

Abstract
Disturbances in lipid homeostasis are hallmarks of severe metabolic disorders and their long-term complications, including obesity, diabetes, and atherosclerosis. Whereas elevation of triglyceride (TG)-rich very-low-density lipoproteins (VLDL) has been identified as a risk factor for cardiovascular complications, high-density lipoprotein (HDL)-associated cholesterol confers atheroprotection under obese and/or diabetic conditions. Here we show that hepatocyte-specific deficiency of transcription factor transforming growth factor β 1-stimulated clone (TSC) 22 D1 led to a substantial reduction in HDL levels in both wild-type and obese mice, mediated through the transcriptional down-regulation of the HDL formation pathway in liver. Indeed, overexpression of TSC22D1 promoted high levels of HDL cholesterol in healthy animals, and hepatic expression of TSC22D1 was found to be aberrantly regulated in disease models of opposing energy availability. The hepatic TSC22D1 transcription factor complex may thus represent an attractive target in HDL raising strategies in obesity/diabetes-related dyslipidemia and atheroprotection.
Keywords: Obesity, Lipid metabolism, Liver, Transcription, High density lipoprotein
1. Introduction
Obesity and diabetes have reached pandemic dimensions and are associated with severe disturbances in systemic lipid handling [1]. Whereas elevation of triglyceride (TG)-rich very-low-density lipoproteins (VLDL) has been identified as an obesity-related risk factor for cardiovascular disease, increases in high-density lipoprotein (HDL)-associated cholesterol confers atheroprotection [2], and mechanisms raising systemic HDL consequently have become major targets in recent drug discovery efforts [3]. Indeed, HDL cholesterol concentration is inversely proportional to the risk for cardiovascular disease and future type 2 diabetes [2,4]. Also, patient cohorts with relatively high HDL cholesterol levels display a low prevalence of type 2 diabetes [5]. Accordingly, HDL deficiency and hypoalphalipoproteinemia as associated with mutations in the Tangier disease gene, ATP-binding cassette (ABC) transporter A1, correlate with early-onset atherosclerosis and fatal complications in affected subjects, prototypically underlining the importance of the HDL formation pathway for vascular integrity [6].
Recent studies in mice and men have identified a number of transcriptional regulators as critical checkpoints in liver lipoprotein homeostasis as exemplified by nuclear receptor co-factor networks, including the liver X receptor (LXR), peroxisome proliferator-activated receptor co-activator (PGC) 1, receptor interacting protein (RIP) 140, and transducin β-like (TBL) 1 [7–10]. In this context, we have recently identified the transcription factor TSC22D4 as an important determinant of hepatic VLDL production and systemic VLDL availability during tumor-induced energy wasting conditions [11]. The fact that TSC22D4 represents a member of a larger transcription factor family, TSC22D1-4 [12], with similar but non-redundant functions in other biological contexts (i.e. cellular senescence and growth control) [13], initially prompted us to explore a potential role of its related family member TSC22D1 in hepatic energy homeostasis. Here we show that TSC22D1 represents a regulatory checkpoint for HDL-associated cholesterol levels under both healthy and disease conditions.
2. Materials and methods
2.1. Recombinant viruses
Adenoviruses expressing a TSC22D1 or a non-specific shRNA under the control of the U6 promoter, or the TSC22D1 cDNA under the control of the CMV promoter were cloned using the BLOCK-iT Adenoviral RNAi expression system (Invitrogen). Propagated viruses were purified by the cesium chloride method. For cesium chloride gradient PBS-TOSH was added to the crude cell lysate to a final volume of 20 ml. All solutions were adjusted to pH 7.2. The first gradient was layered with 9 ml 4.4 M CsCl, 9 ml 2.2 M CsCl and 20 ml virus in PBS-TOSH. After ultracentrifugation (2 h, 4 °C, 24,000 rpm, SW 28 rotor) a clear virus band was visible. The virus band was removed by piercing the tube with a 5 ml syringe. To the obtained virus (3–3.5 ml) the same volume of saturated CsCl was added. The second gradient was layered with 6–7 ml virus in CsCl, 2 ml 4.4 M CsCl und 2 ml 2.2 M CsCl. Following the second ultracentrifugation (3 h, 4 °C, 35,000 rpm, SW 40Ti rotor) the virus band was removed by syringe in the smallest possible volume and dialyzed twice against PBS-glycerol, 1 h and overnight at 4 °C. 10% glycerol was added to the purified virus and aliquots were stored at −80 °C. The Tissue Culture Infectious Dose 50 (TCID50) assay was used to titrate adenovirus. For that purpose, 104 HEK293A cells were seeded in 100 μl virus titration medium in each well of a 96 well plate and infected with decreasing amounts of virus after 2–4 h of adhesion. Serial dilutions of the virus from 10−7 to 10–14 were prepared in titration medium and added to the cells. Double measurements were performed for each virus. 10–12 days after infection and incubation at 37 °C the plaques were counted using a microscope and the titer was calculated by the following formula: T=10×1+(s−0.5)×10 pfu/ml (s=sum of positive wells starting from the 10−1 dilution; 10 positive wells per dilution=1) [8,14,15]. The BLOCKiT™ Pol II miR RNAi Expression System was used to generate adeno-associated viruses expressing miRNA sequences against murine TSC22D1 as well non-specific miRNAs. Oligonucleotide sequences were chosen using Invitrogen's RNAi Designer tool. Forward and reverse oligonucleotides against the target gene sequence were annealed and cloned into the pcDNA6.2-GW/EmGFP-miR vector according to the manufacturer's instructions. Following, they were transferred into the previously described double stranded pdsAAV-LP1-EGFPmut AAV vector [10] using the restriction enzymes BglII and SalI. The plasmids encoding the miRNA constructs were cotransfected into HEK293T cells with the pDGΔVP helper plasmid [16] and a mutated p5E18-VD2/8 expression vector [17] encoding AAV2 rep and a mutated AAV8 cap protein (aa 589–592: QNTA to GNRQ).
For virus production, HEK293T cells from six 80–90% confluent 15 cm plates were suspended in 1100 ml medium. 1000 ml of the cell suspension was transferred to a 10× cell-stack chamber and 100 ml were transferred to a 1×-cell stack. 24 h after plating, the cells were approximately 70–80% confluent and were transfected with the plasmids encoding the viral genes using the PEI method. Once the cell monolayer was approximately 90% confluent, cells were washed with PBS, before they were released from the plate using 10 ml (1× cell stack) or 100 ml (10× cell stack) trypsin-EDTA for 5 min at 37 °C. Fresh medium was added (40 ml or 350 ml respectively) and the cells were transferred to a 50 ml falcon tube, or a 500 ml conical tube. The chambers were washed with PBS and used for a second round of transfection. Cells were centrifuged at 2000 rpm for 10 min. The supernatant was removed and the pellets were resuspended in 8 ml lysis-buffer containing 150 mM NaCl and 50 mM Tris–HCl, pH 8.5. The lysates were transferred into 15 ml falcons, vortexed, snap-frozen in liquid nitrogen and stored at −80 °C. AAV lysates were thawed at 37 °C under vigorous vortexing and then centrifuged at 3500g for 10 min. The supernatant was collected and the pellets were resuspended in 4 ml lysis buffer and snap-frozen. The freeze–thaw cycle was repeated three times. The final pellet was solubilised using a sonicator in a water bath at 48 W for 1 min. The pooled suspension was then digested with benzonase (50 U/ml) for 30 min at 37 °C. This solution was then centrifuged at 4 °C and 3500g for 10 min. The virus was stored at −80 °C until further use. Virus was purified by a two-step iodixanol gradient protocol in a Sorvall WX Ultra 80 Ultracentrifuge. The obtained viral solution was concentrated using a VivaSpin concentrator. The solution was consecutively centrifuged at 3000 rpm and 10 °C for 3–6 min until a final volume of about 1 ml. Viral DNA was isolated by mixing 5 μl of virus suspension with 5 μl H2O and 10 μl 2 M NaOH. The mixture was incubated at 56 °C for 30 min and the reaction was then neutralized by adding 10 μl 2 M HCL. After adding 970 μl H2O, the titer was determined by qPCR using an EGFP standard curve.
2.2. Animal experiments
Male 8–12-week old C57Bl/6, Balb/C mice, JAX® Mice Strain: B6.V-Lep(ob)/J (000632) and BKS. Cg-Dock7m+/+Leprdb/J (000642) were obtained from Charles River Laboratories (Brussels, Belgium), and maintained on a 12 h light–dark cycle with regular unrestricted diet. For hepatic VLDL release animals were fasted for 16 h, for VLDL clearance mice were fasted for 4 h and for fasting experiments mice were fasted for 24 h. Otherwise animals were fed ad libitum and had free access to water. For adenovirus injections, 1–2×109 plaque-forming units (pfu) per mouse were administered via tail vein injection. For AAV experiments 5×1011 virus particles per mouse were injected via the tail vein. For tumor induction in cachexia models, 1.5×106 C26 cells in phosphate buffered saline were injected subcutaneously into 10-week-old Balb/C mice (Charles River Laboratories, Brussels, Belgium). In high-fat diet experiments, C57Bl/6 mice were either fed a low-fat control diet (10% energy from fat, Research diets D12450B, New Brunswick, USA) or a high-fat diet (60% energy from fat, Research diets D12492, New Brunswick, USA) for a period of 12 weeks. For MCD diet experiments, C57Bl/6 mice were either fed a standard control diet or a methionine, choline deficient (MCD) diet (Research diets, New Brunswick, USA) for a period of 4 weeks. In each experiment, 4–10 animals received identical treatments and were analyzed under fasted or refed conditions as indicated. For TGFβ experiments, C57Bl/6 mice were injected intravenously with either 150 µl PBS or 5 µg human recombinant TGFβ1 in 150 µl PBS and organs were taken 2, 8 or 24 h after injection.
Diabetes was induced in male LDL-receptor knockout mice (B6.12957-LdlrtmHer/J, Jackson Laboratories) by intraperitoneal administration of streptozotocin. Diabetes was maintained over a period of 6 weeks by monitoring blood glucose levels. Organs including liver, kidney, epididymal fat pads, and gastrocnemius muscles were collected after specific time periods, weighed, snap-frozen and used for further analysis. Animal handling and experimentation was done in accordance with NIH guidelines and approved by local authorities.
2.3. ApoB clearance assay
Blood was drawn from a fasted individual and human VLDL was isolated by ultracentrifugation. 3.5 ml serum was put in a polyallomer tube (SW40Ti) and mixed with 1.39 g KBr, over-layered with 3×2.8 ml of a NaCl/KBr solution (D=1.063, 1.019 and 1.006 g/ml) and run for 18 h at 40,000 rpm. 20 μg of human VLDL was injected into each animal and serum samples were taken at 2, 10, 30, 60 and 120 min. Human ApoB-100 levels were measured using a human-specific ApoB ELISA. For the ELISA, a primary coating antibody generated against human ApoB-100 (mAb47, kindly supplied by J. Witztum, University of San Diego, USA), at a concentration of 2 μg/well and a secondary biotinylated polyclonal antibody raised in goat against human ApoB at a concentration of 4 μg/well in 1.5% BSA/TBS/0.1% tween were used. To prevent non-specific binding, plates were blocked with 1.5% BSA/TBS/0.1% tween. Samples were diluted 25-fold. Absorbance was read 30 min after addition of TMB and termination of the reaction with 2 M H2SO4 at 450 nm [18].
2.4. Blood metabolites
Serum levels of glucose, triglycerides (TG), total ketone bodies, and non-esterified fatty acids (NEFA) were determined using an automatic glucose monitor (One Touch, Lifescan, Neckargemünd, Germany) or commercial kits (Sigma, Munich, Germany, WAKO, Neuss, Germany, respectively). TGFβ1 levels were determined using a TGFβ1 ELISA (R&D systems, Wiesbaden-Nordenstadt, Germany).
2.5. Fast protein liquid chromatography
The FPLC set-up consisted of a Superose 6 10/300 GL column (GE Healthcare), a fraction collector and an ÄKTA FPLC System (Amersham). During separation, a liquid phase, containing the mixture to be fractionated was pumped over a stationary resin of cross-linked agarose beads with varying surface structure. A pool of 200 μl serum plus 100 µl PBS, from 4–6 animals, was injected into the machine, diluted in 25 ml PBS and fractionated into 500 μl fractions. 40 μl of each fraction was subsequently used for cholesterol and TG analysis using the TG Liquicolor (Human GmbH, Germany) and Cholesterol determination kit (Randox, UK) respectively.
2.6. Hepatic VLDL release
VLDL production was determined after tyloxapol injection. One day before the experiment, tyloxapol was dissolved in saline to obtain a 20% w/v solution. Mice were fasted overnight for 16 h. On the following day, before administration, 40 μl of blood was drawn from each mouse by cutting the tip of the tail. The tyloxapol (20%) volume (in μl) applied per mouse was approx. 3 times that of the weight of the mouse in grams. Specified amounts were administered via the tail vein and 40 μl blood samples were taken every 50 min for 2.5 h. The mice were sacrificed after 300 min. The serum TG values were determined using commercial kit (Sigma, Munich, Germany) [18].
2.7. LPL activity
LPL activity measurements were performed as described [19] using frozen adipose tissue samples.
Enzyme preparation: The frozen tissue samples (50–150 mg wet weight) were immersed in 5 ml acetone at −20 °C and homogenized immediately for 30 s. The precipitate was separated from the organic solvent by vacuum filtration on a pre-weighed glass-fiber filter (Whatmann GF/C, 3.5 cm) and washed once with 3 ml of −20 °C cold diethyl ether. After the filter had been dried in a vacuum at 5 °C, it was weighed and the acetone–ether dry powder was stored at −80 °C for LPL assay on the following day. Acetone-ether dried LPL was reconstituted in a buffer [0.05 M tris(hydroxymethyl)aminomethane (Tris) l HCl (pH 8.6), 20 USP U heparin/ml] by adding 10 mg powder/ml and agitating for 45 min at 5 °C. The protein solution obtained was centrifuged with 12,500g at 5 °C for 10 min (Beckman model 521 B). The supernatant was assayed for LPL activity.
Enzyme assay: LPL activity was determined by glyceryl tri-[l-14C]oleate. In a final volume of 100 ~1 the assay contained 0.14 M Tris HCl (pH 8.6), 5.1 mM glyceryl tri-[l-14C]oleate (44,400 dpm), 12 mM CaCIZ, 2% bovine serum albumin (BSA), 10% human serum, 0.4% gum arabic, 5 USP U/ml heparin, and 1 M NaCl (blanks only). Tris buffers were adjusted with HCl to pH 8.6 at 37 °C. Toluene solutions of 10.2 pmol unlabeled glyceryl trioleate and 14.8 kBq glyceryl tri-[l–14C]oleate (Amersham, sp act 2.09 GBq/mmol) were mixed in a vial, and the solvent was evaporated under nitrogen. The lipids were emulsified four times for 30 s with 30-s intervals at 25 W (Branson sonifier B12) on ice in 400 ~1 stock buffer [0.2 M Tris HCl (pH 8.6), 0.016 M CaCl2] adjusted to 2% gum arabic. After sonification, 400 ~1 stock buffer including 10% BSA (essentially fatty acid free) and 200 ~1 human serum (preincubated 30 min at 37 °C) were mixed with the triglyceride emulsion. This substrate emulsion was incubated for 15 min at 37 °C before use. Duplicate determinations of LPL activity ⁎ were performed by adding 25 ~1 of the supernatant to 25 ~1 stock buffer and 50 ~1 substrate emulsion and incubating at 37 °C for 20 min.
Oleate extraction: The reaction was stopped by the addition of 500 ~1 0.1 M K&Oa-H3B03 buffer (pH 10.5) and 1600 ~1 mixture of methanol, chloroform, and heptanes (1.41:1.25:1, vol/vol/vol). After 5 min of agitation, the two phases were separated by centrifugation at 160g and the radioactivity of the upper phase was determined in a liquid scintillation counter (Beckman model LS 3801).
2.8. Tissue lipid extraction
Lipids were extracted from frozen and pulverized liver tissue using chloroform/methanol (2:1 v/v). About 100 mg (the exact weight was noted) of frozen, pulverized liver were transferred into a 2 ml tube containing 1.5 ml chloroform/methanol and a steel bead. The tissue was homogenized using a tissue lyzer for three times 30 s at a frequency of 30 Hz. For the lipid extraction, samples were incubated on a rotating wheel at room temperature for 20 min. Samples were centrifuged at 13,000 rpm for 30 min at RT and 1 ml of the supernatants were transferred to fresh tubes. The organic layer was mixed 200 µl with 0.9% sodium chloride and the aqueous solution was carefully discarded. The solution was centrifuged for 5 min at 2000 rpm. 750 μl of the lower organic layer was transferred to a fresh tube and stored at −80 °C. For lipid re-suspension 40 μl of triton-X 100/chloroform (1:1 v/v) was pipetted into fresh tubes and 200 µl of the organic lipid sample was added. The reagents were mixed and the solvent was evaporated overnight using a speed vac. The residue containing the hydrophobic contents of the liver was re-suspended in 2000 μl water and stored at −20 °C until further use. TG and total cholesterol content were determined using commercial kits as above. Values were calculated as mg (TG and cholesterol) per gram wet tissue.
2.9. Histochemistry
During preparation of liver tissue, slices were embedded Tissue Tek OCT (Sakura, Torrance, USA) compound and shock frozen in liquid nitrogen. 5 μm cryosections were cut and fixed in Baker's formol. Neutral lipids and TGs were stained with oil red O and nuclei were counterstained with hematoxylin [7].
2.10. Aorta lesion quantification
Mice were sacrificed. Hearts were perfused with 5 ml cold PBS. Tissues surrounding the aorta were cleared under a binocular microscope. The heart was removed by cutting the descending aorta halfway between the aortic root and the brachiocephalic artery and frozen in OCT compound (Tissue-Tek; Sakura Finetek USA Inc.). The remaining aorta was dissected to the iliac bifurcation, opened longitudinally, and fixed between 2 glass slides. The aortas were fixed in 10% buffered formalin, and stained with oil red O [20]. Randomized and blinded quantification of the aortic arch lesion area in diabetic and non-diabetic LDLR KO mice was performed with ImageJ 1.44p (Wayne Rasband, National Institute of Health, USA).
2.11. Quantitative Taqman RT-PCR
Total RNA was extracted from homogenized mouse liver or cell lysates using Qiazol reagent (Qiagen, Hilden, Germany). cDNA was prepared by reverse transcription using the M-MuLV enzyme and Oligo dT primer (Fermentas, St. Leon-Rot, Germany). cDNAs were amplified using assay-on-demand kits and an ABIPRISM 7700 StepONE (Applied Biosystems, Darmstadt, Germany). RNA expression data were normalized to levels of TATA-box binding protein (TBP) RNA.
2.12. Protein analysis
For extraction of nuclear fractions about 50 mg frozen liver samples were grinded in Dounce homogenizer for 15 strokes with 1 ml of lysis buffer (50 mM Tris pH 7.2, 250 mM Sucrose, 10 mM KCL, 1 mM EDTA, 10 mM NaF, 2 mM Na3VO4, 1 mM DTT, 1× Protease Inhibitor Cocktail (Sigma, Heidelberg, Germany), 1× Phosphatase Inhibitor II (Sigma, Heidelberg, Germany)). Homogenates were rotated end over end at 4 °C for 30 min after which they were centrifuged at 4 °C for 30 min at 1000g. The pellet was dissolved in 500 µl lysis buffer (50 mM Tris pH 7.2, 25% glycerol, 420 mM NaCl, 1.5 mM MagCl2, 10 mM NaF, 1 mM DTT, 1× Protease Inhibitor Cocktail, 1× Phosphatase Inhibitor II, 0.1% SDS, 1% NP-40). Pellets were dissolved by grinding with a pestle. Homogenates were rotated end over end at 4 °C for 30 min after which they were centrifuged at 4 °C for 30 min at 9000g. Protein extracts were separated on 15% SDS-polyacrylamide gels and blotted onto PVDF membranes. Western blot assays were performed as described [8] using antibodies specific for TSC22D1 (custom designed from Eurogentec, Seraing, Belgium), Apolipoprotein A1 (Cell Signaling, Danvers, United States), β-actin (Santa Cruz, Heidelberg, Germany) or valosin-containing protein (VCP) (abcam, Cambridge, United Kingdom).
2.13. Plasmids and RNA interference
For shRNA experiments, oligonucleotides targeting mouse TSC22D1 (5'-GCCATTTGATGTATGCGGTGA-3'), were annealed and cloned into the pENTR/U6 shRNA vector (Invitrogen, Karlsruhe, Germany). Non-specific oligonucleotides (5'-GATCTGATCGACACTGTAATG-3') with no significant homology to any mammalian gene sequence were used as non-silencing controls in all experiments. For miRNA experiments, oligonucleotides targeting mouse TSC22D1 (5'-CTCACACGCTGTTCTCGCTTT-3') and non-specific oligonucleotides (5'-AAATGTACTGCGCGTGGAGAC-3') were cloned into the pdsAAV-LP1 vector.
2.14. Cell culture
Primary mouse hepatocytes were isolated and cultured as described [21]. Briefly, male 8–12 week old C57Bl/6 mice were anesthetized by i.p. injection of 100 mg/Kg body weight ketamine hydrochloride and 5 mg/Kg body weight xylazine hydrochloride. After opening the abdominal cavity, the liver was perfused at 37 °C with HANKS I (8 g NaCl, 0.4 g KCl, 3.57 g Hepes, 0.06 g Na2HPO4·2H2O, 0.06 g KH2PO4 in 1 L distilled H2O, 2.5 mM EGTA, 0.1% glucose, adjusted to pH 7.4) via the portal vein for 5 min and subsequently with HANKS II (8 g NaCl, 0.4 g KCl, 3.57 g Hepes, 0.06 g Na2HPO4·2H2O, 0.06 g KH2PO4 in 1 L distilled H2O, 0.1% glucose, 3 mg/ml collagenase CLSII, 5 mM CaCl2, adjusted to pH 7.4) for 5–7 min until disintegration of the liver structure was observed. The liver capsule was removed and the cell suspension was filtered through a 100 µm mesh. The cells were washed and, subsequently, viability of cells was determined by trypan blue staining. 1×106 living cells/well were seeded on collagen I-coated 6-well plates. After 24 h, cells were stimulated with 10 ng of human recombinant TGFβ1 for 30 min, 6 h or 24 h and subsequently harvested for qPCR analysis. Hepa1c1 cells were infected 24 h after seeding, with recombinant adenoviruses at a multiplicity of infection of 500. Cells were harvested 48 h after infection.
2.15. Human liver samples
Normal liver tissue samples were obtained by open liver biopsy from 30 patients, 15 with and 15 without cachexia, who underwent surgery for pancreatic ductal adenocarcinoma as described previously [22]. Since there is no generally accepted definition, cachexia in pancreatic cancer patients was defined as unintended loss of >10% of body weight in the 6 months before diagnosis. Additionally, liver samples were obtained from 10 patients with chronic pancreatitis who underwent pancreatic head resection and from 5 patients with resection of colon cancer. For use as controls, liver biopsies were obtained through an organ donor program from 5 previously healthy individuals who were free of any pancreatic or neoplastic disease. Two of the 15 patients with PDAC and cachexia were excluded after the final histological diagnoses proved to be chronic pancreatitis and mucinous pancreatic carcinoma, respectively. One patient in the group with PDAC without cachexia was determined to have a papillary carcinoma and was also excluded. The study was approved by the local ethics committee of the University of Heidelberg, Germany. All patients gave preoperative written informed consent for the use of their samples.
2.16. Statistical analysis
For each experiment means±SEM were determined. Statistical analyses were performed using student's t-test in one-factorial designs. Correlation was determined using Pearson's correlation coefficient; F-test was applied to determine significance. For multifactorial study designs, Two-way ANOVA and TWO-way ANOVA RM were used when appropriate. Holm–Sidak post hoc was applied when significant differences were found with an overall significance level=0.05.
3. Results
3.1. TSC22D1 controls systemic HDL cholesterol metabolism
To delineate the function of hepatic TSC22D1 in vivo, we disrupted the activity of TSC22D1 in livers of lean wild-type mice by delivering an adenovirus expressing a TSC22D1-specific or a non-specific control shRNA via tail vein injection. TSC22D1 shRNA treatment significantly reduced hepatic TSC22D1 mRNA and protein levels as compared with control shRNA-injected littermates (Figure S1a and b). At day 7 after injection, acute knockdown of TSC22D1 caused no major differences in body-, liver-, and abdominal white adipose tissue weight (Figure S1c–e), hepatic TG levels (Figure S1f), circulating NEFA and ketone bodies (Figure S1g and h), and serum glucose levels (Figure S1i), while serum TG levels were found to be decreased compared with controls (Figure S1j). Also, indirect calorimetry studies revealed no differences in oxygen consumption and energy expenditure in both experimental groups (Figure S1k and l). In contrast, down-regulation of hepatic TSC22D1 resulted in a significant increase in liver cholesterol levels, which was evident particularly during fasting conditions (Figure 1a), and a reduction in HDL-associated cholesterol in the serum (Figure 1b and c), indicating that TSC22D1 fulfills a specific function in hepatic and systemic cholesterol handling in healthy wild-type animals.
Figure 1.
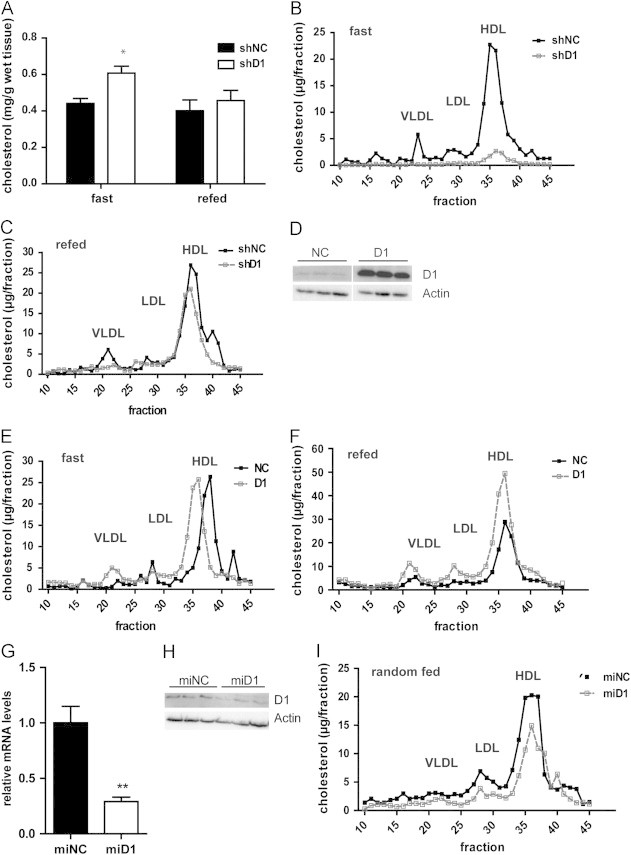
TSC22D1 promotes hypoalphalipoproteinemia in wild-type mice. (a) Liver cholesterol levels of TSC22D1 shRNA adenovirus injected wild-type C57BL/6J mice (means±SEM, n=5). (b) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in 24 h fasted control or TSC22D1 shRNA adenovirus injected wild-type C57BL/6J mice. (c) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in 6 h refed control or TSC22D1 shRNA adenovirus injected wild-type C57BL/6J mice.(d) Representative Western Blot of liver extracts from control or TSC22D1 cDNA adenovirus injected wild-type C57BL/6J mice, using TSC22D1 (D1) or actin specific antibodies. (e) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in 24 h fasted control or TSC22D1 cDNA adenovirus injected wild-type C57BL/6J mice. (f) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in 6 h refed control or TSC22D1 cDNA adenovirus injected wild-type C57BL/6J mice. (g) Quantitative PCR analysis of hepatic TSC22D1 expression levels in random fed control or miRNA TSC22D1 adeno-associated virus injected wild-type C57BL/6N mice (means±SEM, n=5). (h) Representative Western Blot of liver extracts from same mice as in (g) using TSC22D1 (D1) or actin specific antibodies. (i) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in the same mice as in (g). Statistical test: students t-test, ⁎⁎, p<0.01.
To corroborate these findings, we next sought to test the effects of TSC22D1 over-expression (Figures 1d and S1m and n) on systemic lipid handling. To this end, we employed wild-type mice with adenovirus-mediated liver-specific over-expression of TSC22D1 in a fasting-feeding regimen. In line with the results from TSC22D1 loss-of-function experiments indicating a predominantly fasting-associated activity of the endogenous protein (Figure 1b), over-expression of TSC22D1 led to a significant elevation of HDL-associated cholesterol during feeding and lowered hepatic cholesterol content (Figures 1e, f and S1o), while leaving other metabolic parameters mostly unaltered (Figure S1p–v).
To confirm the results from acute knockdown experiments in an independent setting, wild-type mice were injected with an adeno-associated virus (AAV) expressing a TSC22D1-specific miRNA or a non-specific control miRNA under the control of a hepatocyte-specific promoter (LP1), allowing the expression of inhibitory miRNAs specifically in liver parenchymal cells but not in other liver cell types [15]. After 4 weeks, TSC22D1 miRNA delivery led to a 70–80% decrease in hepatic TSC22D1 mRNA levels (Figures 1g and S2a) and also reduced TSC22D1 protein expression in liver as compared with control miRNA-injected littermates (Figure 1h). Inactivation of TSC22D1 again triggered a decrease in HDL cholesterol (Figure 1i), and increased liver cholesterol levels (Figure S2b) in random fed animals, while leaving serum TG levels and other parameters largely unaffected (Figure S2c–g).
Together, these data underlined the notion that hepatic TSC22D1 fulfils a critical and specific checkpoint function for the regulation of circulating HDL cholesterol levels and hepatic cholesterol load under normal health conditions.
3.2. TSC22D1 transcriptionally regulates the HDL formation pathway
To determine the mechanistic basis for TSC22D1 function in systemic lipid metabolism, we tested basic functions of hepatic lipid handling in response to liver-specific TSC22D1-deficiency. In this respect, we initially determined hepatic VLDL production by experimental inhibition of peripheral VLDL clearance. Consistent with mostly unaltered serum TG levels, mice deficient in hepatic TSC22D1 showed no difference in hepatic VLDL release and liver re-uptake of human ApoB as compared with controls (Figures 2a and S3a and b). In addition, adipose tissue LPL activity was also not changed upon impairment of hepatic TSC22D1 activity (Figure 2b), further underlining the notion that TSC22D1 specifically regulates systemic (HDL) cholesterol metabolism while leaving TG-related pathways largely unaffected. Indeed, consistent with an almost exclusive nuclear localization of TSC22D1 in hepatocytes (data not shown), transcriptional profiling of TSC22D1-dependent target gene networks showed that key regulatory genes in the HDL formation and cholesterol loading axis were significantly down-regulated in TSC22D1-deficient wild-type livers, most notably including ATP-binding cassette transporter (ABCA) 1, and ABCG5/8 (Figure 2c and d) [6,23]. In contrast, genes in the lipogenic and bile acid synthesis pathway remained unaltered upon loss of TSC22D1 expression (Figure 2c and data not shown). Of note, hepatic and serum ApoA1 protein levels as well as the protein content of the HDL serum fraction remained unchanged between control and TSC22D1-deficient animals (Figure S3c–e), suggesting that TSC22D1 does not regulate serum HDL cholesterol through changes in lipoprotein generation but rather impacts HDL cholesterol loading. Also, inactivation of TSC22D1 in cultured hepatoma cells by shRNA-mediated gene knockdown resulted in a significant reduction of genes in the HDL formation pathway (Figure 2e), confirming the regulatory impact of TSC22D1 on HDL generating gene networks in a cell autonomous manner.
Figure 2.
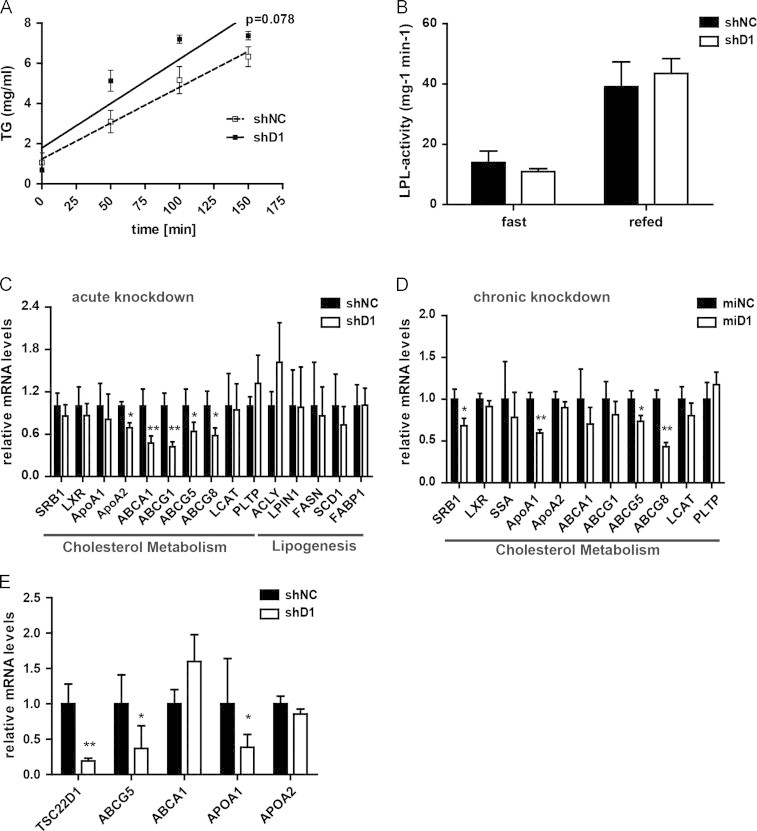
TSC22D1 controls key gene expression in HDL formation. (A) Hepatic VLDL release in control or TSC22D1 shRNA adenovirus injected wild-type C57Bl/6 mice. Time after tyloxapol injection indicated (means=SEM, n=5). (B) White adipose tissue (WAT) lipoprotein lipase (LPL) activity in 24 h fasted or 6 h refed control or TSC22D1 shRNA adenovirus injected wild-type C57BL/6J mice (means±SEM, n=5). (C) Quantitative PCR analysis of scavenger receptor B1 (SRB1), liver X receptor (LXR), Apolipoproteins A1 and A2 (ApoA1, ApoA2), ATP-binding cassette transporter (ABCA1), ATP-binding cassette sub-family G member 1, 5 and 8 (ABCG1, ABCG5, ABCG8), lecithin-cholesterol acyltransferase (LCAT), phospholipid transfer protein (PLTP), ATP citrate lyase (ACLY), lipin 1 (LPIN1), fatty acid synthase (FASN), stearoyl-CoA desaturase-1 (SCD1), Fatty acid-binding protein 1 (FABP1) and RNA levels in livers of refed control or TSC22D1 shRNA adenovirus injected wild-type C57Bl/6 mice (means±SEM, n=5). (D) Quantitative PCR analysis of scavenger receptor B1 (SRB1), liver X receptor (LXR), serum amyloid A (SSA), Apolipoproteins A1 and A2 (ApoA1, ApoA2), ATP-binding cassette transporter (ABCA1), ATP-binding cassette sub-family G member 1, 5 and 8 (ABCG1, ABCG5, ABCG8), lecithin-cholesterol acyltransferase (LCAT), phospholipid transfer protein (PLTP) of hepatic TSC22D1 expression levels in refed control or miRNA TSC22D1 adeno-associated virus injected wild-type C57BL/6N mice (means±SEM, n=5). (E) Quantitative PCR analysis of TSC22D1, ATP-binding cassette sub-family G member 5 (ABCG5), ATP-binding cassette transporter (ABCA1), Apolipoproteins A1 and A2 (ApoA1, ApoA2), (means±SEM, n=4) in Hepa1c1 cells infected with control (shNC) or TSC22D1 shRNA adenovirus. Cells were harvested 48 h post-infection. Statistical tests: for a Two Way Repeated Measures ANOVA, Holm–Sidak post hoc for b–e students t-test, ⁎, p<0.05, ⁎⁎, p<0.01.
3.3. Hepatic TSC22D1 levels are determined by the systemic energy status
HDL is considered to act as an atheroprotective factor, and low HDL cholesterol represents a core component of dyslipidemic conditions as associated with obesity and the Metabolic Syndrome [2]. The differential gene expression of specific transcriptional regulators between healthy and metabolic disease conditions has frequently been found to reflect either a causal, pathogenic or a compensatory, protective role of these factors in disease progression and/or manifestation [24]. The general loss of HDL-associated atheroprotection in obesity [4] next prompted us the screen for the expression of TSC22D1 in various mouse models of obesity-related metabolic dysfunction. Gene expression analysis demonstrated that hepatic TSC22D1 mRNA levels were 2–3-fold elevated in mouse models of both genetic (db/db and ob/ob) as well as diet-induced obesity (12 weeks high fat diet-fed wild-type mice vs. low-fat fed controls) (Figure 3a and b, Sf). Interestingly, obesity has recently been found to promote elevated levels of TGFβ in both human and murine settings, correlating with systemic dyslipidemia [25], and TSC22D1 had originally been identified as a TGFβ-inducible gene in other cells [26]. These findings prompted us to test the response of TSC22D1 gene transcription to TGFβ signaling in liver. Treatment of primary mouse hepatocytes with TGFβ indeed significantly enhanced TSC22D1 mRNA levels after already 6 h, the effect of which was further augmented after 24 h treatment (Figure 3c). Also, treatment of wild-type mice with TGFβ transiently mirrored these effects in vivo (Figure 3d), suggesting that hepatic TSC22D1 indeed is a downstream target of TGFβ signaling, thereby correlating well with elevated TGFβ serum levels in ob/ob mice (Figure S3g [25]) and the up-regulation of TSC22D1 under these conditions as shown above (Figures 3a, b and S3f).
Figure 3.
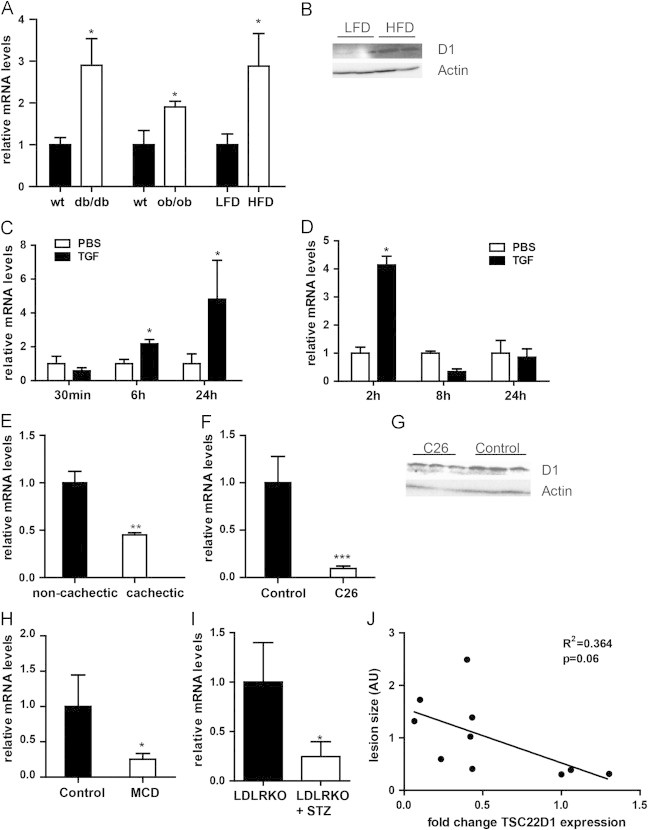
Deregulation of TSC22D1 in conditions of metabolic dysfunction. (A) Quantitative PCR analysis of hepatic TSC22D1 expression of db/db or ob/ob mice compared to wild-type control (wt) or mice fed a high-fat diet (HFD) compared to control mice on a low-fat diet (LFD). (B) Western blot of liver extracts from representative mice fed a high-fat diet (HFD) compared to control mice on a low-fat diet (LFD), using TSC22D1 (D1) or actin specific antibodies. (C) Quantitative PCR analysis of TSC22D1 expression in primary hepatocytes stimulated with 10 ng of human recombinant TGFβ1 or PBS for the indicated time (means±SEM, n=3). (D) Quantitative PCR analysis of hepatic TSC22D1 expression in C57Bl/6 mice treated with PBS or 5 µg human recombinant TGFβ1 for the indicated time points (means±SEM, n=4). (E) Quantitative PCR analysis of hepatic TSC22D1 expression in pancreatic cancer patients with or without cachexia (means±SEM, n=5). (F) Quantitative PCR analysis of hepatic TSC22D1 expression in Balb/C mice treated with PBS or 1.5×106 colon 26 (C26) cells over 3 weeks (means±SEM, n=6). (G) Western blot of liver extracts from representative mice as in (e), using TSC22D1 (D1) or actin specific antibodies. (H) Quantitative PCR analysis of hepatic TSC22D1 in mice fed a choline deficient diet (MCD) or a corresponding control diet for 4 weeks (means±SEM, n=5). (I) Quantitative PCR analysis of TSC22D1 expression levels in LDL-receptor knockout (LDLRKO) mice without or with streptozotocin treatment (STZ). (means±SEM, n=5). (J) Correlation of TSC22D1 RNA expression and lesion size in LDL-receptor knockout (LDLRKO) mice without or with streptozotocin treatment (STZ). The surface lesion area was quantified with ImageJ software and is presented in arbitrary units (n=10). Statistical test for (a)–(h): students t-test, ⁎, p<0.05, ⁎⁎, p<0.01, ⁎⁎⁎, p<0.001, for (i) Pearson correlation coefficient, F-test to determine significance.
Given the promoting effect of TSC22D1 on systemic HDL cholesterol levels (Figure 1b, d and g), these data suggested that the up-regulation of TSC22D1 under obesity conditions reflects a counter-regulatory mechanism to maintain levels of HDL under conditions of energy surplus. Indeed, opposing states of energy availability, i.e. human and murine cancer cachexia, were associated with a marked down-regulation of hepatic TSC22D1 mRNA and protein levels (Figure 3e–g, Sh), correlating with reduced HDL cholesterol levels under these conditions [11]. To extend these findings into an independent model, wild-type C57Bl/6 mice were placed on a methionine-choline-deficient (MCD) diet for 4 weeks which is known to trigger peripheral hyper-metabolism, weight loss [27], and low HDL cholesterol levels [11]. Consistent with the hypothesis of counter-regulatory up-regulation of TSC22D1 in obese animals (Figure 3a), mRNA levels of TSC22D1 were found to be significantly down-regulated in livers of MCD-fed animals as compared to controls (Figure 3h). Furthermore, TSC22D1 levels were also significantly diminished in livers of streptozotocin-induced type 1 diabetic low-density-lipoprotein receptor (LDLR) knockout animals as compared with healthy control littermates (Figure 3i). Consistent with previous reports [20] and a loss of HDL-mediated atheroprotection under these conditions, diabetes exaggerated vascular lesions in these animals as determined by enhanced aortic arch plaque areas after 12 weeks of hyperglycemia as compared with non-diabetic counterparts (Figure S3i). Remarkably, hepatic TSC22D1 mRNA levels tended to negatively correlate with the degree of plaque formation in both diabetic and non-diabetic animals (Figure 3j).
3.4. TSC22D1 is required for the maintenance of HDL cholesterol levels under obese conditions
Our data thus far indicated that the hepatic expression of TSC22D1 adapts to changing states of energy availability and promoted the hypothesis that the up-regulation of TSC22D1 in livers of obese animals is required for the maintenance of basic HDL cholesterol levels under these conditions. To directly test this assumption experimentally, 12 weeks old ob/ob mice were injected with AAV carrying either the TSC22D1-specific or non-specific control miRNA to achieve hepatocyte-specific TSC22D1 deficiency in an obesity setting. 4 weeks after AAV administration, TSC22D1 targeted miRNA delivery led to a more than 90% reduction in hepatic TSC22D1 mRNA levels and substantially blunted TSC22D1 protein expression in these animals (Figure 4a and b). Consistent with an HDL formation-driving function of TSC22D1 even in overweight animals, inhibition of TSC22D1 was sufficient to substantially reduce serum levels of HDL-associated cholesterol mostly under fasting but also during feeding conditions (Figure 4c), and tended to induce hepatic cholesterol retention (Figure S4a), while leaving other metabolic parameters generally unaltered (Figure S4b–g). Consistent with data from lean, healthy animals, TSC22D1 loss-of-function triggered the down-regulation of critical genes in the HDL cholesterol loading pathway in ob/ob mice, (Figure 4d). Together, these data underlined the notion that hepatic TSC22D1 fulfils a critical and specific checkpoint function for the regulation of circulating HDL cholesterol and hepatic cholesterol load, and that high TSC22D1 levels reflect a potentially critical counter-regulatory mechanism in the preservation of residual HDL levels during obesity-related metabolic dysfunction.
Figure 4.
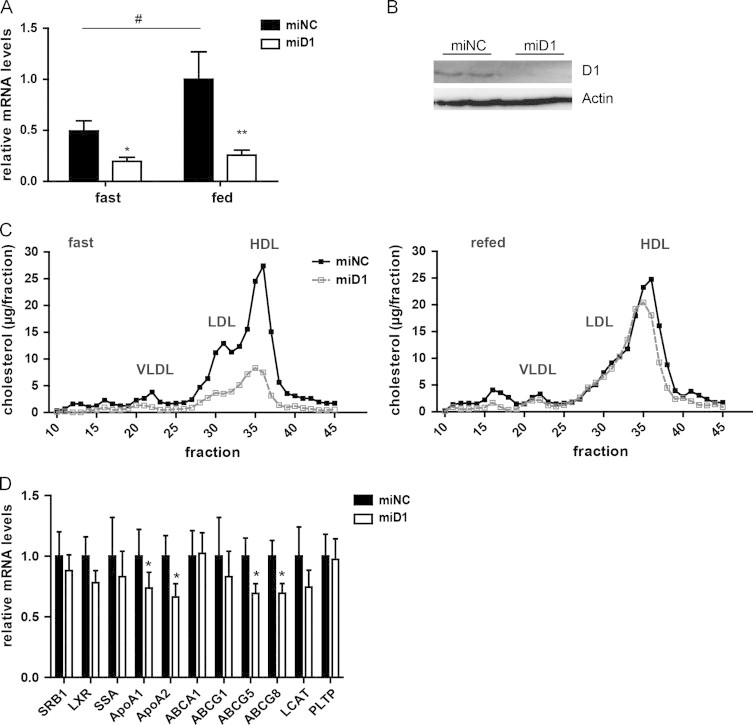
TSC22D1 drives HDL cholesterol levels in obesity. (A) Quantitative PCR analysis of TSC22D1 expression levels of 24 h fasted or 6 h refed control or miRNA TSC22D1 adeno-associated virus injected ob/ob mice (means±SEM, n=7). (B) Representative Western Blot of liver extracts of same mice as in a using TSC22D1 (D1) or actin specific antibodies. (C) Lipoprotein-associated serum cholesterol levels as measured by fast protein liquid chromatography (FPLC) in the same mice as in (a). (D) Quantitative PCR analysis of scavenger receptor B1 (SRB1), liver X receptor (LXR), serum amyloid A (SSA), Apolipoproteins A1 and A2 (ApoA1, ApoA2), ATP-binding cassette transporter (ABCA1), ATP-binding cassette sub-family G member 1, 5 and 8 (ABCG1, ABCG5, ABCG8), lecithin-cholesterol acyltransferase (LCAT), phospholipid transfer protein (PLTP) in 24 h fasted and 6 h refed control or miRNA TSC22D1 adeno associated virus injected ob/ob mice. Statistical tests: for a two-way ANOVA, ⁎, p<0.05, ⁎⁎, p<0.01, shNC vs. shD1, #fast vs. refed; for (d): students t-test, ⁎, p<0.05.
4. Discussion
The TSC22 domain family of transcriptional regulators is conserved from Caenorhabditis elegans to humans and is encoded by four separate genetic loci in mammals, referred to as TSC22D1 to TSC22D4 [28]. Thus far, TSC22 family members had been shown in Drosophila to be part of a growth promoting signaling complex involving the Mlf1 adapter molecule Madm [28], and a TSC22D1/D4 complex has been implicated in BRAF-induced senescence and neoplasia [13]. Furthermore, TSC22D1 was recently found to antagonize Ras/Raf signaling, thereby exerting anti-tumorigenic effects in a Ras/Raf-dependent liver cancer model [29], and TSC22D1 abrogated TGFβ-stimulated growth in intestinal epithelial cells [30]. Given the recently discovered metabolic control function of TSC22D4 for hepatic TG secretion [11], and the non-redundant regulatory impact of TSC22D1 on systemic HDL metabolism as demonstrated by this study, TSC22 proteins may represent a family of transcriptional regulators with dual functionality in both proliferative and metabolic pathways in a broader range of tissues.
Indeed, hepatic TSC22D1 expression seems to be under nutritional/metabolic control as shown by the up- and down-regulation of liver TSC22D1 mRNA and protein levels upon conditions of energy excess and deprivation. Of note, obesity has recently been found to promote elevated levels of TGFβ in both human and murine settings, correlating with systemic dyslipidemia [25], and TSC22D1 had originally been identified as a TGFβ-inducible gene in other cells [26]. In agreement with these findings, TSC22D1 expression was found to be induced under conditions of energy surplus and it is tempting to speculate that these effects are at least partially mediated by the TGFβ pathway under these conditions. In contrast, catabolic situations inhibited TSC22D1 expression in liver, most notably including cancer cachexia and type 1 diabetes. Whether the loss of TGFβ signaling or the induction of catabolic hormonal cues (i.e. glucocorticoids, glucagon, catecholamines) determine TSC22D1 expression under these circumstances remains an open question for future studies. Also, while the regulatory impact of chronic stress conditions is evident from the current results, it remains uncertain whether and how TSC22D1 also exerts more acute functions in short-term metabolic adaptations, i.e. during the fasting-feeding cycle. While a more significant impact of TSC22D1 loss-of-function on cholesterol homeostasis under fasting conditions lines up well with a more pronounced effect of exogenous TSC22D1 overexpression during feeding and would argue for a dominant role in fasting rather than feeding metabolism, the exact role of TSC22D1 in acute metabolic flexibility remains unclear and might rely on hormone-dependent alterations in TSC22D1 expression and/or activity (unpublished results).
With respect to more long-term disease settings, results from this study indicate that chronic loss of hepatic TSC22D1 activity may predispose to atherosclerotic plaque formation and vascular complications as triggered by diabetic hyperglycemia and dyslipidemia. This hypothesis lines up with recent studies in human cohorts, validating the importance of hepatic lipid homeostasis for the prevalence of diabetic late complications, most notably including cardiovascular disease [31,32].
Interestingly, TSC22D1 deficiency predominantly altered the mRNA levels of genes involved in HDL cholesterol loading and/or formation, while leaving lipogenic and bile acid synthesis pathways mostly unaffected (Figure 2 and data not shown). In addition, neither liver nor serum ApoA1 protein levels as well as HDL-associated ApoA1 protein levels were altered upon loss of hepatic TSC22D1 (Figure S3c–e), suggesting that TSC22D1 does not alter circulating HDL cholesterol through an impact on ApoA1 production. Thus, we hypothesize that TSC22D1 does not regulate serum HDL cholesterol through changes in lipoprotein generation but rather impacts HDL cholesterol loading and/or (re-)secretion. Major mechanisms in selective HDL lipid uptake vs. endocytotic HDL holo-particle uptake and re-secretion have not been fully clarified to date, nor the connection and relative importance of these processes are fully understood [33]. Thus, it will be interesting to determine the relative importance of hepatic TSC22D1 for either selective lipid uptake and/or HDL particle endocytosis and re-formation in detail. Preliminary data indicate that TSC22D1 may indeed impact distinct components of these pathways upon liver-specific knockdown (unpublished results).
Of note, normal mice are characterized by constitutively high HDL levels preventing atherosclerotic plaque formation in standard diabetes/obesity models [34]. In humans, however, numerous attempts have been made to increase HDL cholesterol levels to counteract atherosclerotic vascular dysfunction, including the pharmacological inhibition of cholesterol ester transfer protein (CETP), increasing availability of ApoA1, or enhancing ABCA1 activity [35–37]. This becomes particularly important because the majority of cardiovascular events cannot be prevented by focusing on LDL-lowering approaches alone as demonstrated in human studies [38,39]. However, the negative outcome with the CETP inhibitor torcetrapib may also indicate that not every way of raising HDL-cholesterol levels automatically translates into a clinical benefit [40], underlining the necessity to identify as-yet unacquainted approaches in this context.
Specific targeting of the hepatic TSC22D1 complex might thus represent an attractive therapeutic mode to control HDL cholesterol levels under conditions of diabetes/obesity-related energy surplus and loss of atheroprotection.
Author contributions
J.J., V.G., O.S., D.S., K.G., T.S., M.S., A.J., R.G., M.M., G.D.-T., M.B.D., and T.G.H. performed experiments. S.H. designed and directed research. S.H. wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
We thank Kilian Friedrich, Annika Zota, Adam Rose, Yvonne Feuchter, Maria Rohm, Tobias Schafmeier, and Sigrid Stöhr for experimental support and advice on the manuscript. This work was supported by grants from the ZMBH-DKFZ Alliance to T.H. and S.H., and the Deutsche Forschungsgemeinschaft He3260/7-1, and the Helmholtz Portfolio Topic “Metabolic Dysfunction” to SH.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.12.007.
Appendix A. Supporting materials
Supplementary data
References
- 1.Finucane M.M., Stevens G.A., Cowan M.J., Danaei G., Lin J.K., Paciorek C.J., Singh G.M., Gutierrez H.R., Lu Y., Bahalim A.N., Farzadfar F., Riley L.M., Ezzati M. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assmann G., Gotto A.M., Jr. HDL cholesterol and protective factors in atherosclerosis. Circulation. 2004;109:III8–III14. doi: 10.1161/01.CIR.0000131512.50667.46. [DOI] [PubMed] [Google Scholar]
- 3.Luscher T.F., von Eckardstein A., Simic B. Therapeutic targets to raise HDL in patients at risk or with coronary artery disease. Current Vascular Pharmacology. 2012;10:720–724. doi: 10.2174/157016112803520972. [DOI] [PubMed] [Google Scholar]
- 4.Abbasi A., Corpeleijn E., Gansevoort R.T., Gans R.O., Hillege H.L., Stolk R.P., Navis G., Bakker S.J., Dullaart R.P. Role of HDL cholesterol and estimates of HDL particle composition in future development of type 2 diabetes in the general population: the PREVEND study. Journal of Clinical Endocrinology and Metabolism. 2013;98(8):E1352–E1359. doi: 10.1210/jc.2013-1680. [DOI] [PubMed] [Google Scholar]
- 5.Juren A.J., Sarwal G., Al-Sarraf A., Vrablik M., Chan D., Humphries K.H., Frohlich J.J. Low prevalence of type 2 diabetes mellitus among patients with high levels of high-density lipoprotein cholesterol. Journal of Clinical Lipidology. 2013;7:194–198. doi: 10.1016/j.jacl.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Timmins J.M., Lee J.Y., Boudyguina E., Kluckman K.D., Brunham L.R., Mulya A., Gebre A.K., Coutinho J.M., Colvin P.L., Smith T.L., Hayden M.R., Maeda N., Parks J.S. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. Journal of Clinical Investigation. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peet D.J., Turley S.D., Ma W., Janowski B.A., Lobaccaro J.M., Hammer R.E., Mangelsdorf D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 8.Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 9.Diaz M. Berriel, Krones-Herzig A., Metzger D., Ziegler A., Vegiopoulos A., Klingenspor M., Muller-Decker K., Herzig S. Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology. 2008;48:782–791. doi: 10.1002/hep.22383. [DOI] [PubMed] [Google Scholar]
- 10.Kulozik P., Jones A., Mattijssen F., Rose A.J., Reimann A., Strzoda D., Kleinsorg S., Raupp C., Kleinschmidt J., Muller-Decker K., Wahli W., Sticht C., Gretz N., von Loeffelholz C., Stockmann M., Pfeiffer A., Stohr S., Dallinga-Thie G.M., Nawroth P.P., Diaz M. Berriel, Herzig S. Hepatic deficiency in transcriptional cofactor TBL1 promotes liver steatosis and hypertriglyceridemia. Cell Metabolism. 2011;13:389–400. doi: 10.1016/j.cmet.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Jones A., Friedrich K., Rohm M., Schafer M., Algire C., Kulozik P., Seibert O., Muller-Decker K., Sijmonsma T., Strzoda D., Sticht C., Gretz N., Dallinga-Thie G.M., Leuchs B., Kogl M., Stremmel W., Diaz M.B., Herzig S. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Molecular Medicine. 2013;5:294–308. doi: 10.1002/emmm.201201869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kester H.A., Blanchetot C., den Hertog J., van der Saag P.T., van der Burg B. Transforming growth factor-beta-stimulated clone-22 is a member of a family of leucine zipper proteins that can homo- and heterodimerize and has transcriptional repressor activity. Journal of Biological Chemistry. 1999;274:27439–27447. doi: 10.1074/jbc.274.39.27439. [DOI] [PubMed] [Google Scholar]
- 13.Homig-Holzel C., van Doorn R., Vogel C., Germann M., Cecchini M.G., Verdegaal E., Peeper D.S. Antagonistic TSC22D1 variants control BRAF(E600)-induced senescence. EMBO Journal. 2011;30:1753–1765. doi: 10.1038/emboj.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herzig S., Hedrick S., Morantte I., Koo S.H., Galimi F., Montminy M. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature. 2003;426:190–193. doi: 10.1038/nature02110. [DOI] [PubMed] [Google Scholar]
- 15.Rose A.J., Diaz M.B., Reimann A., Klement J., Walcher T., Krones-Herzig A., Strobel O., Werner J., Peters A., Kleyman A., Tuckermann J.P., Vegiopoulos A., Herzig S. Molecular control of systemic bile acid homeostasis by the liver glucocorticoid receptor. Cell Metabolism. 2011;14:123–130. doi: 10.1016/j.cmet.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Grimm D., Kern A., Rittner K., Kleinschmidt J.A. Novel tools for production and purification of recombinant adenoassociated virus vectors. Human Gene Therapy. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 17.Gao G.P., Alvira M.R., Wang L., Calcedo R., Johnston J., Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandard S., Zandbergen F., van Straten E., Wahli W., Kuipers F., Muller M., Kersten S. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. Journal of Biological Chemistry. 2006;281:934–944. doi: 10.1074/jbc.M506519200. [DOI] [PubMed] [Google Scholar]
- 19.Klingenspor M., Klaus S., Wiesinger H., Heldmaier G. Short photoperiod and cold activate brown fat lipoprotein lipase in the Djungarian hamster. American Journal of Physiology. 1989;257:R1123–R1127. doi: 10.1152/ajpregu.1989.257.5.R1123. [DOI] [PubMed] [Google Scholar]
- 20.Vikramadithyan R.K., Hu Y., Noh H.L., Liang C.P., Hallam K., Tall A.R., Ramasamy R., Goldberg I.J. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. Journal of Clinical Investigation. 2005;115:2434–2443. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klingmuller U., Bauer A., Bohl S., Nickel P.J., Breitkopf K., Dooley S., Zellmer S., Kern C., Merfort I., Sparna T., Donauer J., Walz G., Geyer M., Kreutz C., Hermes M., Gotschel F., Hecht A., Walter D., Egger L., Neubert K., Borner C., Brulport M., Schormann W., Sauer C., Baumann F., Preiss R., MacNelly S., Godoy P., Wiercinska E., Ciuclan L., Edelmann J., Zeilinger K., Heinrich M., Zanger U.M., Gebhardt R., Maiwald T., Heinrich R., Timmer J., von Weizsacker F., Hengstler J.G. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. IEE Proceedings – Systems Biology. 2006;153:433–447. doi: 10.1049/ip-syb:20050067. [DOI] [PubMed] [Google Scholar]
- 22.Martignoni M.E., Dimitriu C., Bachmann J., Krakowski-Rosen H., Ketterer K., Kinscherf R., Friess H. Liver macrophages contribute to pancreatic cancer-related cachexia. Oncology Reports. 2009;21:363–369. [PubMed] [Google Scholar]
- 23.Yu L., Hammer R.E., Li-Hawkins J., Von Bergmann K., Lutjohann D., Cohen J.C., Hobbs H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sommerfeld A., Krones-Herzig A., Herzig S. Transcriptional co-factors and hepatic energy metabolism. Molecular and Cellular Endocrinology. 2011;332:21–31. doi: 10.1016/j.mce.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 25.Yadav H., Quijano C., Kamaraju A.K., Gavrilova O., Malek R., Chen W., Zerfas P., Zhigang D., Wright E.C., Stuelten C., Sun P., Lonning S., Skarulis M., Sumner A.E., Finkel T., Rane S.G. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metabolism. 2011;14:67–79. doi: 10.1016/j.cmet.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta R.A., Sarraf P., Brockman J.A., Shappell S.B., Raftery L.A., Willson T.M., DuBois R.N. Peroxisome proliferator-activated receptor gamma and transforming growth factor-beta pathways inhibit intestinal epithelial cell growth by regulating levels of TSC-22. Journal of Biological Chemistry. 2003;278:7431–7438. doi: 10.1074/jbc.M208076200. [DOI] [PubMed] [Google Scholar]
- 27.Rizki G., Arnaboldi L., Gabrielli B., Yan J., Lee G.S., Ng R.K., Turner S.M., Badger T.M., Pitas R.E., Maher J.J. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. Journal of Lipid Research. 2006;47:2280–2290. doi: 10.1194/jlr.M600198-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Gluderer S., Brunner E., Germann M., Jovaisaite V., Li C., Rentsch C.A., Hafen E., Stocker H. Madm (Mlf1 adapter molecule) cooperates with Bunched A to promote growth in Drosophila. Journal of Biology. 2010;9:9. doi: 10.1186/jbiol216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura M., Kitaura J., Enomoto Y., Lu Y., Nishimura K., Isobe M., Ozaki K., Komeno Y., Nakahara F., Oki T., Kume H., Homma Y., Kitamura T. TSC-22 is a negative-feedback regulator of Ras/Raf signaling: implications for tumorigenesis. Cancer Science. 2012;103(1):26–33. doi: 10.1111/j.1349-7006.2011.02108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta R.A., Sarraf P., Mueller E., Brockman J.A., Prusakiewicz J.J., Eng C., Willson T.M., DuBois R.N. Peroxisome proliferator-activated receptor gamma-mediated differentiation: a mutation in colon cancer cells reveals divergent and cell type-specific mechanisms. Journal of Biological Chemistry. 2003;278:22669–22677. doi: 10.1074/jbc.M300637200. [DOI] [PubMed] [Google Scholar]
- 31.Targher G., Bertolini L., Padovani R., Rodella S., Zoppini G., Pichiri I., Sorgato C., Zenari L., Bonora E. Prevalence of non-alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. Journal of Hepatology. 2010;53:713–718. doi: 10.1016/j.jhep.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Targher G., Pichiri I., Zoppini G., Trombetta M., Bonora E. Increased prevalence of cardiovascular disease in Type 1 diabetic patients with non-alcoholic fatty liver disease. Journal of Endocrinological Investigation. 2011;35:535–540. doi: 10.3275/7875. [DOI] [PubMed] [Google Scholar]
- 33.Rohrl C., Stangl H. HDL endocytosis and resecretion. Biochimica et Biophysica Acta. 2013;1831:1626–1633. doi: 10.1016/j.bbalip.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldberg I.J. Why does diabetes increase atherosclerosis? I don't know! Journal of Clinical Investigation. 2004;114:613–615. doi: 10.1172/JCI22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tall A.R. CETP inhibitors to increase HDL cholesterol levels. New England Journal of Medicine. 2007;356:1364–1366. doi: 10.1056/NEJMe078029. [DOI] [PubMed] [Google Scholar]
- 36.Nissen S.E., Tsunoda T., Tuzcu E.M., Schoenhagen P., Cooper C.J., Yasin M., Eaton G.M., Lauer M.A., Sheldon W.S., Grines C.L., Halpern S., Crowe T., Blankenship J.C., Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. Journal of the American Medical Association. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 37.Bailey D., Jahagirdar R., Gordon A., Hafiane A., Campbell S., Chatur S., Wagner G.S., Hansen H.C., Chiacchia F.S., Johansson J., Krimbou L., Wong N.C., Genest J. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. Journal of the American College of Cardiology. 2010;55(23):2580–2589. doi: 10.1016/j.jacc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 38.Castelli W.P., Garrison R.J., Wilson P.W., Abbott R.D., Kalousdian S., Kannel W.B. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. Journal of the American Medical Association. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 39.Gordon T., Castelli W.P., Hjortland M.C., Kannel W.B., Dawber T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. American Journal of Medicine. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 40.McKenney J.M., Davidson M.H., Shear C.L., Revkin J.H. Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels on a background of atorvastatin. Journal of the American College of Cardiology. 2006;48:1782–1790. doi: 10.1016/j.jacc.2006.06.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data