

Abstract
Aims. We previously reported that kamikihito (KKT), a traditional Japanese medicine, improved memory impairment and reversed the degeneration of axons in the 5XFAD mouse model of Alzheimer's disease (AD). However, the mechanism underlying the effects of KKT remained unknown. The aim of the present study was to investigate the mechanism by which KKT reverses the progression of axonal degeneration. Methods. Primary cultured cortical neurons were treated with amyloid beta (Aβ) fragment comprising amino acid residues (25–35) (10 μM) in an in vitro AD model. KKT (10 μg/mL) was administered to the cells before or after Aβ treatment. The effects of KKT on Aβ-induced tau phosphorylation, axonal atrophy, and protein phosphatase 2A (PP2A) activity were investigated. We also performed an in vivo assay in which KKT (500 mg/kg/day) was administered to 5XFAD mice once a day for 15 days. Cerebral cortex homogenates were used to measure PP2A activity. Results. KKT improved Aβ-induced tau phosphorylation and axonal atrophy after they had already progressed. In addition, KKT increased PP2A activity in vitro and in vivo. Conclusions. KKT reversed the progression of Aβ-induced axonal degeneration. KKT reversed axonal degeneration at least in part through its role as an exogenous PP2A stimulator.
1. Introduction
Nearly 36 million people worldwide are living with dementia, and as the population ages, this number is expected to be more than triple by 2050. The countries with the largest number of people with dementia are China (5.4 million), Japan (4.3 million), the United States (3.9 million), India (3.7 million), Germany (1.5 million), Russia (1.2 million), France (1.1 million), Italy (1.1 million), and Brazil (1.0 million). Dementia has become a prominent social issue [1, 2]. Alzheimer's disease (AD), the most common cause of dementia, is characterized by extracellular deposits of amyloid beta (Aβ) protein and intracellular neurofibrillary tangles (NFT) composed of hyperphosphorylated tau. Acetylcholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and an N-methyl-d-aspartate (NMDA) receptor antagonist (memantine) are currently the only approved drugs for the treatment of AD [3]. However, because these drugs do not target Aβ and hyperphosphorylated tau, it is difficult to prevent, halt, or reverse the disease [4].
In spite of efforts to discover drugs targeting Aβ and hyperphosphorylated tau, no truly effective therapy has been developed to date [5]. Solanezumab, a humanized monoclonal antibody to Aβ (16–24) that preferentially binds soluble Aβ, is one of the most notable immunotherapeutic agents for AD. In the phase II clinical trials, solanezumab increased plasma and CSF levels of Aβ40 and Aβ42, thus suggesting that the amount of Aβ plaques was decreased in the brain; however, solanezumab had no effect on behavioral tests of AD patients [6]. These results suggest that the reduction of Aβ levels after the appearance of symptoms is not effective for the treatment of AD. Other therapeutic approaches, such as tau aggregation inhibitors and anti-inflammatory drugs, are currently under investigation, but such efforts are on a very small scale. We propose that it is important for new anti-AD drugs to reverse the degeneration of neuronal networks after the appearance of symptoms.
From this point of view, we are investigating drugs used in Japanese traditional medicine (called Kampo), focusing on kamikihito (KKT). KKT is a herbal drug known for its effectiveness in treating insomnia, loss of appetite, amnesia, and depression. We previously reported the effect of KKT in the 5XFAD mouse model of AD [7]. 5XFAD mice exhibit increased Aβ production, Aβ plaques, and gliosis at 2 months of age and synapse loss and memory impairment at 4 months of age [8]. Therefore, 5XFAD mice are considered to be a model of AD after the appearance of symptoms. Our previous study showed that the administration of KKT to 5XFAD mice (4–7 months old) for 15 days improves memory impairment and reverses the degeneration of cortical axons and presynaptic terminals. These effects could be beneficial for AD patients, but the mechanism underlying the effects of KKT remains unknown. The aim of the present study is to investigate the mechanism by which KKT reverses progressive axon degeneration.
2. Materials and Methods
All experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the University of Toyama.
2.1. Preparation of KKT Extract
KKT is composed of 14 crude drugs: Ginseng Radix (P. ginseng C.A. Meyer), Polygalae Radix (P. tenuifolia Willd.), Astragali Radix (A. membranaceus Bunge), Zizyphi Fructus (Zizyphus jujube Mill. var. inermis Rehd.), Zizyphi Spinosi Semen (Z. jujube Mill. var. spinosa), Angelicae Radix (Angelica acutiloba Kitagawa), Glycyrrhizae Radix (Glycyrrhiza uralensis Fisch), Atractylodis Rhizoma (Atractylodes japonica Koidzumi ex Kitamura), Zingiberis Rhizoma (Zingiber officinale Roscoe), Poria (Poria cocos Wolf), Saussureae Radix (Saussurea lappa Clarke), Longanae Arillus (Euphoria longana Lam.), Bupleuri Radix (Bupleurum falcatum Linne), and Gardeniae Fructus (Gardenia jasminoides Ellis) at a ratio of 3 : 1.5 : 2: 1.5 : 3: 2 : 1 : 3 : 1 : 3 : 1 : 3 : 3 : 3, respectively. All crude drugs were purchased from Tochimoto Tenkaido (Osaka, Japan). The mixture of crude drugs (100 g) was extracted with water (500 mL) at 100°C for 50 min, evaporated under reduced pressure, and freeze-dried to obtain a powder extract (12.3 g).
2.2. Primary Culture
Embryos were removed from a pregnant ddY mouse (Japan SLC, Shizuoka, Japan) at 14 days of gestation. The cerebral cortices were dissected, and the dura mater was removed. The tissues were minced, dissociated, and cultured with Neurobasal medium (Gibco BRL, Rockville, MD, USA) containing 12% horse serum, 0.6% D-glucose, and 2 mM L-glutamine. Cells were grown in 8-well chamber slides (Falcon, Franklin Lakes, NJ, USA) or 10 cm dishes coated with 5 μg/mL poly-D-lysine at 37°C in a humidified incubator with 10% CO2. On the next day of culture, the medium was replaced with fresh Neurobasal medium containing 2% B-27 supplement rather than horse serum.
2.3. Aβ-Induced Axonal Atrophy
Mouse cortical neurons (E14) were cultured in 8-well chamber slides (Falcon, Franklin Lakes, NJ, USA) at a density of 1.45 × 104 cells/cm2 for 3 days. To induce axon atrophy, the cells were treated with Aβ (25–35) (10 μM) (Sigma-Aldrich, St. Louis, MO, USA). The Aβ (25–35), an active fragment of Aβ, was previously incubated for 4 days at 37°C to induce aggregation. In experiments involving pretreatment, KKT (10 μg/mL), Kenpaullon (KPL, 1 μM, an inhibitor of glycogen synthase kinase-3β: GSK-3β), or vehicle was administered to the cells, and Aβ (25–35) was added to the cells 1 h later. At 24 h after incubation with Aβ, the medium was replaced with Aβ (25–35)-free medium, and the drug treatments were continued for an additional 72 h. In experiments involving posttreatment, cells were incubated with Aβ (25–35) (10 μM) for 24 h. The medium was then replaced to remove Aβ (25–35). The replacement medium contained KKT (10 μg/mL), KPL (1 μM), or vehicle. After drug treatment for 72 h, the cells were fixed with 4% paraformaldehyde and immunostained axons with a monoclonal antibody against phosphorylated NF-H (pNF-H, dilution 1 : 500, Sternberger Monoclonals, Lutherville, MD, USA) and neuronal cell bodies with a polyclonal antibody against MAP 2a and 2b (MAP2, dilution 1 : 500, Chemicon, Temecula, CA, USA). Secondary antibodies were Alexa Fluor 488-conjugated goat anti-mouse IgG (dilution 1 : 300) and Alexa Fluor 568-conjugated goat anti-rabbit IgG (dilution 1 : 300) (Molecular Probes, Eugene, OR, USA). Fluorescent images were detected by a fluorescence microscope system (BX-61/DP70, Olympus, Tokyo, Japan). The lengths of axons were measured using the Neurocyte image analyzer (Kurabo, Osaka, Japan), which automatically traced and measured neurite lengths without measuring cell bodies. The total length of the axons was divided by the number of cells in an identical area to calculate the average length of axon per neuron.
2.4. Aβ-Induced Tau Phosphorylation
Mouse cortical neurons (E14) were cultured in 8-well chamber slides at a density of 1.45 × 104 cells/cm2 for 3 days. For pretreatment experiments, KKT (10 μg/mL), KPL (1 μM), or vehicle was administered to the cells, and Aβ (25–35) (10 μM) was added 1 h later. At 4 h or 72 h after incubation with Aβ, the cells were fixed. For posttreatment experiments, cells were treated with Aβ (25–35) (10 μM) for 24 h. Then, the medium was replaced with medium lacking Aβ (25–35). KKT (10 μg/mL), KPL (1 μM), or vehicle was included in the replacement medium. At 72 h after drug treatment, the cells were fixed and immunostained with a monoclonal antibody against paired helical filament tau (PHF-tau, AT-8 clone, dilution 1 : 200, Thermo Scientific, Rockford, IL, USA). Alexa Fluor 488-conjugated goat anti-mouse IgG (dilution 1 : 300) was used as the secondary antibody. Images were captured using a fluorescence microscope system (BX-61/DP70, Olympus). The level of tau phosphorylation was measured as the fluorescence intensity in the cell body of each neuron using a CS analyzer (ATTO, Tokyo, Japan).
2.5. PP2A Activity Assay
Mouse cortical neurons (E14) were cultured in a 10 cm dish at a density of 8.67 × 104 cells/cm2 for 3 days. KKT (10 μg/mL) or vehicle was administered to the cells, and Aβ (25–35, 10 μM) was added 1 h later. At 72 hours after incubation with Aβ, the cells were harvested and suspended in phosphate storage buffer (2 mM EGTA, 5 mM EDTA, 0.5 mM PMSF, 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% Triton X-100, and protease inhibitor cocktail (1 : 200)). The cell lysates were sonicated and centrifuged at 16,000 ×g for 10 min, and the supernatants were used for the PP2A activity assay. PP2A activity was measured using a serine/threonine phosphatase assay system (V2460, Promega, Madison, WI, USA) according to the manufacturer's protocol. The cell lysates were passed twice through Sephadex G-25 Spin Columns to remove endogenous free phosphate. The protein concentration was determined using the Pierce 660 nm Protein Assay. PP2A reaction buffer (50 mM imidazole, pH 7.2, 0.2 mM EGTA, 0.02% β-mercaptoethanol, and 0.1 mg/mL bovine serum albumin) and 1 mM phosphopeptide were prepared in a 96-well plate. The enzyme reaction was started by adding 2.5 μg of the protein lysate to each well, and the mixture was incubated for 30 min at 33°C. The reaction was stopped by the addition of 50 μL of molybdate dye-additive mixture, and the plate was incubated at room temperature for 15 min. Phosphate release from the substrate was detected by measuring the absorbance of the molybdate-malachite green-phosphate complex at 630 nm.
2.6. Animals
Transgenic mice (5XFAD) [8] were obtained from Jackson laboratory (Bar Harbor, ME, USA). To investigate the effect of KKT on 5XFAD mice, the study used hemizygous 5XFAD mice (female, 9–11 months) and nontransgenic wild-type mice (female, 9–10 months). All mice were housed with free access to food and water and kept in a controlled environment (22 ± 2°C, 50 ± 5% humidity, 12 h light cycle starting at 7:00 a.m.).
KKT was dissolved in physiological saline. KKT (500 mg/kg/day) or vehicle solution was intragastrically administered once a day for 15 days. On the sixteenth day, the mice were sacrificed, and the cerebral cortex was isolated and homogenized in phosphate storage buffer for the PP2A activity assay.
2.7. GSK-3β Activity Assay
Mouse cortical neurons (E14) were cultured in 8-well chamber slides at a density of 1.45 × 104 cells/cm2 for 3 days. KKT (10 μg/mL) or vehicle was administered to the cells, and Aβ (25–35) (10 μM) was added 1 h later. At 4 h after incubation with Aβ, the cells were fixed with 4% paraformaldehyde and immunostained with a monoclonal antibody against phosphorylated GSK-3β at Tyr-216 (pY216, dilution 1 : 200, BD Biosciences, Franklin Lakes, NJ, USA) as a marker of activated GSK-3β and a polyclonal antibody against GSK-3β (dilution 1 : 500, Santa Cruz Biotechnology, Santa Cruz, CA, USA) as a marker of total GSK-3β. Alexa Fluor 488-conjugated goat anti-mouse IgG (dilution 1 : 300) and Alexa Fluor 568-conjugated goat anti-rabbit IgG (dilution 1 : 300) were used as secondary antibodies. Images were captured using a fluorescence microscope system (BX-61/DP70, Olympus). Expression of phosphorylated GSK-3β and total GSK-3β was measured by determining the fluorescence intensity in the cell body of each neuron using a CS analyzer (ATTO). The ratio of GSK-3β (pY216) to GSK-3β (total) was calculated.
2.8. Statistical Analysis
Statistical comparisons were performed through a One-way analysis of variance (ANOVA) with post hoc Dunnett's test or Bonferroni's multiple comparison test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered significant. The data are presented as the means ± SE.
3. Results
3.1. KKT Decreases Aβ (25–35)-Induced Phosphorylation of Tau
We first focused on the effectiveness of KKT against Aβ (25–35)-induced phosphorylation of tau. Full-length Aβ (1–42) and its active fragment Aβ (25–35) have been reported to induce phosphorylation of tau [9, 10]. Phosphorylation of tau leads to axon transport deficits, synaptic loss, and mitochondrial dysfunction, resulting in axonal dysfunction [11–13]. Among the various kinases involved in phosphorylation of tau, GSK-3β plays a critical role in AD pathology [14]. We therefore investigated the effect of KKT (10 μg/mL) and KPL (inhibitor of GSK-3β; Ki = 0.23 μM) (1 μM) on Aβ (25–35)-induced phosphorylation of tau. Drugs were administered before or after treatment with Aβ (25–35).
As shown in Figure 1(a), treatment of cortical neurons with Aβ (25–35) for 4 h resulted in a 3.7-fold increase in phosphorylation of tau compared with the control. However, the phosphorylation of tau was attenuated to 2.6-fold by 1 h of pretreatment with KKT (10 μg/mL). With KPL pretreatment, phosphorylation of tau was remarkably prevented with 1.4-fold (Figure 1(a)). Tau remained phosphorylated at 72 h after Aβ (25–35) treatment, although the degree of phosphorylation was lower than at 4 h after treatment (Figure 1(b)). Phosphorylation of tau in the late phase was remarkably prevented by pretreatment with KKT, but pretreatment with KPL resulted in only a slight attenuation of tau phosphorylation.
Figure 1.
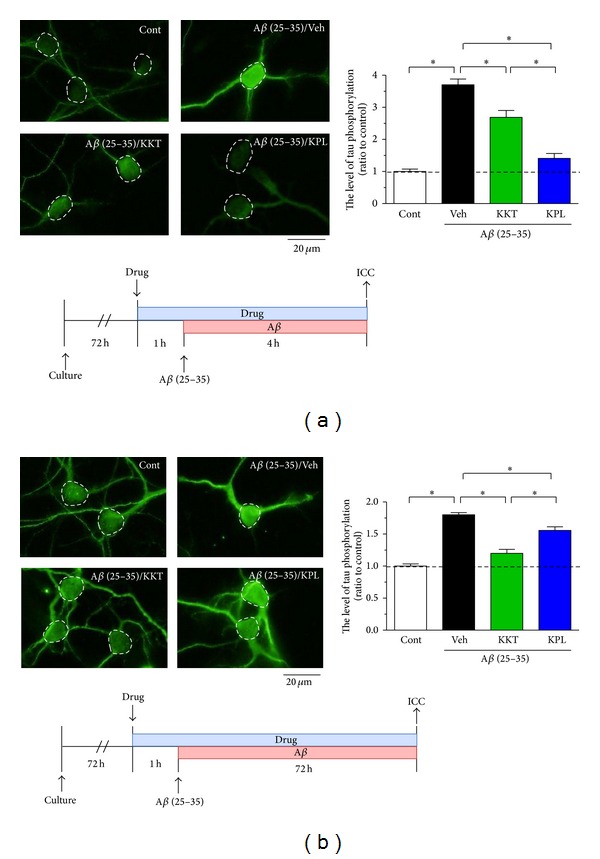
Effect of KKT and KPL pretreatment on Aβ-induced phosphorylation of tau. Cortical neurons were cultured for 3 days and then treated with vehicle solution, KKT (10 μg/mL), or KPL (1 μM). At 1 h after each treatment, the cells were treated with Aβ (25–35) (10 μM) for 4 h (a) or 72 h (b). After Aβ treatment, the cells were fixed and immunostained with an antibody against paired helical filament tau (PHF-tau). The PHF-tau fluorescence intensity was quantified for each treatment. *P < 0.05 (n = 15–20, One-way ANOVA, post hoc Bonferroni's multiple comparison test).
We next investigated the effects of posttreatment with KKT (10 μg/mL) and KPL (1 μM) on Aβ (25–35)-induced phosphorylation of tau (Figure 2). We added Aβ (25–35) to the cells for 24 h, and the cells were incubated with each drug for the next 72 h without Aβ (25–35). KPL no longer affected the Aβ-induced phosphorylation of tau under these conditions, whereas KKT posttreatment significantly inhibited the phosphorylation of tau.
Figure 2.
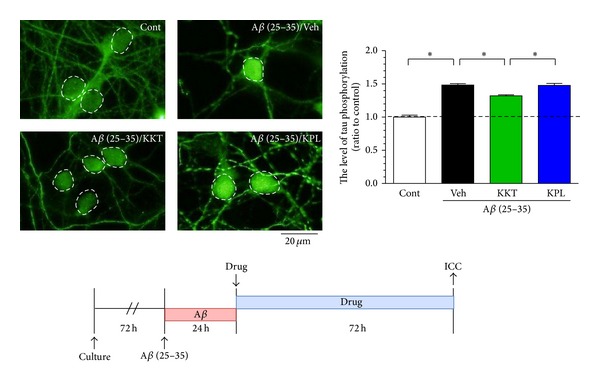
Effect of KKT and KPL posttreatment on Aβ-induced phosphorylation of tau. Cortical neurons were cultured for 3 days and then treated with Aβ (25–35) (10 μM). At 24 h after Aβ treatment, Aβ was removed from the medium, and the cells were treated with vehicle solution, KKT (10 μg/mL), or KPL (1 μM). At 72 h after each treatment, the cells were fixed and immunostained with an antibody against paired helical filament tau (PHF-tau). The PHF-tau fluorescence intensity was quantified for each treatment. *P < 0.05 (n = 15–17, One-way ANOVA, post hoc Bonferroni's multiple comparison test).
These results suggest that KKT reduces the phosphorylation of tau at both early and late phases and that increased tau phosphorylation can be reversed by KKT. In contrast, KPL may inhibit the phosphorylation of tau only in the early phase.
3.2. KKT Reverses Aβ (25–35)-Induced Axonal Atrophy
Figure 1 shows that KKT remarkably reduced the phosphorylation of tau in the late phases (Figure 1(b)), and KPL prevented the phosphorylation of tau more completely in the early phases (Figure 1(a)). Inhibition of tau phosphorylation by KPL no longer occurred when phosphorylation had already progressed (Figure 2). Because it is known that Aβ induces axonal atrophy through phosphorylation of tau, we investigated the effects of KKT and KPL on Aβ (25–35)-induced axonal atrophy.
Treatment of cortical neurons with Aβ (25–35) (10 μM) for 24 h significantly decreased the axonal density (Figure 3(a)), which did not recover even 72 h after the removal of Aβ (25–35) (Figure 3(b)). In the pretreatment protocol, administration of KKT (10 μg/mL) and KPL (1 μM) significantly inhibited Aβ (25–35)-induced axonal atrophy (Figure 3(a)). The protection of axonal atrophy was equivalent in neurons that were pretreated with KKT or with KPL. In contrast, in the posttreatment protocol, only administration of KKT reversed Aβ (25–35)-induced axonal atrophy (Figure 3(b)). These results suggest that KKT, but not KPL, increases axonal density even after axonal atrophy has already progressed.
Figure 3.
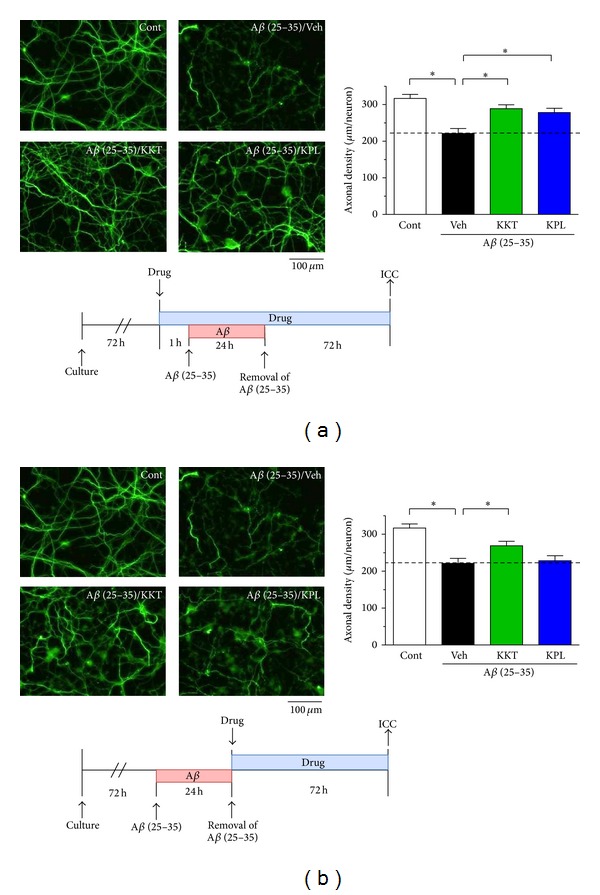
Effect of KKT and KPL pre- and posttreatment on Aβ-induced axonal atrophy. (a) For pretreatment, cortical neurons were cultured for 3 days and then treated with vehicle solution, KKT (10 μg/mL), or KPL (1 μM). After 1 h, the cells were treated with Aβ (25–35) (10 μM) for 24 h, and Aβ was removed from the medium. Each drug treatment was continued for 72 h. (b) For posttreatment, cortical neurons were cultured for 3 d and then treated with Aβ (25–35) (10 μM). At 24 h after Aβ treatment, Aβ was removed from the medium and the cells were treated with vehicle solution, KKT (10 μg/mL), or KPL (1 μM) for 72 h. After each drug treatment, the cells were fixed and immunostained with an antibody against phosphorylated NF-H and MAP2. The lengths of phosphorylated NF-H-positive neurites were quantified for each treatment. *P < 0.05 (n = 23–31, One-way ANOVA, post hoc Bonferroni's multiple comparison test).
3.3. KKT Reverses the Aβ-Induced Decrease in PP2A Activity
KKT remarkably reduced the phosphorylation of tau in the late phase (Figure 1(b)). To explain the long-lasting inhibitory effect of KKT on the phosphorylation of tau, we proposed that KKT may play a role in the dephosphorylation of tau. It was previously reported that Aβ increases the phosphorylation of tau by decreasing PP2A activity [15]; we therefore investigated the effect of KKT on PP2A activity under Aβ (25–35) treatment. KKT (10 μg/mL) was added to the cells, and Aβ (25–35) (10 μM) was added 1 h later. PP2A activity was measured 72 h after Aβ addition. As shown in Figure 4(a), Aβ treatment decreased PP2A activity to 79.6% compared with the control group. However, pretreatment with KKT completely prevented the decrease in PP2A activity (Figure 4(a)).
Figure 4.
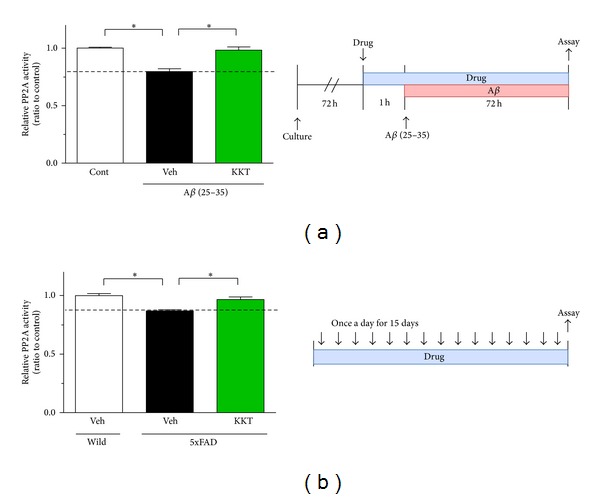
KKT enhances PP2A activity in vitro and in vivo. (a) Cortical neurons were cultured for 3 d and then treated with vehicle solution or KKT (10 μg/mL). At 1 h after each treatment, the cells were treated with Aβ (25–35) (10 μM) for 72 h. The cell lysates were prepared for the PP2A activity assay. PP2A activity was measured using a serine/threonine phosphatase assay system. (b) Vehicle solution or KKT (500 mg/kg/day) was administered once a day for 15 days to wild-type or 5XFAD mice. Cerebral cortex homogenates were used for the PP2A activity assay. *P < 0.05 versus Veh (n = 4–8, One-way ANOVA, post hoc Dunnett's test).
PP2A activity was also measured in vivo. KKT (500 mg/kg/day) was administered orally to the 5XFAD mice, and cerebral cortex lysates were prepared for measurement of PP2A activity. Our previous data showed that KKT administration for 15 days completely restored memory impairment in 5XFAD mice [7]. In the present experiment, we further confirmed that memory was improved by KKT treatment (data not shown). Vehicle-treated 5XFAD mice showed a significant decrease in PP2A activity compared with wild-type mice. However, KKT treatment of 5XFAD mice significantly increased PP2A activity compared with vehicle treatment (Figure 4(b)). These results demonstrate that KKT enhances the activity of PP2A under Aβ (25–35)-induced downregulation of PP2A activity.
3.4. KKT Has No Effect on GSK-3β Activity
Figures 1 and 2 show that KKT inhibits the Aβ (25–35)-induced phosphorylation of tau in both the early and late phases. However, the effect of KKT in the early phase was lower than that of KPL. Because the effects of KKT and KPL were different, we next investigated whether KKT was also an inhibitor of GSK-3β or not. GSK-3β activity was shown to be regulated by phosphorylation at specific residues; for example, phosphorylation of Ser9 and Ser389 inhibited its kinase activity, whereas phosphorylation at Tyr216 was required for its activation [16]. In this experiment, we measured the ratio of GSK-3β (pY216) to GSK-3β (total) to evaluate the level of GSK-3β activation. As shown in Figure 5, Aβ (25–35) (10 μM) treatment for 4 h significantly increased the amount of phosphorylated GSK-3β (Tyr216). However, pretreatment with KKT (10 μg/mL) had no effect on this increase (Figure 5). These results suggest that KKT may not be involved in the regulation of GSK-3β activity.
Figure 5.
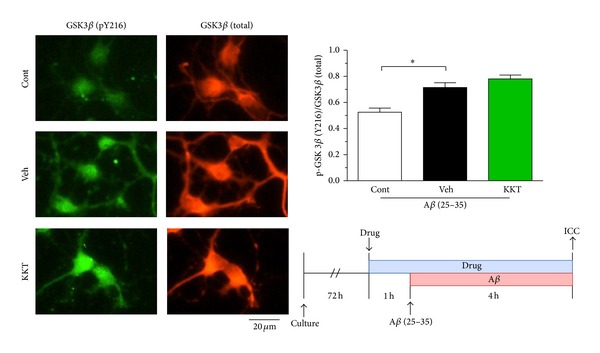
Effect of KKT on Aβ-induced activation of GSK-3β. Cortical neurons were cultured for 3 d and then treated with vehicle solution or KKT (10 μg/mL). At 1 h after each treatment, the cells were treated with Aβ (25–35) (10 μM) for 4 h. After Aβ treatment, the cells were fixed and immunostained with antibodies against phosphorylated GSK-3β at Tyr-216 (pY216), a marker of activated GSK-3β, and against total GSK-3β. The fluorescence intensity was quantified for each treatment. The ratio of GSK-3β (pY216) to GSK-3β (total) is shown. *P < 0.05 versus Veh (n = 15-16, One-way ANOVA, post hoc Dunnett's test).
4. Discussion
Senile plaques and neurofibrillary tangles are two hallmark pathologies of AD. Senile plaques contain extracellular deposits of fibrillar Aβ, and neurofibrillary tangles are composed of hyperphosphorylated tau. It has been reported that Aβ induces phosphorylation of tau [9, 10] and that the phosphorylation of tau leads to axon transport deficits, synaptic loss, and mitochondrial dysfunction [11–13]. These disturbances eventually lead to axonal degeneration and neuronal death. Consequently, therapeutic approaches targeting tau have been growing steadily in recent years. For example, 2-methyl-5-(3-4-[(S)-methylsulfinyl]phenyl-1-benzofuran-5-yl)-1,3,4-oxadiazole (MMBO) and NP12 have been reported to decrease tau phosphorylation and reverse cognitive deficits in AD mouse models, which are novel GSK-3 inhibitors [17, 18]. In patients with mild to moderate AD, methylene blue (a tau aggregation inhibitor) is effective in treating cognitive deficits [19]. Phase II clinical trials of NP12 (tideglusib) and phase III clinical trials of LMTX, a derivative of methylene blue, are ongoing.
In this study, we investigate the mechanism underlying the effect of KKT. KKT reversed memory impairment in 5XFAD mice in our previous study [7], and here we demonstrate three properties of the effects of KKT. First, posttreatment with KKT reverses the progression of tau phosphorylation; however, posttreatment with KPL has no effect on tau phosphorylation (Figure 2). Second, posttreatment with KKT restores atrophied axons, whereas KPL does not (Figure 3(b)). Third, KKT enhances the activity of PP2A (Figure 4) but is not involved in the regulation of GSK-3β activity (Figure 5).
Recent reports of therapeutic strategies targeting Aβ, such as solanezumab and semagacestat, demonstrate that decreasing Aβ deposits or plasma Aβ levels after the appearance of symptoms does not improve cognitive dysfunction [6, 20]. In contrast, several reports have shown that tau is essential for Aβ-induced neuronal dysfunction in vitro and in vivo [21, 22]. Therefore, a decrease in tau phosphorylation may improve the symptoms of AD. From this point of view, it is very interesting that KKT reverses the progression of tau phosphorylation and axonal atrophy.
Our data suggest that KKT decreases the phosphorylation of tau through the activation of PP2A; however, the mechanism by which KKT increases PP2A activity remains unknown. The PP2A subtype accounts for more than 70% of all phosphatases in the human brain. In addition, the activity of PP2A decreases more than that of other phosphatases in the brains of AD patients [23]. Thus, PP2A likely plays a critical role in the dephosphorylation of tau. There are reports showing that PP2A activators decrease the phosphorylation of tau and reverse memory deficits in an AD model. For example, memantine enhances PP2A activity indirectly, and 1 year of treatment with memantine significantly decreases the levels of phosphorylated tau in the CSF of AD patients [24, 25]. Other reports have shown that sodium selenite, which induces dephosphorylation of tau via PP2A activation, improves memory deficits in tau transgenic mice [26]. Therefore, activation of PP2A likely plays an important role in the effects of KKT for AD treatment. However it has also been reported that KKT ameliorates scopolamine-induced spatial memory deficits and methyl-β-carboline-3-carboxylate(β-CCM)-induced-anxiogenic behavior [27, 28]. These reports suggest that KKT may stimulate the cholinergic system and antagonize the benzodiazepine receptor system. Multiple constituents of KKT may therefore contribute to its pharmacological effects.
Our present data indicating that KKT is effective even after Aβ-induced degeneration has occurred are important. Future studies should clarify which compounds in KKT are related to the restorative effect. KKT extract contains many constituents. Among them, at least ginsenoside Rg1, ginsenoside Rb1, saikosaponin a, saikosaponin b1, saikosaponin b2, geniposide, liquiritin, apioliquiritin, glycyrrhizic acid, 6-gingerol, and ligustilide were identified in our previous report [7]. Ginsenoside Rb1, a constituent in Ginseng Radix, attenuated spatial memory deficits in Aβ (25–35)-injected mice [29] and extended axons under Aβ (25–35) treatment (our unpublished data). Therefore ginsenoside Rb1 may be one of the active constituents in KKT.
Also we previously showed that posttreatment with Polygalae Radix extract reversed Aβ (25–35)-induced axonal atrophy [30]; therefore, Polygalae Radix may be at least partially responsible for the effect of KKT.
AD is characterized by two pathological hallmarks: Aβ aggregates and phosphorylated tau. However, AD is a multifactorial disease caused by protein misfolding and aggregation, oxidative stress, metal dyshomeostasis, altered protein phosphorylation, and mitochondrial dysfunction. One of the reasons for the failure of Aβ-targeted immune therapy in clinical trials is that the drugs have only one pharmacological target. In light of the multifactorial pathology of AD, it has been suggested that a multitarget drug, such as Memoquin, is necessary for AD treatment [31]. Memoquin acts as an acetylcholinesterase inhibitor, BACE1 inhibitor, and antioxidant, and it has been reported to improve scopolamine- and Aβ-induced cognitive impairment [32].
KKT has been used for a long time and is based on traditional philosophy (called Kampo in Japan) and offers the particular advantage of not being composed of only one or two active compounds. It is likely that the synergistic and additive effects of multiple compounds are necessary to obtain the best treatment performance. This multitarget treatment paradigm is inline with the concept of systems pharmacology [33].
5. Conclusion
KKT reverses axonal degeneration, in part through its action as an exogenous PP2A stimulator. In Japan, Korea, and China, KKT is already prescribed as a traditional formula for clinical medication. The usefulness of KKT as an anti-AD drug with the crucial property of reversing memory dysfunction must be confirmed by clinical studies.
Acknowledgment
This work was partially supported by the Regional Innovation Cluster Program, a Global Type (II) grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1. Ministry of Health, Labour and Welfare, http://www.mhlw.go.jp/houdou_kouhou/kaiken_shiryou-/2013/-130607-01.html.
- 2.WHO. Dementia: a public health priority. 2012.
- 3.Gauthier S, Molinuevo JL. Benefits of combined cholinesterase inhibitor and memantine treatment in moderate-severe Alzheimer's disease. Alzheimer's & Dementia. 2013;9(3):326–331. doi: 10.1016/j.jalz.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullane K, Williams M. Alzheimer's therapeutics: continued clinical failures question the validity of the amyloid hypothesis—but what lies beyond? Biochemcal Pharmacology. 2012;85(3):289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 6.Farlow M, Arnold SE, van Dyck CH, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimer's & Dementia. 2012;8(4):261–271. doi: 10.1016/j.jalz.2011.09.224. [DOI] [PubMed] [Google Scholar]
- 7.Tohda C, Nakada R, Urano T, Okonogi A, Kuboyama T. Kamikihi-to (KKT) rescues axonal and synaptic degeneration associated with memory impairment in a mouse model of Alzheimer’s disease, 5XFAD. International Journal of Neuroscience. 2011;121(12):641–648. doi: 10.3109/00207454.2011.602809. [DOI] [PubMed] [Google Scholar]
- 8.Oakley H, Cole SL, Logan S, et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. The Journal of Neuroscience. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Q-L, Yang F, Rosario ER, et al. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. The Journal of Neuroscience. 2009;29(28):9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ittner LM, Fath T, Ke YD, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(41):15997–16002. doi: 10.1073/pnas.0808084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mocanu M-M, Nissen A, Eckermann K, et al. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous tau in inducible mouse models of tauopathy. The Journal of Neuroscience. 2008;28(3):737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.David DC, Hauptmann S, Scherping I, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. The Journal of Biological Chemistry. 2005;280(25):23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- 14.Giese KP. GSK-3: a key player in neurodegeneration and memory. IUBMB Life. 2009;61(5):516–521. doi: 10.1002/iub.187. [DOI] [PubMed] [Google Scholar]
- 15.Park SS, Jung HJ, Kim YJ, et al. Asp664 cleavage of amyloid precursor protein induces tau phosphorylation by decreasing protein phosphatase 2A activity. Journal of Neurochemistry. 2012;123(5):856–865. doi: 10.1111/jnc.12032. [DOI] [PubMed] [Google Scholar]
- 16.Salcedo-Tello P, Ortiz-Matamoros A, Arias C. GSK3 function in the brain during development, neuronal plasticity, and neurodegeneration. International Journal of Alzheimer’s Disease. 2011;2011:12 pages. doi: 10.4061/2011/189728.189728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onishi T, Iwashita H, Uno Y, et al. A novel glycogen synthase kinase-3 inhibitor 2-methyl-5-(3-{4-[(S)-methylsulfinyl]phenyl}-1-benzofuran-5-yl)-1,3,4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer’s disease. Journal of Neurochemistry. 2011;119(6):1330–1340. doi: 10.1111/j.1471-4159.2011.07532.x. [DOI] [PubMed] [Google Scholar]
- 18.Serenó L, Coma M, Rodríguez M, et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo . Neurobiology of Disease. 2009;35(3):359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 19.Wischik C, Staff R. Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of tau-aggregation inhibitor therapy. The Journal of Nutrition, Health and Aging. 2009;13(4):367–369. doi: 10.1007/s12603-009-0046-5. [DOI] [PubMed] [Google Scholar]
- 20.Fleisher AS, Raman R, Siemers ER, et al. Phase 2 safety trial targeting amyloid β production with a γ-secretase inhibitor in Alzheimer disease. Archives of Neurology. 2008;65(8):1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(9):6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 23.Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. European Journal of Neuroscience. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 24.Chohan MO, Khatoon S, Iqbal I-G, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by memantine. FEBS Letters. 2006;580(16):3973–3979. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 25.Gunnarsson MD, Kilander L, Basun H, Lannfelt L. Reduction of phosphorylated tau during memantine treatment of Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2007;24(4):247–252. doi: 10.1159/000107099. [DOI] [PubMed] [Google Scholar]
- 26.van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(31):13888–13893. doi: 10.1073/pnas.1009038107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egashira N, Manome N, Kurauchi K, et al. Kamikihi-to, a Kampo medicine, ameliorates impairment of spatial memory in rats. Phytotherapy Research. 2007;21(2):126–129. doi: 10.1002/ptr.2034. [DOI] [PubMed] [Google Scholar]
- 28.Nishizawa K, Yamashita A. Effects of Kamikihi-to, a traditional Chinese medicine, on behavioral changes induced by methyl-β-carboline-3-carboxylate in mice and rats. Japanese Journal of Pharmacology. 1997;75(4):391–397. doi: 10.1254/jjp.75.391. [DOI] [PubMed] [Google Scholar]
- 29.Tohda C, Matsumoto N, Zou K, Meselhy MR, Komatsu K. Aβ(25–35)-induced memory impairment, axonal atrophy, and synaptic loss are ameliorated by M1, a metabolite of protopanaxadiol-type saponins. Neuropsychopharmacology. 2004;29(5):860–868. doi: 10.1038/sj.npp.1300388. [DOI] [PubMed] [Google Scholar]
- 30.Naito R, Tohda C. Characterization of anti-neurodegenerative effects of Polygala tenuifolia in Aβ(25–35)-treated cortical neurons. Biological and Pharmaceutical Bulletin. 2006;29(9):1892–1896. doi: 10.1248/bpb.29.1892. [DOI] [PubMed] [Google Scholar]
- 31.Cavalli A, Bolognesi ML, Mìnarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. Journal of Medicinal Chemistry. 2008;51(3):347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 32.Capurro V, Busquet P, Lopes JP, et al. Pharmacological characterization of memoquin, a multi-target compound for the treatment of Alzheimer's disease. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056870.e56870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang C, Zheng C, Li Y, Wang Y, Lu A, Yang L. Systems pharmacology in drug discovery and therapeutic insight for herbal medicines. Briefings in Bioinformatics. 2013 doi: 10.1093/bib/bbt035. [DOI] [PubMed] [Google Scholar]