

Abstract
Skeletal muscle is a major component of the human body. Age-related loss of muscle mass and function contributes to some public health problems such as sarcopenia and osteoporosis. Skeletal muscle, mainly composed of appendicular lean mass (ALM), is a heritable trait. Copy number variation (CNV) is a common type of human genome variant which may play an important role in the etiology of many human diseases. In this study, we performed genome-wide association analyses of CNV for ALM in 2,286 Caucasian subjects. We then replicated the major findings in 1,627 Chinese subjects. Two CNVs, CNV1191 and CNV2580, were detected to be associated with ALM (p = 2.26×10−2 and 3.34×10−3, respectively). In the Chinese replication sample, the two CNVs achieved p-values of 3.26×10−2 and 0.107, respectively. CNV1191 covers a gene, GTPase of the immunity-associated protein family (GIMAP1), which is important for skeletal muscle cell survival/death in humans. CNV2580 is located in the Serine hydrolase-like protein (SERHL) gene, which plays an important role in normal peroxisome function and skeletal muscle growth in response to mechanical stimuli. In summary, our study suggested two novel CNVs and the related genes that may contribute to variation in ALM.
Introduction
Loss and function impairment of skeletal muscle, especially in the elderly, are related to a number of public health problems (such as sarcopenia, osteoporosis) and increased mortality [1], [2]. Whole lean body mass (LBM) is composed of skeletal muscle (∼60%), viscera, and some other connective tissues. Appendicular lean mass (ALM) is sum of skeletal muscle mass in arms and legs which is the primary portion of skeletal muscle involved in ambulation and physical activities. ALM is considered to be an ideal measure for skeletal muscle mass [3], [4], [5], [6]. ALM can be measured accurately by dual energy X-ray absorptiometry (DXA).
Skeletal muscle is under strong genetic control, with heritability estimates of 30–85% for muscle strength and 50–80% for muscle mass [7], [8]. Genome wide association studies have identified a number of variants that may account for variation in ALM [9], [10]. However, collectively, the identified loci/genes/variants only explain a small fraction of genetic variation in ALM, and the majority of the genetic determination remains to be revealed. Traditional association studies have focused on single nucleotide polymorphisms (SNPs). Studies on other types of genetic variants, which may account for the “missing” heritability, have been relatively rare.
Recent studies have shown that copy number variation (CNV) plays an important role in human diseases, such as schizophrenia [11], [12], Parkinson’s disease [13], and autism [14]. CNV is a common type of genomic variability with the size of DNA fragments ranging from one kilobase to several megabases and presents at variable copy numbers in comparison with reference genome [15]. CNV may influence gene expression, phenotypic variation and adaptation by disrupting coding or altering gene dosage [16], [17], [18], [19]. Furthermore, it may affect gene expression indirectly through position effects, predispose to deleterious genetic changes, or provide substrates for chromosome change in evolution [15], [20], [21], [22]. A recent GWAS of CNVs in Chinese identified the gremlin1 gene that was associated with LBM variation [23]. However, to date, no study has been performed to investigate whether CNVs contribute to ALM in other ethnic groups such as Caucasians.
In this study, we performed a CNV-based GWAS to identify genetic loci influencing variation in ALM in 2,286 Caucasian subjects. Follow-up replication analyses were performed in a Chinese population consists of 1,627 subjects.
Materials and Methods
Ethics Statement
The study was approved by Institutional Review Boards of Creighton University, University of Missouri-Kansas City, Hunan Normal University of China and Xi’an Jiaotong University of China. Signed informed-consent documents were obtained from all study participants before they entered the study.
Subjects
The discovery sample consisted of 2,286 unrelated Caucasian subjects that were of European origin recruited in Midwestern US (Kansas City, Missouri and Omaha, Nebraska). The inclusion and exclusion criteria were described in our previous publications [24].
Replication sample is an independent Chinese sample containing 1,627 unrelated subjects. All subjects were recruited from the cities of Xi’an and Changsha and their neighboring areas in China.
Phenotyping
Anthropometric measures and a structured questionnaire covering lifestyle, diet, family information, medical history, etc. were obtained for all the study subjects. ALM and fat body mass (FBM) were measured using a dual-energy X-ray absorptiometry scanner Hologic QDR 4500 W (Hologic Inc., Bedford, MA, USA), for the all study samples. ALM (kg) was calculated as the sum of lean soft tissue (nonfat, non-bone) mass in the arms and legs. Weight was measured in light indoor clothing, using a calibrated balance beam scale, and height was measured as without shoes using a calibrated stadiometer.
Genotyping
Genomic DNA was extracted from peripheral blood leukocytes using standard protocols. Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA), which includes 906,600 SNPs and 940,000 copy number probes, was used to genotype each subject from the discovery sample, according to the Affymetrix protocol. Briefly, approximately 250 ng of genomic DNA was digested with restriction enzyme NspI or StyI. Digested DNA was adaptor-ligated and PCR-amplified for each sample. Fragment PCR products were then labeled with biotin, denatured, and hybridized to the arrays. Arrays were then washed and stained using Phycoerythrin on Affymetrix Fluidics Station, and scanned using the GeneChip Scanner 3000 7 G to quantitate fluorescence intensities. Data management and analyses were conducted using the Genotyping Command Console Software. For sample quality control (QC), a contrast QC threshold was set at a default value of greater than 0.4. The final average contrast QC across the entire sample reached a high level of 2.76 for our Caucasian cohort and 2.62 for our Chinese cohort.
Copy Number Analysis
Common CNVs were identified using the CANARY algorithm implemented in the Birdsuite software [25], which utilized a previously defined copy number polymorphism (CNP, namely CNV with frequency greater than 1%) map based on HapMap samples [26]. In total, 1,216 CNPs were genotyped for the subjects of the discovery sample and 1280 CNPs in the replication sample, respectively.
QC
We conducted QC filtering both at the sample level and the CNV level, according to the previously reported methods [27].
First, for the sample level QC, we used three quality metrics reported by the Birdseye method to evaluate the initial 2,286 subjects for quality in copy number genotyping. The following procedures were adopted: 1) we removed any sample that was greater or less than three standard deviations (SD) from the average estimate of copy number, which was approximate two copies at genome-wide level; 2) we calculated the variability in copy number and SNP probe intensities with each standardized per chromosome. We removed any sample with more than three SD than these estimates on the average genome-wide level; 3) we removed any sample in which more than two chromosomes failed any of these three metrics, i.e. more than three SD in estimated copy number or excessive CNV or SNP variability for chromosome. According to above criteria, 71 subjects were discarded. The copy numbers of the remaining 2,215 subjects were successfully genotyped using the CANARY software.
Second, we conducted QC filtering at the CNV level. Out of the initially called CNVs, we excluded those with uncertain or missing copy call of >5% or with a minor variant frequency of <1%. We discarded the CNVs with allele frequency of <1%. With the above QC criteria, a total of 410 CNVs remained in the subsequent analyses for the Caucasian sample.
Statistical Analyses
Association analyses of CNV were performed using a linear regression model in R package “glm” [28]. For both the initial GWAS and subsequent replication studies, stepwise regression was performed to screen the effects of covariates on ALM variation. Age, sex, height, and FBM were significant effectors (p<0.05) and raw ALM values were adjusted for these factors. We adjusted for covariates by a 2-stage procedure where the outcomes were regressed on covariates only, and then the resulting residuals were regressed on CNVs. To correct for the effect of potential population stratification, we conducted a principal component analysis on genome-wide SNP data with EIGENSTRAT [29] and included the top ten principal components as covariates. Fisher’s method [30] was used to combine the p-values from the discovery sample and replication sample.
Results
The basic characteristics of the subjects used in both discovery and replication samples are summarized in Table 1.
Table 1. Basic characteristics of the study subjects.
| Discovery Sample (Caucasian) | Replication Sample (Chinese) | |||||
| Total | Male | Female | Total | Male | Female | |
| No. of subjects | 2,286 | 558 | 1,728 | 1,627 | 802 | 825 |
| Age | 51.37 (13.75) | 50.71 (16.05) | 51.59 (12.92) | 34.49 (13.24) | 31.43 (7.97) | 37.46 (13.77) |
| Height (cm) | 164.81 (43.03) | 171.66 (70.64) | 162.61 (28.65) | 164.25 (8.16) | 170.27 (5.96) | 158.38 (5.22) |
| Weight (kg) | 73.86 (42.67) | 83.23 (67.08) | 70.83 (30.33) | 65.72 (9.61) | 65.74 (9.64) | 54.63 (8.09) |
| FBM (kg) | 19.34 (5.65) | 20.67 (9.30) | 20.92 (13.20) | 14.01 (2.54) | 11.86 (5.10) | 16.13 (4.90) |
| ALM (kg) | 22.58 (2.68) | 29.92 (4.84) | 20.22 (3.54) | 19.79 (1.94) | 23.95 (3.20) | 15.74 (2.10) |
Note: The numbers within parentheses are standard deviation (SD).
In the discovery sample, 20 CNVs showed evidence for association with ALM at a p value of 0.05 (Table 2). CNV1191 and CNV2580 were replicated in the Chinese sample. The p values of CNV1191 in the discovery and replication samples were 2.26×10−2 and 3.26×10−2, respectively, and p values of CNV2580 in the discovery and replication samples were 3.34×10−3 and 0.107, respectively (Table 2). The combined p values of the two CNVs were 6.05×10−3 and 3.27×10−3, respectively.
Table 2. CNVs achieved a p value of 0.05 or less in the discovery and replication samples.
| CNV | Chr | Start | End | p-value1 | p-value2 |
| CNV2563 | 22 | 23,993,985 | 24,248,712 | 1.70×10−5 | 0.810 |
| CNV11821 | 11 | 5,228,247 | 5,230,232 | 4.66×10−5 | 0.995 |
| CNV148 | 1 | 195,089,940 | 195,168,372 | 2.32×10−4 | 0.864 |
| CNV160 | 1 | 213,560,092 | 213,565,727 | 7.34×10−4 | 0.614 |
| CNV1610 | 10 | 58,880,511 | 58,880,997 | 1.50×10−3 | 0.698 |
| CNV2057 | 15 | 19,803,370 | 20,089,386 | 1.63×10−3 | 0.173 |
| CNV11449 | 8 | 36,194,697 | 36,197,883 | 2.09×10−3 | 0.731 |
| CNV2580 | 22 | 41,234,550 | 41,276,824 | 3.34×10 −3 | 0.107 |
| CNV575 | 4 | 34,455,420 | 34,500,578 | 3.71×10−3 | 0.485 |
| CNV2546 | 22 | 20,055,998 | 20,175,294 | 5.29×10−3 | 0.647 |
| CNV2694 | 23 | 139,324,076 | 139,328,860 | 2.16×10−2 | 0.963 |
| CNV1191 | 7 | 149,916,734 | 149,932,502 | 2.26×10 −2 | 3.26×10 −2 |
| CNV770 | 5 | 176,44,656 | 17,698,273 | 2.61×10−2 | 0.810 |
| CNV1004 | 6 | 126,225,385 | 126,228,469 | 2.89×10−2 | 0.659 |
| CNV200 | 2 | 24,460,486 | 24,464,632 | 2.97×10−2 | 0.706 |
| CNV825 | 5 | 86,151,134 | 86,154,902 | 3.12×10−2 | 0.463 |
| CNV2529 | 21 | 43,794,765 | 43,797,240 | 3.16×10−2 | 0.542 |
| CNV2417 | 19 | 47,986,230 | 48,149,894 | 3.18×10−2 | 0.695 |
| CNV1282 | 8 | 24,201,375 | 24,207,011 | 4.01×10−2 | 0.311 |
| CNV2430 | 19 | 56,834,427 | 56,840,009 | 4.66×10−2 | 0.721 |
Notes:
in discovery samples.
in replication samples.
Chr: chromosome.
We further tested association between normal (CN = 2) and deletion (CN = 0, 1) groups, and between normal and duplication (CN = 3, 4) groups, separately. The results showed that while the direction of effect of CNV2580 was consistent in discovery and replication samples, it was not the case for CNV1191 (Table 3). However, both CNVs remained to be significant in the combined analyses.
Table 3. Association of normal_deletion and normal_duplication with CNVs.
| pair | β | SE | p-value | Combinedp-value | |
| CNV2580 | |||||
| Caucasian | normal_deletion | −5.53×10−3 | 71.82 | 0.94 | 0.99 |
| normal_duplication | −4.16×10−2 | 15.59 | 7.71×10−3 | 6.84×10−3 | |
| Chinese | normal_deletion | −9.27×10−3 | 152.84 | 0.95 | |
| normal_duplication | −4.40×10−2 | 27.23 | 0.11 | ||
| CNV1191 | |||||
| Caucasian | normal_deletion | −1.20×10−2 | 10.43 | 0.25 | 0.04 |
| normal_duplication | −21.3×10−2 | 72.84 | 3.46×10−3 | 0.02 | |
| Chinese | normal_deletion | 4.70×10−2 | 21.88 | 0.03 | |
| normal_duplication | 9.84×10−3 | 94.49 | 0.92 |
Notes:
β, the standardized regression coefficient, was estimated in kilograms for ALM.
SE: Standard error.
In addition to the 2-step adjustment procedure for covariates aforementioned, we performed association analyses where CNVs and covariates were included in a single model. The results were quite similar to those of the 2-step procedure (Table S1).
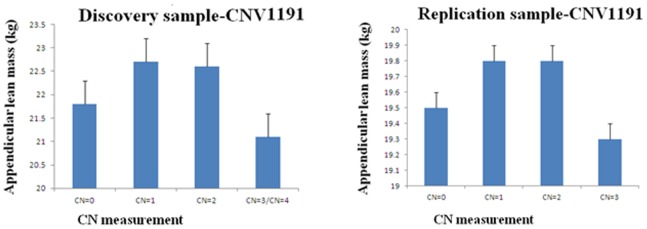
According to the UCSC Genome Browser on Human February 2009 (GRCh37/hg19) Assembly, CNV1191 is located at the chromosome region 7q36.1 with physical position ranging from 149,916,734 bp to 149,932,502 bp, within the gene GTPase IMAP family member 1 (GIMAP1). The number of carriers with CN = 0, 1, 2, 3 and 4 was 126, 855, 1273, 28 and 4, respectively, in the discovery sample. Due to the limited number of subjects with CN = 4, we merged CN = 3 and CN = 4 into a single group. The number of carriers with CN = 0, 1, 2, 3 was 42, 465, 1093 and 27, respectively, in the replication sample. In the discovery sample, carriers with CN = 1 and CN = 2 had higher ALM (22.7 kg and 22.6 kg) and carriers with CN = 3 had the lowest ALM (21.1 kg) (Figure 1). Consistently, in the replication sample, carriers with CN = 1 and CN = 2 had higher ALM (19.8 kg) and carriers with CN = 3 had the lowest ALM (19.3 kg) (Figure 1).
Figure 1. ALM in groups with different copy number (CN) of CNV1191 in the discovery and replication samples.
CNV2580 is located at the chromosome region 22q13.2 with physical position ranging from 41,234,550 bp to 41,276,824 bp, within the gene serine hydrolase-like protein (SERHL). The number of carriers with CN = 1, 2, 3, and 4 was 11, 1763, 455, and 57, respectively, in the discovery sample, and was 5, 1257, 314 and 50, respectively, in the replication sample. Due to the limited number of subjects with CN = 1, we merged CN = 1 and CN = 2 into a single group. In the discovery sample, carriers with CN = 2 and CN = 3 had higher ALM (22.6 kg and 22.7 kg, respectively) and carrier with CN = 4 had the lowest ALM (21.7 kg) (Figure 2), with the estimated β to be −5.24×10−2 (ALM in kg) for each copy number. Consistently, in the replication sample, carriers with CN = 2 and CN = 3 had ALM of 19.8 kg and19.9 kg, respectively, and carrier with CN = 4 had ALM of 18.5 kg (Figure 2), with the estimated β to be −4.34×10−2 (ALM in kg) for each copy number.
Figure 2. ALM in groups with different CN of CNV2580 in the discovery and replication samples.
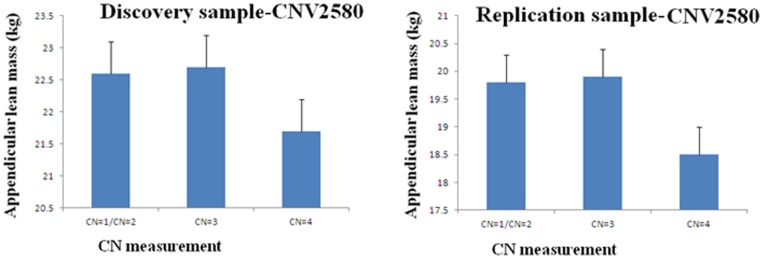
Table 4 lists the proportion of subjects for each copy of CNV2580. The table also includes theoretical proportion calculated based on empirical CN frequencies and random mating assumption. Goodness-of-fit (GOF) test showed that empirical distribution did not deviate from the theoretical distribution (p = 0.22 for both populations).
Table 4. The proportion of the subjects in each CN category of CNV2580.
| Caucasian | Chinese | |||
| Theoretical | Actual | Theoretical | Actual | |
| CN = 0 | 1.16×10−5 | 0 | 4.72×10−6 | 0 |
| CN = 1 | 4.00×10−3 | 5.00×10−3 | 3.00×10−3 | 3.00×10−3 |
| CN = 2 | 0.76 | 0.77 | 0.76 | 0.77 |
| CN = 3 | 0.22 | 0.20 | 0.22 | 0.19 |
| CN = 4 | 0.02 | 0.02 | 0.02 | 0.03 |
| GOF | 0.22 | 0.22 | ||
Note:
GOF: Goodness-of-fit.
There are two SNPs that are located in the region of CNV1191 and eight SNPs outside the CNV1191 boundaries but inside the gene of GIMAP1. None of these ten SNPs was significantly associated with ALM in the discovery sample, but rs11769150 was associated with ALM in the replication sample with p-value of 0.02 (Table 5).
Table 5. SNPs located in the two CNVs regions or outside the two CNVs boundaries and their association signals with ALM.
| CNV region | Chr | SNP | Position | Discovery sample | Replication sample | ||||
| Allele | MAF | p-value | Allele | MAF | p-value | ||||
| CNV1191 | 7 | rs6969250 | 149,923,478 | C/T | 0.23 | NA | C/T | 0.17 | NA |
| 7 | rs11984138 | 149,931,668 | G/A | 0.24 | 0.77 | G/A | 0.33 | 0.9 | |
| 7 | rs10271154 | 150,413,004 | C/T | 0.25 | 0.82 | C/T | 0.37 | 0.46 | |
| 7 | rs4725388 | 150,413,550 | G/A | 0.28 | NA | G/A | 0.55 | NA | |
| 7 | rs13239507 | 150,415,340 | T/C | 0.28 | NA | T/C | 0.42 | NA | |
| 7 | rs6464126 | 150,416,254 | C/T | 0.28 | NA | C/T | 0.42 | NA | |
| 7 | rs11769150 | 150,416,686 | C/T | 0.18 | 0.82 | C/T | 0.14 | 0.02 | |
| 7 | rs10270107 | 150,417,979 | G/A | 0.26 | NA | G/A | 0.35 | NA | |
| 7 | rs6957724 | 150,419,237 | C/T | 0.24 | NA | C/T | 0.12 | NA | |
| 7 | rs11773852 | 150,421,052 | C/T | 0.25 | 0.63 | C/T | 0.22 | 0.98 | |
| CNV2580 | 22 | rs4820470 | 41,236,248 | A/G | 0.15 | NA | A/G | 0.05 | - |
| 22 | rs12485160 | 41,237,563 | C/T | 0.13 | NA | C/T | 0.11 | NA | |
| 22 | rs5758786 | 41,256,609 | A/A | 0 | – | A/G | 0.02 | – | |
| 22 | rs4822160 | 41,262,261 | A/G | 0.20 | 0.56 | A/G | 0.19 | 0.99 | |
| 22 | rs9614207 | 42,898,768 | A/G | 0.42 | NA | A/G | 0.23 | NA | |
| 22 | rs9614208 | 42,899,411 | G/A | 0.41 | 0.67 | G/A | 0.14 | 0.34 | |
| 22 | rs7292425 | 42,900,747 | C/A | 0.18 | NA | C/A | 0.34 | NA | |
| 22 | rs139107 | 42,901,313 | G/A | 0.14 | NA | G/A | 0.16 | NA | |
| 22 | rs9614362 | 42,903,911 | G/A | 0.30 | 0.96 | G/A | 0.13 | 0.46 | |
| 22 | rs12483834 | 42,904,365 | C/G | 0.02 | 0.67 | C/G | 0.16 | 0.37 | |
| 22 | rs139116 | 42,904,875 | T/C | 0.36 | 0.28 | T/C | 0.20 | 0.02 | |
| 22 | rs2092351 | 42,905,737 | G/A | 0.36 | 0.31 | G/A | 0.33 | 0.18 | |
| 22 | rs139120 | 42,905,880 | G/A | 0.34 | 0.32 | G/A | 0.17 | 0.02 | |
| 22 | rs878406 | 42,906,306 | A/T | 0.02 | 0.82 | A/T | 0.19 | 0.32 | |
| 22 | rs3747208 | 42,907,201 | A/G | 0.10 | NA | A/G | 0.08 | – | |
| 22 | rs139123 | 42,907,418 | G/A | 0.37 | NA | G/A | 0.36 | NA | |
| 22 | rs139124 | 42,907,465 | G/A | 0.34 | NA | G/A | 0.15 | NA | |
| 22 | rs7287117 | 42,907,988 | G/A | 0.02 | – | G/A | 0.18 | NA | |
| 22 | rs9306473 | 42,908,483 | G/A | 0.002 | – | G/A | 0 | – | |
Notes:
NA: the failure of passing the quality control procedure (QC).
–: not available due to low minor allele frequency.
Chr: chromosome.
MAF: minor allele frequency.
There are four SNPs that are located in the region of CNV2580 and fifteen SNPs outside the CNV2580 boundaries but inside the gene of SERHL. None of these nineteen SNPs was significantly associated with ALM in the discovery sample, but two SNPs rs139116 and rs139120 were associated with ALM in the replication sample with p-values of 0.02 (Table 5).
Discussion
This is the first CNV–based GWAS for ALM in Caucasians. Two CNVs, CNV1191 and CNV2580, were identified to be associated with ALM.
CNV1191 is located in the gene GIMAP1, which encodes GTPase, IMAP family member 1. GIMAP (GTPase of the immunity–associated protein family) proteins are a family of putative GTPases believed to be regulators of cell death in lymphomyeloid cells. GIMAP1 was the first reported member of this gene family [31]. This gene was involved in the differentiation of T helper (Th) cells of the Th1 lineage, and the related mouse gene has been shown to be critical for the development of the mature B and T lymphocytes [32].
Culturing myotubes from skeletal muscle-biopsies found coordinated reduced expression of five members of the GIMAP family GIMAP1, GIMAP4, GIMAP5, GIMAP6 and GIMAP7, which form a cluster on chromosome 7 and participate in SM cell survival/death [33]. A study in pig skeletal muscle indicated that GIMAP1 was correlated with meat quality and regulation of biological processes involved in the induction of apoptosis [34]. This gene was also involved in regulation of lipid catabolic process, defense response and positive regulation of calcium ion transport [35]. Our findings, combined with the above evidence, support the potential contribution of GIMAP1 to variation in skeletal muscle.
SERHL is a gene coding for a new member of the family of serine hydrolases that is located within peroxisomes [36]. In vivo studies showed that mRNA expression of SERHL increased in response to passive stretch imposed upon skeletal muscle [36].
The association directions of CNV1191 in the discovery and replication studies were different. This inconsistency may be explained by the following reasons. First, genetic variants may have different effects in different populations. A genetic variant may have different allele frequencies among diverse populations because of different evolution histories, which result in different modes of genotype-phenotype association [37]. Second, significant associations are usually found at molecular markers that are in linkage disequilibrium (LD) with causal variant, rather than the causal variant itself. Therefore, the inconsistency in direction could be a result of opposite patterns of LD between the two populations.
Within the two CNVs regions, we did not identify any significant SNPs that were associated with ALM in the discovery sample. A possible explanation is that, different from SNP, CNV is a structural genetic variant that generally covers a larger genomic region and thus CNV may influence phenotypic variation by mechanisms that are different from SNP.
In summary, we identified CNV1191 and CNV2580 that were associated with ALM. The relevant genes, GIMAP1 and SERHL, may play roles in skeletal muscle metabolism. Our findings may provide useful information for molecular functional studies of candidate genes for ALM.
Supporting Information
Association of normal_deletion and normal_duplication with CNVs.
(DOCX)
Acknowledgments
We thank all the participants who joined this study.
Funding Statement
The study was partially supported by startup funding from Shanghai University of Science and Technology and Shanghai Leading Academic Discipline Project (S30501). The investigators of this work were partially supported by grants from the National Institutes of Health (R01AG026564, RC2DE020756, R01AR057049, R01AR050496 and R03TW008221), a SCOR (Specialized Center of Research) grant (P50AR055081) supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the Office of Research on Women’s Health (ORWH), and the Edward G. Schlieder Endowment and the Franklin D. Dickson/Missouri Endowment. Lei Zhang was also supported by the National Natural Science Foundation of China project (31100902). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sipila S, Heikkinen E, Cheng S, Suominen H, Saari P, et al. (2006) Endogenous hormones, muscle strength, and risk of fall-related fractures in older women. J. Gerontol. A Biol. Sci. Med. Sci. 61: 92–96. [DOI] [PubMed] [Google Scholar]
- 2. Karakelides H, Nair KS (2005) Sarcopenia of aging and its metabolic impact. Curr. Top Dev. Biol. 68: 123–148. [DOI] [PubMed] [Google Scholar]
- 3. Baumgartner RN (2000) Body composition in healthy aging. Ann.N Y.Acad.Sci. 904: 437–48. [DOI] [PubMed] [Google Scholar]
- 4. Gallagher D, Visser M, De Meersman RE, Sepulveda D, Baumgartner RN, et al. (1997) Appendicular skeletal muscle mass: effects of age, gender, and ethnicity. J Appl. Physiol. 83: 229–239. [DOI] [PubMed] [Google Scholar]
- 5. Newman AB, Kupelian V, Visser M Simonsick E, Goodpaster B, et al. (2003) Sarcopenia: alternative definitions and associations with lower extremity function. J. Am. Geriatr. Soc. 51: 1602–1609. [DOI] [PubMed] [Google Scholar]
- 6. Delmonico MJ, Harris TB, Lee JS, Visser M, Nevitt M, et al. (2007) Alternative definitions of sarcopenia, lower extremity performance, and functional impairment with aging in older men and women. J. Am. Geriatr. Soc. 55: 769–74. [DOI] [PubMed] [Google Scholar]
- 7. Thomis MA, Huygens W, Heuninckx S, Chagnon M, Maes HHM, et al. (2004) Exploration of myostatin polymorphisms and the angiotensin-converting enzyme insertion/deletion genotype in responses of human muscle to strength training. Eur J Appl Physiol 92: 267–274. [DOI] [PubMed] [Google Scholar]
- 8.Arden NK, Spector TD (1997) Genetic influences on muscle strength, lean body mass, and bone mineral density: a twin study. J Bone Miner Res, 12, 2076–2081. [DOI] [PubMed]
- 9. Sun L, Tan LJ, Lei SF, Chen XD, Li X, et al. (2011) Bivariate genome-wide association analyses of femoral neck bone geometry and appendicular lean mass. PLoS One 6: e27325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han YY, Pei YF, Liu YZ, Zhang L, Wu SY, et al. (2012) Bivariate genome-wide association study suggests fatty acid desaturase genes and cadherin DCHS2 for variation of both compressive strength index and appendicular lean mass in males. Bone 51: 1000–1007. [DOI] [PubMed] [Google Scholar]
- 11. Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, et al. (2008) Rare structural variants disrupt multiple genes in neuro developmental pathways in schizophrenia. Science 320: 539–543. [DOI] [PubMed] [Google Scholar]
- 12. The International Schizophrenia Consortium (2008) Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, et al. (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364: 1169–1171. [DOI] [PubMed] [Google Scholar]
- 14. Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feuk L, Carson AR, Scherer SW (2006) Structural variation in the human genome. Nat Rev Genet 7: 85–97. [DOI] [PubMed] [Google Scholar]
- 16. Repping S, van Daalen SK, Brown LG, Korver CM, Lange J, et al. (2006) High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet 38: 463–467. [DOI] [PubMed] [Google Scholar]
- 17. McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, et al. (2006) Common deletion polymorphisms in the human genome. Nat Genet 38: 86–92. [DOI] [PubMed] [Google Scholar]
- 18. Buckland PR (2003) Polymorphically duplicated genes: their relevance to phenotypic variation in humans. Ann Med 35: 308–315. [DOI] [PubMed] [Google Scholar]
- 19. Nguyen DQ, Webber C, Ponting CP (2006) Bias of selection on human copy number variants. PLoS Genet 2: e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, et al. (2006) Copy number variation: new insights in genome diversity. Genome Res 16: 949–961. [DOI] [PubMed] [Google Scholar]
- 21. Lupski JR, Stankiewicz P (2005) Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PloS Genet 1: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feuk L, Marshall CR, Wintle RF, Scherer SW (2006) Structural variants: changing the landscape of chromosomes and design of disease studies. Hum Mol Genet 15 Spec No 1: R57–66. [DOI] [PubMed] [Google Scholar]
- 23. Hai R, Pei YF, Shen H, Zhang L, Liu XG, et al. (2011) Genome-wide association study of copy number variation identified gremlin1 as a candidate gene for lean body mass. J Hum Genet 57: 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deng HW, Deng HY, Liu YJ, Liu YZ, Xu FH, et al. (2002) A genomewide linkage scan for quantitative-trait loci for obesity phenotypes. Am J Hum Genet. 70(5): 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, et al. (2008) Integrated enotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 40: 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, et al. (2008) Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40: 1166–1174. [DOI] [PubMed] [Google Scholar]
- 27. Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, et al. (2009) Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 41: 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Development Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. Available: http://www.R-project.org/.
- 29. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, et al. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. [DOI] [PubMed] [Google Scholar]
- 30. Fisher KA, Miles R (2008) Modeling the acoustic radiation force in microfluidic chambers. J Acoust Soc Am 123: 1862–1865. [DOI] [PubMed] [Google Scholar]
- 31. Saunders A, Lamb T, Pascall J, Hutchings A, Dion C, et al. (2009) Expression of GIMAP1, a GTPase of the immunity-associated protein family, is not upregulated in malaria. Malar J. 8: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwefel D, Daumke O (2011) GTP-dependent scaffold formation in the GTPase of Immunity Associated Protein family. Small GTPases 2 1: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raymond F, Metairon S, Kussmann M, Colomer J, Nascimento A, et al. (2010) Comparative gene expression profiling between human cultured myotubes and skeletal muscle tissue. BMC Genomics 11: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liaubet L, Lobjois V, Faraut T, Tircazes A, Benne F, et al. (2011) Genetic variability of transcript abundance in pig peri-mortem skeletal muscle: eQTL localized genes involved in stress response, cell death, muscle disorders and metabolism. BMC Genomics 12: 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ravnik-Glavač M, Hrašovec S, Bon J, Dreu J, Glavač D (2012) Genome-wide expression changes in a higher state of consciousness. Consciousness and Cognition 21(3): 1322–1344. [DOI] [PubMed] [Google Scholar]
- 36. Sadusky TJ, Kemp TJ, Simon M, Carey N, Coulton GR (2001) Identification of Serhl, a new member of the serine hydrolase family induced by passive stretch of skeletal muscle in vivo. Genomics 73: 38–49. [DOI] [PubMed] [Google Scholar]
- 37. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP (2004) Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol 99: 2393–2404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Association of normal_deletion and normal_duplication with CNVs.
(DOCX)