

Abstract
Paget's disease of bone (PDB) is a common disorder which may affect one or many bones. Although many patients are asymptomatic, a variety of symptoms and complications may occur. PDB is a focal disorder of bone turnover characterized by excessive bone resorption coupled with bone formation. PDB begins with a period of increased osteoclastic activity and bone resorption, followed by increased osteoblast production of woven bone that is poorly mineralized. In the final phase of the disease process, dense cortical and trabecular bone deposition predominates, but the bone is sclerotic and poorly organized and lacks the structural integrity and strength of normal bone. This article briefly reviews the etiopathogenesis, clinical radiographic and histological features of Paget's disease.
KEYWORDS: Osteoclast, osteoprotegerin, receptor activator of NF-κB, receptor activator of NF-κB ligand
INTRODUCTION
Paget's disease of bone (PDB) also known as osteitis deformans, was first described by Sir James Paget in 1877.[1] PDB is a common focal skeletal disorder that may involve a single bone (monostotic) or multiple bones (polyostotic). PDB is characterized by focal areas of increased osteoclastic bone resorption and a coupled but disorganized increase in osteoblastic bone formation. It occurs as the result of derangement in osteoclast formation and activity. Thus, there is excessive and abnormal remodeling of bone leading to bones that are extensively vascularized, weak, enlarged and deformed. The disease is characterized by focal regions of highly exaggerated bone remodeling, with abnormalities in all phases of the remodeling process. The majority of patients with PDB are elderly, with the age at diagnosis usually more than 50 years.[2]
EPIDEMIOLOGY
PDB is the second most common bone remodeling disease after osteoporosis. This condition occurs in 1-2% of white adults older than 55 years. An analysis of archeological skeletons from Northern England found overall prevalence of PDB of 2.1% in cases older than 40 years. The prevalence before 1500 was 1.7% and after 1500 was 3.1%.[3] Epidemiological studies in Europe and North America have revealed that PDB shows an increasing frequency of occurrence with age and is more prevalent among men than women.[4] The prevalence differs amongst various ethnic/geographic groups. This disease is most common in the United Kingdom and Western Europe, but is also common in British immigrants to Australia, New Zealand, South Africa and South America. The disease is uncommon in African blacks, Scandinavia, China, Japan, Southeast Asia and the Indian subcontinent.[5] Furthermore, there is evidence of decreasing incidence and severity of PDB in the United Kingdom and New Zealand over the past 25-30 years. First-degree relatives of patients with PDB have an increased risk of PDB, particularly if the patient has an early age of diagnosis or deforming bone disease. Family history is positive in approximately 15-30% of patients with PDB and first degree relatives of patients with PDB have about a sevenfold greater risk for the development of Paget's disease. Familial PDB also tends to be diagnosed at a younger age and involve more of the skeleton than sporadic disease.[2,6]
NORMAL BONE REMODELING
The normal adult skeleton undergoes constant remodeling, with osteoclasts removing bone and osteoblasts forming new bone at sites of previous bone resorption in a closely coupled fashion Bone remodeling occurs in discrete areas, termed basic multicellular units and it is estimated that the entire adult skeleton is remodeled every 2-4 years.[7,8]
Osteoclasts are derived from mononuclear precursor cells in the monocyte-macrophage lineage, which fuse to form multinucleated osteoclasts that are then activated to resorb bone Normal remodeling of bone is under the control of receptor activator of NF-κB ligand (RANKL)/receptor activator of NF-κB (RANK)/osteoprotegerin (OPG) system and this system regulates the osteoclast formation and function. RANK present on osteoclast precursors. RANKL, which is expressed in the marrow stroma and on osteoblasts, binds RANK on osteoclast precursors promoting osteoclast proliferation and differentiation. RANKL then binds to RANK on osteoclast precursors, leading to activation of a number of downstream signaling pathways, including the nuclear factor κB (NF-κB), protein kinase B, c-jun N-terminal kinase, p38 mitogen-activated protein kinase and ERK pathways. Each of these pathways has been implicated in osteoclast differentiation, function, or survival. OPG is a decoy receptor, which binds RANKL, thus preventing RANKL from binding RANK. OPG, therefore, inhibits osteoclast differentiation. Most of the osteotropic factors, including 1, 25-(OH)2 D3, interleukin (IL)-1, IL-11 and parathyroid hormone (PTH), promote osteoclast formation indirectly by binding to marrow stromal cells and inducing expression of RANKL on their surface [Figure 1].[2,9]
Figure 1.
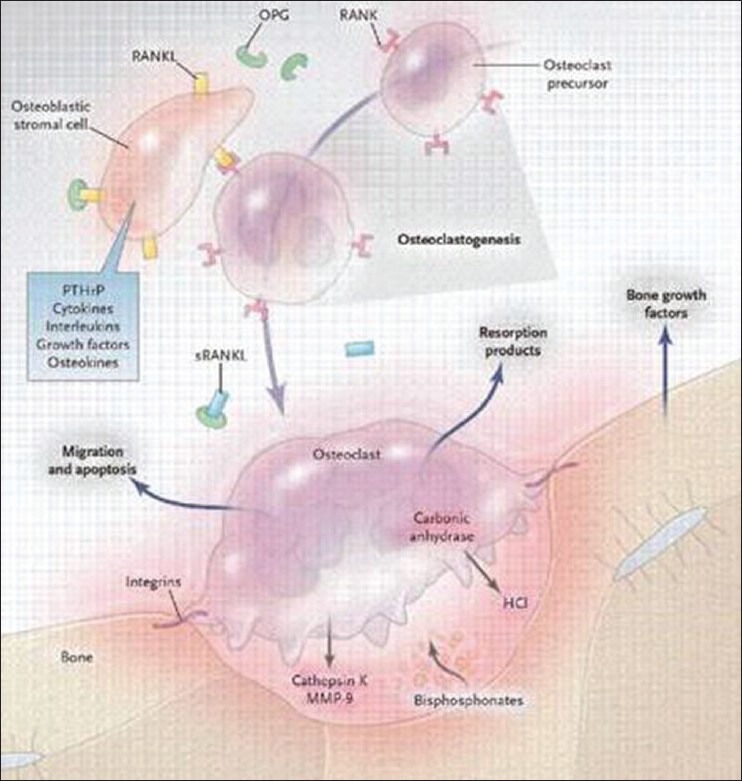
Normal remodeling of bone
Cells affected in PDB
The osteoclast is the primary cell affected by PDB. The osteoclasts show both morphological and physiological abnormalities.
Morphologically osteoclasts in PDB are increased in both number and size and contain up to 100 nuclei, in contrast to normal osteoclasts, which contain 3-20 nuclei.[10]
Another feature of pagetic osteoclasts is the characteristic nuclear inclusions, which consist of paracrystalline arrays that appear similar to nucleocapsids of paramyxoviruses.[11]
Physiological abnormalities include increased hypersensitivity of osteoclast precursors to several osteoclastogenic factors, including 1, 25-(OH)2D3 and RANKL.[12,13]
Osteoblasts are also increased in numbers at pagetic sites, but they are morphologically normal and are not considered to be a primary pathophysiologic factor in PDB.[14]
Etiopathogenesis
The initial lesion is characterized by focal increase in osteoclastic bone resorption. Subsequently followed by accelerated bone formation and with increased numbers of osteoblasts that appear hyperactive, but normal morphologically. The increased population of osteoblasts rapidly deposits new bone. Due to the accelerated bone turnover, new collagen fibers are not laid down in an orderly linear fashion but rather in a disorganized manner. The resultant bone is a mosaic of woven and lamellar bone that is mechanically insufficient and at increased risk for fracture or deformity.[15]
The main etiological factors include: Genetic factors, viral infections, autoimmune, connective tissue and vascular disorders.
Genetic factors
The loci for susceptibility to PDB and related syndromes have been identified by genome-wide scans and recent evidence suggests that mutations in genes that encode components of the RANK [RANK]/NF-κB signaling pathway play a vital role in the pathogenesis of this group of diseases.
There are spectrums of rare genetic bone disorders that have clinical, radiological or histological features in common with classic PDB. These include
Familial expansile osteolysis (FEO).
Expansile skeletal hyperphosphatasia (ESH).
Juvenile PDB (also known as idiopathic hyperphosphatasia).
Early onset PDB.
Inclusion body myopathy with early onset PDB and fronto-temporal dementia (IBMPFD).
The clinical features of some of these diseases, such as ESH and FEO, overlap with classical PDB, but the age at onset is much earlier and the distribution of involvement is different. For IBMPFD, the clinical features and age at onset seem to be similar to that in non-syndromic PDB.
The genes involved in PDB and related syndromes are Sequestosome 1 gene (SQSTM1), tumor necrosis factor receptor superfamily member IIA (TNFRSF11A), valosine containing protein (VCP) and TNFRSF11B.
Classical PDB is caused due to mutations in SQSTM1 that encodes for a protein known as p62 that plays an important role in osteoclast regulation. These mutations are only in 5-10% of sporadic cases and in 40-50% of familial cases.
SQSTM1 acts as a scaffold protein and has various binding domains for atypical protein kinase-C (aPKC), TNFR associated factor 6 (TRAF6), ubiquitin associated domain (UBA) and links the RANK/TRAF6 signaling complex to aPKC and probably regulates this step through the UBA domain interaction with polyubiquitylated (Ub) TRAF6. Binding of RANKL to RANK leads to activation of RANK/TRAF6 signaling complex leading to activation of IκB kinase (IKK). The activated IKK complex phosphorylates IκB, which undergoes degradation, mediated by VCP, in the ubiquitin/proteasome degradation pathway allowing for dissociation of NF-κB and its translocation to the nucleus with consequent activation of response genes leading to osteoclastogenesis [Figure 2]. Mutations affecting the UBA domain of Sequestosome 1 cause classic PDB.[16]
Figure 2.
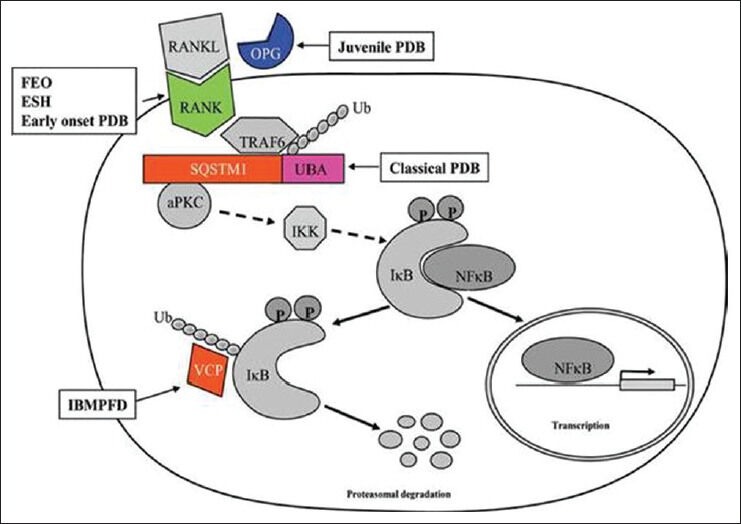
Binding of receptor activator of NF-κB ligand to receptor activator of NF-κB activates the nuclear factor κB signaling cascade, which leads to increased osteoclastogenesis
TNFRSF11A encodes for RANK, thus, activating mutations in this gene causes FEO, ESH and early onset PDB. TNFRSF1B encodes for OPG, inactivating mutations in this gene cause increased binding of RANKL to RANK leading to increased osteoclastic differentiation and activity resulting in Juvenile PDB. Mutations in VCP cause the syndrome of IBMPFD.[2]
Viral etiology
Viral infections have been suggested as one of the etiological factors for PDB because the nuclear inclusion bodies in osteoclasts appear to represent viral nucleocapsids.[17]
The ultrastructural features of the osteoclast inclusions were similar to the nucleocapsids of the viruses of the paramyxoviridae family. The paramyxoviridae viruses found to be associated with the PDB include measles virus, mumps virus, respiratory syncytial virus, canine distemper virus and the parainfluenza viruses.[18]
Measles virus consists of six genes that constitute the viral polymerase: The nucleocapsid, matrix, fusion, hemagglutinin, P protein and L protein. The Pittsburgh group demonstrated that transfection of the MVNP gene (but not of the matrix or the fusion gene) into the normal osteoclast precursors results in the formation of osteoclasts that express many of the features of pagetic osteoclasts. The pagetic features of the osteoclasts include increased rate of osteoclast formation, increased number and size of osteoclasts, increased bone resorbing capacity and hyper-responsivity to 1, 25-dihydroxy vitamin D3.[19,20]
PERSISTENCE OF MEASLES VIRUS INFECTION
Measles virus infection generally occurs in children, whereas Paget's disease predominates in elderly patients. If measles virus plays a role in the etiology of Paget's disease, there must be a mechanism whereby the measles virus remains viable in the human body into adulthood. However, osteoclasts are not self-renewing. Therefore, in patients with Paget's disease, cell types other than osteoclasts must serve as a reservoir for measles virus, enabling the virus to persist for long periods of time.[20]
PITTSBURG GROUP MODEL FOR PAGET'S ETIOLOGY
This model involves both genetic and non-genetic factors. It is divided into three stages:[20,21]
Stage I: In this stage, the measles virus infects pluripotent stem cells in patients with an acquired or inherited genetic predisposition for increased osteoclast activity or persistence of the measles virus. The stem cells differentiate and proliferate, giving rise to all hematopoietic lineages. The measles virus undergoes specific mutations that result in persistence of the virus in hematopoietic stem cells. When the cells differentiate toward the osteoclast lineage, a pagetic osteoclast forms [Figure 3].
Figure 3.
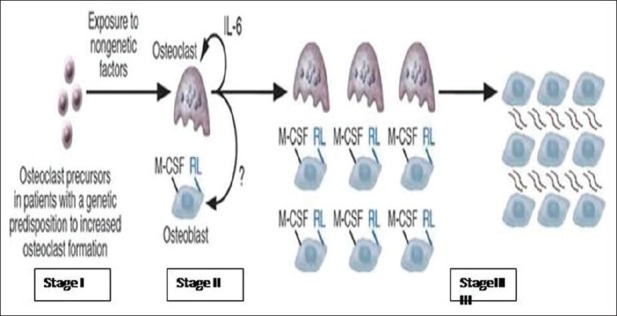
Pittsburg group model for Paget's etiology
Stage II: Over time, the pagetic osteoclasts produce increased amounts of IL-6 and other cytokines, which enhance osteoclast formation and lead to changes in the marrow microenvironment, further increasing osteoclast formation. Since osteoclastic and osteoblastic activity remains coupled in PDB thus increased osteoclastic activity results in increased osteoblast number and formation of new bone [Figure 3].
Stage III: As the lesion progresses, the marrow microenvironment is permanently changed: Increased number of osteoblasts express high levels of RANKL and other osteoclastogenetic factors Macrophage colony stimulating factor that further enhance the formation of osteoclast [Figure 3]. The following conditions are required for the development of Paget's disease: Mutations in the measles virus nucleocapsid protein (MVNP) gene, presence of the MVNP gene in osteoclasts and a genetic predisposition for enhanced osteoclastogenesis. As more and more bone is formed, the lesion would eventually become sclerotic.
Clinical features
Paget's disease is characterized by progressive enlargement and deformity of the affected bone resulting in structural weakness. The signs, symptoms and overall morbidity are largely determined by the sites involved. The most commonly involved bones include the pelvis, femur, spine, skull and tibia. The upper extremities, clavicles, ribs and scapulae are less frequently affected and the hands and feet are rarely involved.[22] Focal disease, in a vertebra or the articular surface of a joint, may produces severe symptoms, whereas relatively extensive involvement of the jaws may be functionally asymptomatic. Bone pain may be present and may be worse at night. Bone pain may be worse with weight-bearing if there is an osteolytic lesion. Bone enlargement or deformity may be present. There may be enlargement of the skull or frontal bossing and long bones may be bowed. Deformity in the long bones is more likely to occur in untreated long-standing disease and may be associated with pain. A bowed limb is typically shortened, resulting in specific gait abnormalities that can lead to abnormal mechanical stresses. Fissure fractures may occur on the convex aspect of bones.[2] The classic symptom of Paget's disease of the skull is enlarging head size or flattening of the occipital region (platybasia). Facial bones are more commonly involved leading to leontiasis ossea (lion like facies). Involved bones are warm to touch due to increased vascularity. Skeletal deformities and increased size of skull bones result in change in the patient's hat size. The patient may also complain of non-specific headache due to cranial nerve entrapment. Changes in vision may occur secondary to the optic nerve involvement. Osteosarcomas or other sarcomas (chondrosarcoma, fibrosarcoma) are the most serious complications of PDB. Although these sarcomas are more common than in age-matched controls, they occur in less than 1% of patients with PDB.[2,23]
Oral manifestations
The most frequent complication of Paget's disease of the jaws is associated with dental extractions. Hypercementosis and ankylosis may necessitate surgical extraction of teeth. Jaw involvement is present in approximately 17% of patients diagnosed with PDB. Maxillary involvement is far more common than mandibular involvement. The alveolar ridges tend to remain symmetric but become grossly enlarged. If the patient is dentulous, the enlargement causes spacing of the teeth. Edentulous patients may complain that their dentures no longer fit because of the increased alveolar size.[23]
Radiographic features
There is an initial phase of de-ossification and softening followed by bizarre dysplastic re-ossification occurring simultaneously or alternatively.
Radiographically, the early stages of Paget's disease reveal a decreased radiodensity of the bone and alteration of the trabecular pattern. Particularly in the skull, large circumscribed areas of radiolucency may be present (osteoporosis circumscripta). During the osteoblastic phase of the disease, patchy areas of sclerotic bone are formed, which tend to become confluent. The patchy sclerotic areas often are described as having a “cotton wool" appearance.[24]
A total body bone scan or a radiographic skeletal survey is often performed to assess the extent of disease. The enhanced skeletal uptake of radionuclide reflects increased bone formation and increased blood flow and may detect PDB before X-ray findings can be appreciated. Alternatively, some lesions in which osteoblastic activity has become inactive will be negative on bone scanning despite findings of PDB on plain radiographs.[2]
Histology
In the early phase of the disease, bone resorption predominates with abnormally large osteoclasts containing multiple pleomorphic nuclei and microfilamentous inclusion bodies. Following this initial osteolytic phase, there is a mixed osteolytic-osteoblastic phase with an abundance of osteoblasts forming new matrix in the form of woven bone. Repeated episodes of bone resorption and deposition results in appearance of many small irregular bony fragments that appear to be joined in a jigsaw or mosaic pattern with deeply staining hematoxyphillic reversal lines [Figure 4].[25] Macroscopically, the bones are thickened and enlarged with reduced medullary spaces and the marrow is replaced by highly vascularized fibrous tissue. Eventually, bone formation predominates, but the osteosclerotic bone is thickened, architecturally disorganized and less able to withstand stress. There is a corresponding reduction in vascular fibrous tissue and hemopoietic function returns to the marrow cavity with no abnormality seen in hematological indices.[26]
Figure 4.
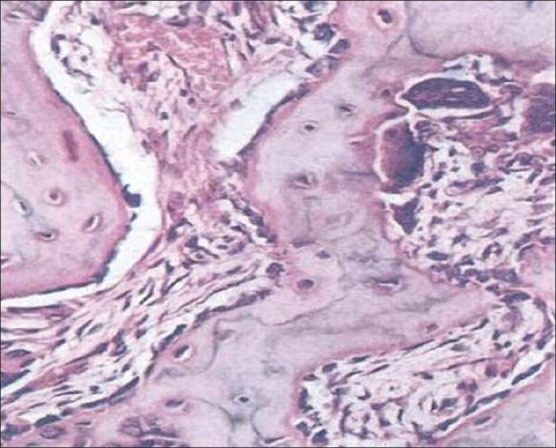
Histopathological picture of Paget's disease
Laboratory findings
The increased osteoclastic activity in the disease results in collagen or bone matrix degradation, producing an abnormally high excretion of urinary hydroxyproline and proline; both are reliable markers of the activity of the disease.[27]
Compensatory osteoblastic activity results in abnormally high serum alkaline phosphatase which may be raised up to 20 folds in patients with active disease (level as high as 250 Bodansky units in osteoblastic phase and in case of polyostotic involvement while its level seldom exceeds 50 Bodansky units in case of monostotic involvement). Increases in serum alkaline phosphatase and urinary hydroxyproline reflect the extent and severity of the disease. Alkaline phosphatase is the more sensitive and hydroxyproline is more accurate of the indices. Extra-cellular calcium homeostasis is usually maintained, though increased serum calcium levels may result from immobility. Several other biochemical markers of bone metabolism are also altered, including osteocalcin, acid phosphatase and α2 HS-glycoproteins.[23]
Treatment
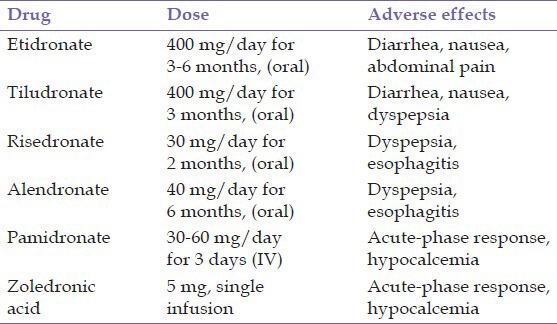
Although Paget's disease is chronic and slowly progressive, it is seldom the cause of death. In patients with more limited involvement and no symptoms treatment often is not required. In asymptomatic patients, systemic therapy is usually not initiated unless the alkaline phosphatase is more than 25-50% above normal. When symptomatic, bone pain is noted most frequently and often may be controlled by aspirin or other analgesics. Use of PTH antagonists, such as calcitonin and bisphosphonates, can reduce bone turnover and improve the biochemical abnormalities. Salmon calcitonin has been available for many years. It is generally given a dose of 100 IU subcutaneously daily. followed by the maintenance dose is often subsequently decreased to 50-100 IU 3 times weekly.[24] Simple bisphosphonates such as etidronate and tiludronate and the more potent nitrogen-containing bisphosphonates such as pamidronate, alendronate, risedronate and zoledronic acid (ZA).[28] For PDB, etidronate is typically given as 400 mg daily for 6 months followed by 6 months without therapy. Tiludronate is another ‘less potent’ bisphosphonate. It is given in a dosage of 400 mg daily for 3 months. The more potent amino bisphosphonates include alendronate, risedronate, pamidronate and ZA and are the preferred drugs. The potency of nitrogen-containing bisphosphonates in inhibiting osteoclastic bone resorption may be related to their ability to inhibit farnesyl diphosphate synthase.[29] Alendronate and risedronate are given orally while pamidronate and ZA are given intravenously. In the treatment of PDB, alendronate is given at a dosage of 40 mg daily for 6 months. Risedronate is given in a dosage of 30 mg daily for 2 months to treat PDB. Pamidronate is FDA approved for PDB in a dosage of 30 mg intravenously over 4 h on 3 consecutive days; however, other regimens are more commonly used. Patients with mild disease may be treated with a single 60-90 mg infusion while patients with more severe disease may receive multiple 90 mg infusions. ZA was FDA approved for PDB in 2007. It is given in a dosage of 5 mg intravenous (IV) over 15-30 min. In summary, when specific antipagetic therapy is needed, the amino bisphosphonates (alendronate, risedronate, pamidronate and ZA) are first line therapy and provide both oral and IV options. IV therapy may be preferred in patients with significant upper gastrointestinal problems or esophageal disease, patients in whom compliance to oral therapy is problematic, patients in whom a rapid antiresorptive effect is desired (e.g. neurologic symptoms or the urgent need for surgery on pagetic bone), or by patient preference. ZA appears to be the most potent drug and with the most sustained therapeutic effect for at least 2 years.[2,30]
IV bisphosphonates often cause transient bone pain, myalgia, headache, nausea, pyrexia and fatigue within 1 to 3 days after the infusion (acute-phase response). These symptoms can be ameliorated if acetaminophen is administered before and for a few days after the infusion, but they almost always subside within 7 days, even without treatment. Patients taking oral bisphosphonates must fast for 30 min (in the case of risedronate and alendronate) or 120 min (in the case of etidronate and tiludronate) before and after dosing to achieve adequate absorption. For this reason, it is customary to advise patients to take the medications in the morning. The most common adverse effects are dyspepsia (with risedronate and alendronate) and diarrhea (with tiludronate and etidronate). Uncommon side-effects of bisphosphonates include uveitis, rash and atrial fibrillation; osteonecrosis of the jaw and atypical subtrochanteric fractures have also been reported as rare complications. Bisphosphonates can cause kidney injury and are contraindicated in patients with clinically significant renal impairment.[30] Table 1 summarizes the bisphosphonates currently used in treatment and their adverse effects. In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone.[31]
Table 1.
Bisphosphonates in the Paget's disease of bone and their adverse effects
The Paget's Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2-5 years in patients treated with bisphosphonates versus those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < 0.001).[32]
In the observational extension study of ZA described above, 58 three of four fractures occurred in the group treated with ZA, echoing the findings of the PRISM study.[33] Denosumab is a monoclonal antibody to RANKL, which is currently being used for treating Osteoporosis and PDB. Denosumab acts by inhibiting RANKL binding to RANK thus inhibits osteoclastic activity.[34] Surgery including fracture stabilization, corrective osteotomy and total joint arthroplasty is sometimes needed for patients with PDB.[35] Pagetic bone is very vascular and it is recommended that patients receive specific antipagetic therapy prior to elective surgery on pagetic bone. Patients with spinal involvement may have radiculopathy or myelopathy.[36]
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Britton C, Walsh J. Paget disease of bone-An update. Aust Fam Physician. 2012;41:100–3. [PubMed] [Google Scholar]
- 2.Shaker JL. Paget's disease of bone: A review of epidemiology, pathophysiology and management. Ther Adv Musculoskelet Dis. 2009;1:107–25. doi: 10.1177/1759720X09351779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogers J, Jeffrey DR, Watt I. Paget's disease in an archeological population. J Bone Miner Res. 2002;17:1127–34. doi: 10.1359/jbmr.2002.17.6.1127. [DOI] [PubMed] [Google Scholar]
- 4.Cooper C, Harvey NC, Dennison EM, van Staa TP. Update on the epidemiology of Paget's disease of bone. J Bone Miner Res. 2006;21(Suppl 2):3–8. doi: 10.1359/jbmr.06s201. [DOI] [PubMed] [Google Scholar]
- 5.Altman RD. Epidemiology of Paget's disease of bone. Clin Rev Bone Miner Metab. 2002;1:99–102. [Google Scholar]
- 6.Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget's disease of bone: The New England Registry for Paget's Disease of Bone. J Bone Miner Res. 2003;18:1519–24. doi: 10.1359/jbmr.2003.18.8.1519. [DOI] [PubMed] [Google Scholar]
- 7.Jilka RL. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med Pediatr Oncol. 2003;41:182–5. doi: 10.1002/mpo.10334. [DOI] [PubMed] [Google Scholar]
- 8.Parfitt AM. Targeted and nontargeted bone remodeling: Relationship to basic multicellular unit origination and progression. Bone. 2002;30:5–7. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 9.Raisz LG. Physiology and pathophysiology of bone remodeling. Clin Chem. 1999;45:1353–8. [PubMed] [Google Scholar]
- 10.Hosking DJ. Paget's disease of bone. Br Med J (Clin Res Ed) 1981;283:686–8. doi: 10.1136/bmj.283.6293.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rebel A, Basle M, Pouplard A, Malkani K, Filmon R, Lepatezour A. Towards a viral etiology for Paget's disease of bone. Metab Bone Dis Relat Res. 1981;3:235–8. doi: 10.1016/0221-8747(81)90038-2. [DOI] [PubMed] [Google Scholar]
- 12.Menaa C, Barsony J, Reddy SV, Cornish J, Cundy T, Roodman GD. 1,25-Dihydroxyvitamin D3 hypersensitivity of osteoclast precursors from patients with Paget's disease. J Bone Miner Res. 2000;15:228–36. doi: 10.1359/jbmr.2000.15.2.228. [DOI] [PubMed] [Google Scholar]
- 13.Neale SD, Smith R, Wass JA, Athanasou NA. Osteoclast differentiation from circulating mononuclear precursors in Paget's disease is hypersensitive to 1,25-dihydroxyvitamin D (3) and RANKL. Bone. 2000;27:409–16. doi: 10.1016/s8756-3282(00)00345-8. [DOI] [PubMed] [Google Scholar]
- 14.Singer FR, Mills BG, Gruber HE, Windle JJ, Roodman GD. Ultrastructure of bone cells in Paget's disease of bone. J Bone Miner Res. 2006;21(Suppl 2):51–4. doi: 10.1359/jbmr.06s209. [DOI] [PubMed] [Google Scholar]
- 15.Siris E, Roodman GD. Paget's disease of bone. In: Rosen CJ, Compston JE, Lian JB, editors. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008. pp. 335–43. [Google Scholar]
- 16.Daroszewska A, Ralston SH. Genetics of Paget's disease of bone. Clin Sci (Lond) 2005;109:257–63. doi: 10.1042/CS20050053. [DOI] [PubMed] [Google Scholar]
- 17.Mills BG, Singer FR. Nuclear inclusions in Paget's disease of bone. Science. 1976;194:201–2. doi: 10.1126/science.959849. [DOI] [PubMed] [Google Scholar]
- 18.Singer FR. Update on the viral etiology of Paget's disease of bone. J Bone Miner Res. 1999;14(Suppl 2):29–33. doi: 10.1002/jbmr.5650140207. [DOI] [PubMed] [Google Scholar]
- 19.Kurihara N, Reddy SV, Menaa C, Anderson D, Roodman GD. Osteoclasts expressing the measles virus nucleocapsid gene display a pagetic phenotype. J Clin Invest. 2000;105:607–14. doi: 10.1172/JCI8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roodman GD. Recent developments in Paget's disease. Adv Stud Med. 2003;3:286–92. [Google Scholar]
- 21.Roodman GD, Windle JJ. Paget disease of bone. J Clin Invest. 2005;115:200–8. doi: 10.1172/JCI24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siris E. Indications for medical treatment of Paget's disease of bone. In: Singer FR, Wallach S, editors. Paget's Disease of Bone: Clinical Assessment, Present and Future Therapy. New York: Elsevier; 1991. [Google Scholar]
- 23.Thomas DW, Shepherd JP. Paget's disease of bone: Current concepts in pathogenesis and treatment. J Oral Pathol Med. 1994;23:12–6. doi: 10.1111/j.1600-0714.1994.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 24.Navell BW, Damm DD, Allen CM, Bouquot JE. Bone Pathology. 2nd ed. New York: Elsevier; 2002. Oral and Maxillofacial Pathology; pp. 555–87. [Google Scholar]
- 25.Smith BJ, Eveson JW. Paget's disease of bone with particular reference to dentistry. J Oral Pathol. 1981;10:233–47. doi: 10.1111/j.1600-0714.1981.tb01270.x. [DOI] [PubMed] [Google Scholar]
- 26.Ooi CG, Fraser WD. Paget's disease of bone. Postgrad Med J. 1997;73:69–74. doi: 10.1136/pgmj.73.856.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russell RG, Beard DJ, Cameron EC, Douglas DL, Forrest AR, Guilland-Cumming D, et al. Biochemical markers of bone turnover in Paget's disease. Metab Bone Dis Relat Res. 1981;3:255–62. doi: 10.1016/0221-8747(81)90041-2. [DOI] [PubMed] [Google Scholar]
- 28.Licata AA. Discovery, clinical development, and therapeutic uses of bisphosphonates. Ann Pharmacother. 2005;39:668–77. doi: 10.1345/aph.1E357. [DOI] [PubMed] [Google Scholar]
- 29.Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, et al. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J Pharmacol Exp Ther. 2001;296:235–42. [PubMed] [Google Scholar]
- 30.Ralston SH. Clinical practice. Paget's disease of bone. N Engl J Med. 2013;368:644–50. doi: 10.1056/NEJMcp1204713. [DOI] [PubMed] [Google Scholar]
- 31.Seton M. Paget disease of bone: Diagnosis and drug therapy. Cleve Clin J Med. 2013;80:452–62. doi: 10.3949/ccjm.80a.12142. [DOI] [PubMed] [Google Scholar]
- 32.Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH, et al. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget's disease of bone. J Bone Miner Res. 2010;25:20–31. doi: 10.1359/jbmr.090709. [DOI] [PubMed] [Google Scholar]
- 33.Reid IR, Lyles K, Su G, Brown JP, Walsh JP, del Pino-Montes J, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: Data to 6.5 years. J Bone Miner Res. 2011;26:2261–70. doi: 10.1002/jbmr.438. [DOI] [PubMed] [Google Scholar]
- 34.Hirao M, Hashimoto J. Denosumab as the potent therapeutic agent against Paget's disease of bone. Clin Calcium. 2011;21:1231–8. [PubMed] [Google Scholar]
- 35.Parvizi J, Klein GR, Sim FH. Surgical management of Paget's disease of bone. J Bone Miner Res. 2006;21(Suppl 2):75–82. doi: 10.1359/jbmr.06s214. [DOI] [PubMed] [Google Scholar]
- 36.Rongstad KM, Wheeler DL, Enneking WF. A comparison of the amount of vascularity in pagetic and normal human bone. Clin Orthop Relat Res. 1994;306:247–9. [PubMed] [Google Scholar]