIDP patients
| Patient | Blocking function | Contactin epitope | Reactivity against |
||||
|---|---|---|---|---|---|---|---|
| Caspr | Tunicamycin-treated | Complex N-glycans | Mannose-rich | N457Q/N473Q/N494Q | |||
| 1 | + | Ig | − | − | + | + | + |
| 2 | + | Ig | − | − | + | + | − |
| 3 | + | Full-length | + | − | − | + | + |
| 4 | + | Ig | − | − | + | + | + |
A Cluster of Oligosaccharides on the Ig5 Domain of Contactin Is Implicated in Interaction with Neurofascin-155
We identified a cluster of three N-glycans located on contactin Ig5 sites specifically required for NF155 binding. Strikingly, these sites are also specifically required for recognition by function-blocking autoantibodies in one CIDP patient. NF155 may directly bind the oligosaccharide chains on Ig5 of contactin. Alternatively, the presence of N-glycans on contactin may support an optimal conformation of the polypeptide for the binding of NF155.
Contactin constructs with mutations of N-glycosylation sites were detected at the cell surface as an indication that they passed the primary quality control in the ER. Moreover, all of these contactin mutants associate with Caspr and trigger its export to the plasma membrane. The unglycosylated protein cores of contactin and Caspr can be co-immunoprecipitated (13). Thus, it was expected that the contactin mutants missing only one or few N-linked oligosaccharide chains, could associate with Caspr. We previously showed that the ER retention motif of Caspr consists of a short sequence with PGY motifs in the ectodomain of the protein and that association of contactin may induce a switch toward a transport-permissive conformation (2). This is consistent with the view that mutations of the different N-glycosylation sites of contactin did not induce gross structural alteration. However, the absence or presence of oligosaccharide chains on the Ig5 domain may induce a local structural variation that may be critical for NF155 binding.
Structural information on the six Ig domains of contactin is still incomplete. The three-dimensional structure of contactin-2 was the first to be reported, showing that the Ig1–4 domains adopt a horseshoe conformation (27, 28). It seems that this structural motif is a hallmark of contactin family members (25, 29). The crystal structure of contactin-4 has been resolved for the Ig1–4 domains with the four N-glycosylation sites exposed to the molecular surface (29).
Functional Relevance of Contactin Processed with Mannose-rich N-Linked Carbohydrates
There is great structural variety based on the type, length, and linkage of the carbohydrate components, and this glycan structural diversity can provide insights into potential biological recognition events. We previously demonstrated that the glycan composition of contactin is of importance for modulating its interaction with NF155 (2). Here, we showed that the sera of three patients bind the Ig domains of contactin bearing either complex or mannose-rich N-glycans. In contrast, the serum of one patient reacts specifically with the mannose-rich glycoform of contactin. In addition, it reacts independently with Caspr, which also bears oligomannose type sugars (13). It must be pointed out that only the full-length and not the truncated contactin and Caspr are recognized by such autoantibodies. This means that the overall molecular structure with specific post-translational modification is required for the independent recognition of Caspr and contactin, whereas their cis-association is not a prerequisite for antibody recognition. Unlike other human IgG subclasses, the IgG4 isotype has been reported to have two different antigen-combining sites (30). This bispecific activity may explain why IgG4 of patient 3 binds contactin and Caspr glycoproteins.
In autoimmune disease, the fact that autoantibodies may preferentially target conformational epitopes is well documented. The question is not resolved of whether the autoantibodies recognize a glycopeptidic motif or a conformational epitope, which may depend on the presence of N-oligosaccharide residues. We determined that the autoantibodies may not react with N-linked glycans alone because their binding on contactin is not blocked by preincubation with oligomannose-5. With this respect, it is interesting to note that a neutralizing antibody isolated from a patient has been reported, which is directed against the carbohydrates of the envelope glycoprotein gp120 of HIV-1 virus. The unusual configuration of this antibody allows targeting of a cluster of three oligomannose type sugars through multivalent interaction (31).
Nodal and Paranodal CAMs as Immune Targets in CIDP
CIDP is the most frequent chronic inflammatory neuropathy but includes a remarkably heterogeneous group of clinical features and pathogenic mechanisms. The nodal and paranodal antigens, including gliomedin, contactin, NrCAM, NF186, and NF155, have been identified recently as targets for autoimmunity in CIDP and Guillain-Barré syndrome patients (1, 14, 15, 17). Intriguingly, both anti-contactin and anti-NF155 antibodies in CIDP patients are of the IgG4 isotype. The reason why IgG4 is the main subclass induced in these CIDP subgroups, as also observed in other IgG4-mediated diseases, is unknown (32). IgG4 plays a role in tolerization of persistent antigens in IgE-mediated diseases (33), but its role in non-allergic immune responses has not been extensively studied so far. An important question is whether the pathogenicity of the autoantibodies may rely on recognition of functional epitopes. In myasthenia gravis, for example, it has recently been shown that binding of IgG4 antibodies to MuSK prevents the physiological role of specific proteins of the neuromuscular junctions (34).
In CIDP, autoantibodies may bind specific functional modules of CAMs and act by perturbating cell adhesion. Interestingly, antibodies raised against the extracellular domain of NF155 have been reported to inhibit myelination in a co-culture system of brain neurons and oligodendrocytes. The anti-NF155 antibodies added before the onset of myelination for a 2-week period strongly reduced the number of myelinated segments, presumably by blocking the adhesive relationship between axons and oligodendrocytes (6). Different conditions were used in the present study, with CIDP autoantibodies added for a short period of time (36 h) in a mature co-culture of neurons and Schwann cells. Therefore, we did not observe any decrease in the number of myelinated segments but alteration of the paranodal junctions. In addition to perturbating adhesive function, antibodies to contactin may have an additional effect on cell signaling or internalization. We did not observe any endocytosis of contactin after cross-linking with antibodies in transfected COS-7 cells. It is interesting to note that antibody-mediated cross-linking of contactin has been shown to elicit an increase in the Fyn activity in neurons and oligodendrocytes, regulating positively their survival (35–37). Further experiments will be required to examine the effect of CIDP anti-contactin autoantibodies in the modulation of glycoprotein signaling at the paranodes.
Antibodies specific to NF155 have been reported in two patients with CIDP that are mainly of the IgG4 subclass and recognize the Fn3-Fn4 domains in both patients (15). The domains of NF155 interacting with contactin have been mapped to the Ig5-Ig6, whereas the Fn1-Fn2 domains shared with NF186 interact with the nodal gliomedin (22, 38). The Fn3-Fn4 region is specific to the NF155 isoform, and the way it may participate in the formation of paranodal junctions is unknown. We showed in the present study that the Ig domains of contactin, which are implicated in adhesive interaction, are specifically targeted in CIDP patients. Moreover, the N-glycosylation-specific recognition of contactin is also compatible with perturbation of paranodal complex. Importantly, the anti-contactin autoantibodies display functional blocking activity in cell aggregation assays and perturbating effects on paranodal junctions of myelinated axons in culture. These data strongly suggest that anti-contactin antibodies may be pathogenic through the disruption of paranodal architecture and nodal demyelination.
Acknowledgments
We thank Dr. Jérôme Devaux and Dr. Marie-Jeanne Papandréou for helpful discussions and critical reading of the manuscript.
This work was supported by the Association pour la Recherche sur la Sclérose en Plaques (to C. F.-S.) and the European E-rare-2 project (to C. F.-S. and I. I.).
- CAM
- cell adhesion molecule
- CIDP
- chronic inflammatory demyelinating polyradiculoneuropathy
- DRG
- dorsal root ganglia
- Fn
- fibronectin type III-like domain
- NF
- neurofascin
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Querol L., Nogales-Gadea G., Rojas-Garcia R., Martinez-Hernandez E., Diaz-Manera J., Suárez-Calvet X., Navas M., Araque J., Gallardo E., Illa I. (2013) Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 73, 370–380 [DOI] [PubMed] [Google Scholar]
- 2. Bonnon C., Bel C., Goutebroze L., Maigret B., Girault J. A., Faivre-Sarrailh C. (2007) PGY repeats and N-glycans govern the trafficking of paranodin and its selective association with contactin and neurofascin-155. Mol. Biol. Cell 18, 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eshed Y., Feinberg K., Poliak S., Sabanay H., Sarig-Nadir O., Spiegel I., Bermingham J. R., Jr., Peles E. (2005) Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 47, 215–229 [DOI] [PubMed] [Google Scholar]
- 4. Sherman D. L., Tait S., Melrose S., Johnson R., Zonta B., Court F. A., Macklin W. B., Meek S., Smith A. J., Cottrell D. F., Brophy P. J. (2005) Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48, 737–742 [DOI] [PubMed] [Google Scholar]
- 5. Feinberg K., Eshed-Eisenbach Y., Frechter S., Amor V., Salomon D., Sabanay H., Dupree J. L., Grumet M., Brophy P. J., Shrager P., Peles E. (2010) A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 65, 490–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Charles P., Tait S., Faivre-Sarrailh C., Barbin G., Gunn-Moore F., Denisenko-Nehrbass N., Guennoc A. M., Girault J. A., Brophy P. J., Lubetzki C. (2002) Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr. Biol. 12, 217–220 [DOI] [PubMed] [Google Scholar]
- 7. Bhat M. A., Rios J. C., Lu Y., Garcia-Fresco G. P., Ching W., St Martin M., Li J., Einheber S., Chesler M., Rosenbluth J., Salzer J. L., Bellen H. J. (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30, 369–383 [DOI] [PubMed] [Google Scholar]
- 8. Boyle M. E., Berglund E. O., Murai K. K., Weber L., Peles E., Ranscht B. (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30, 385–397 [DOI] [PubMed] [Google Scholar]
- 9. Poliak S., Salomon D., Elhanany H., Sabanay H., Kiernan B., Pevny L., Stewart C. L., Xu X., Chiu S. Y., Shrager P., Furley A. J., Peles E. (2003) Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 162, 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Traka M., Goutebroze L., Denisenko N., Bessa M., Nifli A., Havaki S., Iwakura Y., Fukamauchi F., Watanabe K., Soliven B., Girault J. A., Karagogeos D. (2003) Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J. Cell Biol. 162, 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation. Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faivre-Sarrailh C., Gauthier F., Denisenko-Nehrbass N., Le Bivic A., Rougon G., Girault J. A. (2000) The glycosylphosphatidyl inositol-anchored adhesion molecule F3/contactin is required for surface transport of paranodin/contactin-associated protein (caspr). J. Cell Biol. 149, 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonnon C., Goutebroze L., Denisenko-Nehrbass N., Girault J. A., Faivre-Sarrailh C. (2003) The paranodal complex of F3/contactin and caspr/paranodin traffics to the cell surface via a non-conventional pathway. J. Biol. Chem. 278, 48339–48347 [DOI] [PubMed] [Google Scholar]
- 14. Devaux J. J., Odaka M., Yuki N. (2012) Nodal proteins are target antigens in Guillain-Barre syndrome. J Peripher. Nerv. Syst. 17, 62–71 [DOI] [PubMed] [Google Scholar]
- 15. Ng J. K., Malotka J., Kawakami N., Derfuss T., Khademi M., Olsson T., Linington C., Odaka M., Tackenberg B., Prüss H., Schwab J. M., Harms L., Harms H., Sommer C., Rasband M. N., Eshed-Eisenbach Y., Peles E., Hohlfeld R., Yuki N., Dornmair K., Meinl E. (2012) Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 79, 2241–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prüss H., Schwab J. M., Derst C., Görtzen A., Veh R. W. (2011) Neurofascin as target of autoantibodies in Guillain-Barre syndrome. Brain 134, e173. [DOI] [PubMed] [Google Scholar]
- 17. Querol L., Nogales-Gadea G., Rojas-Garcia R., Diaz-Manera J., Pardo J., Ortega-Moreno A., Sedano M. J., Gallardo E., Berciano J., Blesa R., Dalmau J., Illa I. (2014) Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lonigro A., Devaux J. J. (2009) Disruption of neurofascin and gliomedin at nodes of Ranvier precedes demyelination in experimental allergic neuritis. Brain 132, 260–273 [DOI] [PubMed] [Google Scholar]
- 19. Devaux J. J. (2012) Antibodies to gliomedin cause peripheral demyelinating neuropathy and the dismantling of the nodes of Ranvier. Am. J. Pathol. 181, 1402–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Menegoz M., Gaspar P., Le Bert M., Galvez T., Burgaya F., Palfrey C., Ezan P., Arnos F., Girault J. A. (1997) Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron 19, 319–331 [DOI] [PubMed] [Google Scholar]
- 21. Stanley P., Ioffe E. (1995) Glycosyltransferase mutants. Key to new insights in glycobiology. FASEB J. 9, 1436–1444 [DOI] [PubMed] [Google Scholar]
- 22. Labasque M., Devaux J. J., Lévêque C., Faivre-Sarrailh C. (2011) Fibronectin type III-like domains of neurofascin-186 protein mediate gliomedin binding and its clustering at the developing nodes of Ranvier. J. Biol. Chem. 286, 42426–42434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zisch A. H., D'Alessandri L., Ranscht B., Falchetto R., Winterhalter K. H., Vaughan L. (1992) Neuronal cell adhesion molecule contactin/F11 binds to tenascin via its immunoglobulin-like domains. J. Cell Biol. 119, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Volkmer H., Zacharias U., Nörenberg U., Rathjen F. G. (1998) Dissection of complex molecular interactions of neurofascin with axonin-1, F11, and tenascin-R, which promote attachment and neurite formation of tectal cells. J. Cell Biol. 142, 1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lamprianou S., Chatzopoulou E., Thomas J. L., Bouyain S., Harroch S. (2011) A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc. Natl. Acad. Sci. U.S.A. 108, 17498–17503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Labasque M., Faivre-Sarrailh C. (2010) GPI-anchored proteins at the node of Ranvier. FEBS Lett. 584, 1787–1792 [DOI] [PubMed] [Google Scholar]
- 27. Freigang J., Proba K., Leder L., Diederichs K., Sonderegger P., Welte W. (2000) The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell 101, 425–433 [DOI] [PubMed] [Google Scholar]
- 28. Mörtl M., Sonderegger P., Diederichs K., Welte W. (2007) The crystal structure of the ligand-binding module of human TAG-1 suggests a new mode of homophilic interaction. Protein Sci. 16, 2174–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouyain S., Watkins D. J. (2010) The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc. Natl. Acad. Sci. U.S.A. 107, 2443–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schuurman J., Van Ree R., Perdok G. J., Van Doorn H. R., Tan K. Y., Aalberse R. C. (1999) Normal human immunoglobulin G4 is bispecific. It has two different antigen-combining sites. Immunology 97, 693–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calarese D. A., Scanlan C. N., Zwick M. B., Deechongkit S., Mimura Y., Kunert R., Zhu P., Wormald M. R., Stanfield R. L., Roux K. H., Kelly J. W., Rudd P. M., Dwek R. A., Katinger H., Burton D. R., Wilson I. A. (2003) Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300, 2065–2071 [DOI] [PubMed] [Google Scholar]
- 32. Querol L., Illa I. (2013) Myasthenia gravis and the neuromuscular junction. Curr. Opin. Neurol. 26, 459–465 [DOI] [PubMed] [Google Scholar]
- 33. Nirula A., Glaser S. M., Kalled S. L., Taylor F. R. (2011) What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr. Opin. Rheumatol. 23, 119–124 [DOI] [PubMed] [Google Scholar]
- 34. Huijbers M. G., Zhang W., Klooster R., Niks E. H., Friese M. B., Straasheijm K. R., Thijssen P. E., Vrolijk H., Plomp J. J., Vogels P., Losen M., Van der Maarel S. M., Burden S. J., Verschuuren J. J. (2013) MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc. Natl. Acad. Sci. U.S.A. 110, 20783–20788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zisch A. H., D'Alessandri L., Amrein K., Ranscht B., Winterhalter K. H., Vaughan L. (1995) The glypiated neuronal cell adhesion molecule contactin/F11 complexes with src-family protein tyrosine kinase Fyn. Mol. Cell Neurosci. 6, 263–279 [DOI] [PubMed] [Google Scholar]
- 36. Cervello M., Matranga V., Durbec P., Rougon G., Gomez S. (1996) The GPI-anchored adhesion molecule F3 induces tyrosine phosphorylation. Involvement of the FNIII repeats. J. Cell Sci. 109, 699–704 [DOI] [PubMed] [Google Scholar]
- 37. Laursen L. S., Chan C. W., ffrench-Constant C. (2009) An integrin-contactin complex regulates CNS myelination by differential Fyn phosphorylation. J. Neurosci. 29, 9174–9185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thaxton C., Pillai A. M., Pribisko A. L., Labasque M., Dupree J. L., Faivre-Sarrailh C., Bhat M. A. (2010) In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J. Neurosci. 30, 4868–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]