

Abstract
Melanocortins and their receptors are critical components of energy homeostasis and the paraventricular nucleus of the hypothalamus (PVH) is an important site of melanocortin action. Although best known for its role in osmoregulation, arginine vasopressin (AVP) has been implicated in feeding and is robustly expressed in the PVH. Since the anorectic melanocortin agonist MTII activates PVH-AVP neurons, we hypothesized that PVH-AVP neurons contribute to PVH-mediated anorexia. To test this, we used an AVP-specific Cre-driver mouse in combination with viral vectors to acutely manipulate PVH-AVP neuron function. Using designer receptors exclusively activated by designer drugs (DREADDs) to control PVH-AVP neuron activity, we show that activation of PVH-AVP neurons acutely inhibits food intake, whereas their inhibition partially reverses melanocortin-induced anorexia. We further show that MTII fails to fully suppress feeding in mice with virally-induced PVH-AVP neuron ablation. Thus PVH-AVP neurons contribute to feeding behaviors, including the acute anorectic response to MTII.
Keywords: Arginine vasopressin, Paraventricular nucleus, Melanocortins, Anorexia, Food intake
1. Introduction
The importance of the paraventricular nucleus of the hypothalamus (PVH) in regulating energy balance is well established [1,2]. Lesions of the PVH produce hyperphagia and obesity in rodents [3]. Inactivation of one copy of Sim1, a key hypothalamic transcription factor required for PVH development, disrupts PVH function and produces hyperphagic obesity with associated glucose dysregulation in both rodents and humans [4–6]. A detailed understanding of the molecular mechanisms and neural pathways used by the PVH to modulate energy homeostasis has however been complicated by the cellular complexity of this nucleus. Although some PVH cell types have been neurochemically-defined, the specific roles of these PVH subsets in energy balance are incompletely understood [7].
The central melanocortin system is critical for energy homeostasis. Melanocortin agonists such as melanotan-II (MTII) promote energy expenditure and suppress food intake [8,9] largely through the action of melanocortin-4 receptors (MC4R) [10,11]. MC4Rs are densely expressed in the PVH and MC4R action in the PVH is particularly important for restraining feeding [12]. Although oxytocin neurons have been proposed as important regulators of feeding, mice lacking oxytocin neurons display normal feeding and a normal anorectic response to MTII [13]. Together these findings suggest that neurons other than oxytocin neurons within the PVH are important for feeding regulation.
Arginine vasopressin (AVP) is robustly expressed in parvocellular cells of the PVH, but its physiologic function within the brain, and specifically in energy balance, has not been fully defined [14]. AVP has been implicated in feeding, blood glucose regulation and locomotor activity [15–18], but the role of PVH-AVP in melanocortin signaling and energy regulation remains unclear. In this study, we demonstrate that the anorectic melanocortin agonist MTII activates PVH-AVP neurons and that pharmacogenetic activation of PVH-AVP neurons blunts food intake acutely. Moreover, acute inhibition or deletion of PVH-AVP neurons partially reverses melanocortin-induced anorexia. Thus PVH-AVP neurons appear to be involved in the regulation of food intake and may function as an important regulatory “node” in melanocortin-controlled feeding pathways.
2. Materials and methods
2.1. Materials
Melanotan-II (MTII) was purchased from Bachem (Torrance, CA). Donkey and goat serum, biotinylated goat anti-rabbit IgG and donkey anti-rabbit IgG were from Jackson ImmunoResearch (West Grove, PA). ABC Vectastain Elite kit was purchased from Vector Laboratories (Burlingame, CA). Metal Enhanced DAB Substrate Kit was from Thermo scientific (Rockford, IL).
2.2. Animals
AVP-iCre mice were generated using recombineering techniques as previously described [13,19,20]. All mice were housed in a 12 h light/12 h dark cycle and cared by Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan. Animals had ad libitum access to food and water, except during experiments in which mice were fasted before perfusion or during timed feeding studies. All animal care and procedures are approved by and in accordance with the guidelines of the University Committee on the Care and Use of Animals at the University of Michigan.
2.3. Immunohistochemistry and immunofluorescence
Mice were anesthetized with an overdose of intraperitoneal (IP) pentobarbital and transcardially perfused with sterile PBS followed by 10% formalin. Brains were removed, postfixed, and cryoprotected before sectioning into 30 μm coronal slices (four representative series) and stored at −20 °C. For immunocytochemistry, sections were pretreated in 0.3% H2O2, blocked in 3% goat or donkey serum and incubated in primary antibodies. For DAB staining, detection was performed following incubation with biotinylated secondary antibodies using the ABC Vectastain kit and a Metal Enhanced DAB Substrate Kit. For immunofluorescent detection, species-specific Alexa 488 or 568 antibodies (Invitrogen, 1:200) were used to identify primary antibody targets. The following antibodies were used: c-Fos (Calbiochem, rabbit, 1:20,000), GFP (Abcam, chicken, 1:1000), AVP (Peninsula Laboratories, guinea pig, 1:4000 or Millipore, Rabbit, 1:10,000), oxytocin (Peninsula Laboratories, rabbit, 1:4000), Dsred (Clontech, rabbit, 1:1000).
2.4. Stereotaxic virus injections
For intracranial virus injections, adult AVP-iCre (>8-week-old) mice were anesthetized with vaporized isofluorane, and fixed in a stereotaxic apparatus. PVH coordinates relative to Bregma were A/P, −0.54; M/L, −0.2; and D/V, −4.85. An access hole was drilled in this spot and virus was injected into the brain using a 500 nl Hamilton syringe at a rate of 100 nl per min. Following virus infusion the syringe was removed after 2 min, the craniotomy was sealed with bone wax and the surgical incision was sutured. Mice were then allowed to recover for 5–14 days, after which they were used in physiologic experiments or perfused and processed for immunohistochemistry.
2.5. Neuronal activation by melanocortin agonist (MTII)
To identify melanocortin-responsive cells in the PVH, mice were habituated to daily intraperitoneal (IP) injection for 5 days with sterile PBS. Following habituation, 6–8-week-old mice were fasted for 24 h, injected with 10 mg/kg MTII, and then perfused 2 h later. Nuclear c-Fos immunoreactivity was then used as a marker for neuronal activation in PVH subsets.
2.6. Temporal control of PVH-AVP neuron activity alters feeding
AVP-iCre mice were injected bilaterally in the PVH with either stimulatory (DIO-hM3Dq-mCherry-AAV10; 3Dq) or inhibitory (DIO-hM4Di-mCherry-AAV10; 4Di) DREADD AAV-vectors. After recovery (10–14 days), mice were habituated to IP injections for 5 days. To determine the feeding response following PVH-AVP neuron activation, food was removed 9 h before lights out; then, at the onset of the dark cycle (1800 h), mice were injected IP with vehicle or CNO (0.3 mg/kg) and at the same time, food was provided. Cumulative food intake was then measured over the following 4 h. For PVH-AVP inhibition experiments, food was removed from the cage in the morning and then, 1 h before the onset of dark (1700 h), mice were injected IP with vehicle or CNO (0.3 mg/kg). One hour later, just prior to the onset of the dark cycle (1800 h), mice were injected IP with PBS or 5 mg/kg MTII and food was given back. Cumulative food intake was measured over the next 4 h. In both experiments, each mouse served as its own control.
2.7. MTII-induced anorexia in mice lacking PVH-AVP neurons
To specifically ablate AVP neurons from the PVH, we placed small bilateral injections of a Cre-dependent cellular toxin, lox-mCherry-lox-DTA-AAV (DTA-AAV) into the PVH of AVP-iCre mice. Matching volume injections of GFP-AAV were placed into additional mice to serve as vector injection controls. After 14–21 days, to allow recovery and DTA-mediated cell death, the mice were habituated to IP injections and fed ad libitum. On the day of the experiment, food was removed from the cage in the morning. At the onset of the dark cycle (1800 h), mice were injected IP with PBS or 5 mg/kg MTII in PBS and food was given back. Cumulative food intake was measured for 4 h following the injection.
3. Results
3.1. AVP-iCre knock-in mice express Cre recombinase in AVP neurons
To selectively manipulate AVP neurons, we generated knock-in mice that express Cre recombinase in AVP neurons. Using homologous recombination techniques, a DNA cassette containing an internal ribosomal sequence, Cre coding sequence and polyadenylation sequence (-iCre) was inserted downstream of the stop codon of the endogenous AVP gene. This results in Cre recombinase being expressed by the targeted, endogenous neuropeptide promoter and prevents the problem of ectopic Cre expression often seen with transgenic lines. To confirm the specific expression of Cre, we crossed AVP-iCre mice with a Cre-dependent loxGFP reporter line. Immunostaining of brain sections from the double transgenic mice (AVP-iCre, loxGFP) showed a nearly complete colocalization of AVP (red) with GFP (green) in the PVH and essentially no Cre activity in neighboring oxytocin neurons. Therefore, this strain can be used for genetic manipulation of AVP neurons (Supplemental Figure 1).
3.2. PVH-AVP neurons are activated by the melanocortin agonist (MTII)
Injection of melanocortin agonist (MTII) suppresses food intake and melanocortin receptors in the paraventricular hypothalamus are important for this effect [21]. Since MTII induces c-Fos expression in PVH-Sim1 neurons [22], we determined if PVH-AVP neurons are responsive to melanocortin action. Because MTII treatment acutely reduces AVP immunoreactivity in the PVH (data not shown), we used AVP-iCre, loxGFP reporter mice to assess MTII-induced activation of PVH-AVP neurons. As shown in Figure 1A, MTII administration induced nuclear c-Fos immunoreactivity in the majority of GFP-expressing PVH-AVP neurons (~65%), but only a small percentage (~15%) of PVH-oxytocin neurons. Quantification of MTII-activated PVH-AVP and PVH-OXT neurons is represented in Figure 1B.
Figure 1.
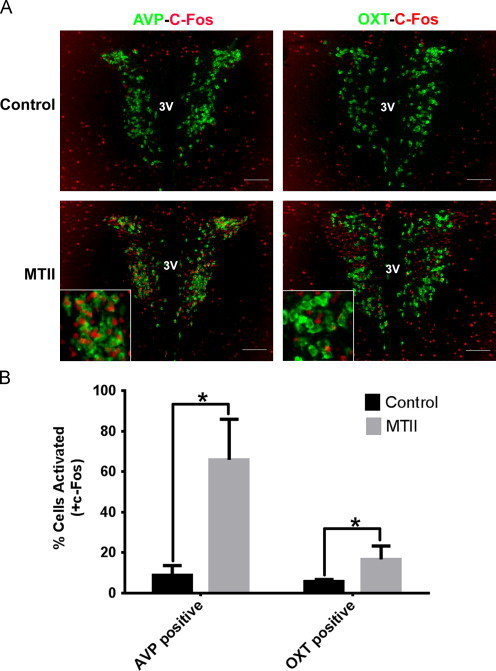
MTII induces c-Fos expression in PVH-AVP neurons. AVP-iCre, loxGFP mice were treated with PBS or MTII. (A) Representative images of PVH showing the colocalization between c-Fos (red) and AVP (green), left; colocalization of c-Fos (red) and oxytocin (green), right, following MTII treatment. (B) Quantification of colocalization between c-Fos and AVP or oxytocin in the PVH after MTII treatment. (n=4, SEM; ⁎P<0.05) Scale bar=100 µm. 3V: third ventricle.
3.3. Activation of PVH-AVP neurons can decrease food intake
Since melanocortin agonists both suppress food intake and activate PVH-AVP neurons, we hypothesized that direct PVH-AVP activation would be sufficient to blunt food intake. To test our hypothesis, we took advantage of new technology that allows for acute manipulation of neural activity. Designer receptors exclusively activated by designer drugs (DREADDs) are modified human muscarinic receptors that are coupled to stimulatory G-proteins (Gq) or inhibitory G-proteins (Gi). Upon binding the otherwise pharmacologically inert ligand clozapine-N-oxide (CNO), the stimulatory (3Dq) or inhibitory (4Di) DREADD couples through the Gq or Gi pathway to depolarize or hyperpolarize neurons, respectively. In order to directly test the ability of PVH-AVP neurons to modulate feeding, AVP-iCre, loxGFP transgenic mice were stereotaxically injected in the PVH with the Cre-dependent, stimulatory AAV-3Dq-mCherry vector. Transduction of AVP-iCre neurons with the stimulatory 3Dq-mCherry was confirmed by co-localization of the mCherry fusion protein with the GFP reporter protein showing that DREADD expression was limited to the PVH-AVP neurons (Figure 2A). Administration of CNO greatly increased c-Fos immunoreactivity in the PVH-AVP neurons demonstrating that this approach acutely and specifically stimulated neuronal activity (Figure 2B). Importantly, DREADDs expression and neuronal activation did not extend to AVP neurons within the suprachiasmatic nucleus ventral to the PVH (Supplemental Figure 2). To assess the physiologic effect of PVH-AVP neural activation, AVP-iCre mice with PVH stimulatory 3Dq injections were treated with either vehicle or CNO at the onset of dark and food intake was measured during refeeding. As shown in Figure 2C, the activation of PVH-AVP neurons by CNO blunted food intake through 4 h of refeeding (2 h feeding suppressed by 40%; 4 h feeding suppressed by 30%) relative to the vehicle control, suggesting that PVH-AVP neuron activation directly suppressed food intake.
Figure 2.
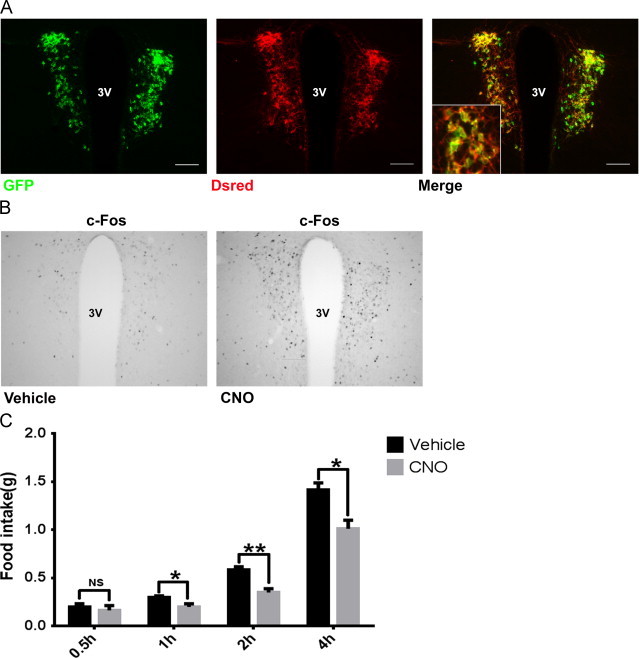
Activation of PVH-AVP neurons suppresses food intake. AVP-iCre, loxGFP mice were injected bilaterally into PVH with stimulatory 3Dq-DREADD vector. (A) Costaining of DREADD (dsred) and AVP (GFP) showed that DREADD expression was limited to PVH-AVP neurons. (B) CNO treatment of AVP-iCre mice expressing 3Dq-DREADDs drives increased c-Fos expression in the PVH. (C) Dark cycle food intake in AVP-iCre mice with PVH-3Dq-DREADDs treated with vehicle or CNO; food intake was measured over the next 4 h. Each animal serves as its own control. (SEM, n=6; ⁎P<0.05, ⁎⁎P<0.01, NS: not significant). Insets: magnified view of merged images between GFP (AVP) and DREADD. Scale bar=100 µm. 3V: third ventricle.
3.4. Inhibition of PVH-AVP neurons blunts MTII-mediated anorexia
To confirm the contribution of PVH-AVP neuron activity to melanocortin-induced anorexia, AVP-iCre, loxGFP transgenic mice were stereotaxically injected bilaterally in the PVH with the inhibitory AAV-4Di-mCherry vector. Immunostaining showed that the inhibitory DREADD expression was limited to PVH-AVP neurons (Figure 3A). Furthermore, CNO-pretreatment of AVP-iCre mice with PVH-directed inhibitory 4Di-DREADD blunted c-Fos activation following MTII administration (Figure 3B). If PVH-AVP activation is required for a full anorectic response to MTII, then blocking this activation should ameliorate the MTII-associated anorexia. To test this hypothesis, the PVH-AVP-4Di-DREADD mice were pretreated with vehicle or CNO and then given MTII at the onset of dark cycle feeding. We found that MTII decreased food intake as expected (~65%) following vehicle pretreatment. However, when these same mice were pretreated with CNO to “pre-inhibit” PVH-AVP neurons, MTII only suppressed food intake by ~25% compared to CNO-treated controls (Figure 3C). Thus PVH-AVP neuron activation is an important component of the acute anorectic response to MTII.
Figure 3.
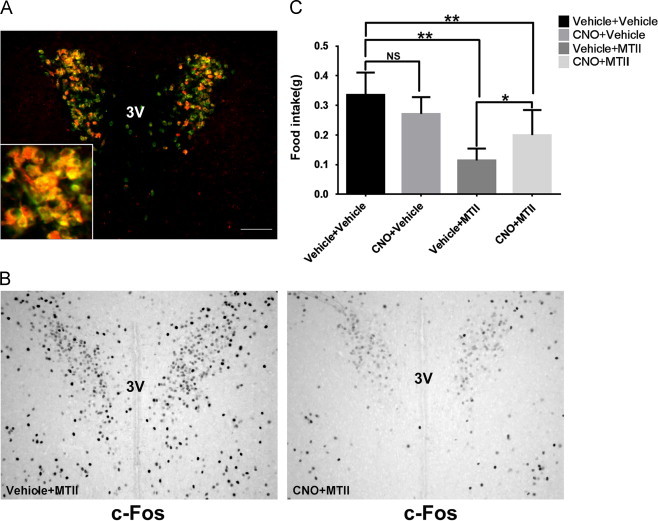
Inhibition of PVH-AVP neurons reverses partly MTII-induced anorexia. AVP-iCre, loxGFP mice were injected bilaterally into PVH with inhibitory 4Di-DREADD. (A) Merged image between GFP and DREADD showed that 4Di-DREADD expression was limited to PVH-AVP neurons. (B) CNO treatment of AVP-iCre mice with PVH-4Di-DREADDs inhibits c-Fos expression in the PVH. (C) AVP-iCre mice expressing 4Di-DREADD were injected with PBS or MTII after an hour vehicle or CNO pretreatment at the onset of dark cycle. Cumulative food intake was measured 2 h after injection. (SEM, n=7; ⁎P<0.05, ⁎⁎P<0.01, NS: not significant) Scale bar=100 µm. 3V: third ventricle.
3.5. Deletion of PVH-AVP neurons blunts MTII-anorexia
To extend and confirm our findings from the DREADD-injected animals, we selectively ablated PVH-AVP neurons using a Cre-dependent “toxic” virus, lox-mCherry-lox-DTA-AAV (DTA-AAV) (Figure 4A). This virus expresses the cellular toxin diphtheria toxin subunit A (DTA) only in the presence of Cre recombinase. Stereotaxic delivery of the DTA-AAV virus limits neuronal death to Cre-expressing cells in the field of injection without affecting neurons in adjacent brain regions. To demonstrate the efficacy of DTA-AAV in inducing cell death within the PVH, we injected it into AVP-iCre, loxGFP mice. PVH-directed DTA-AAV injection results in selective deletion of AVP neurons only in the PVH while leaving AVP neurons of supraoptic nucleus intact (Figure 4B and C). Importantly, the injection of DTA virus had little effect on neighboring PVH-oxytocin neurons (Figure 4D).
Figure 4.
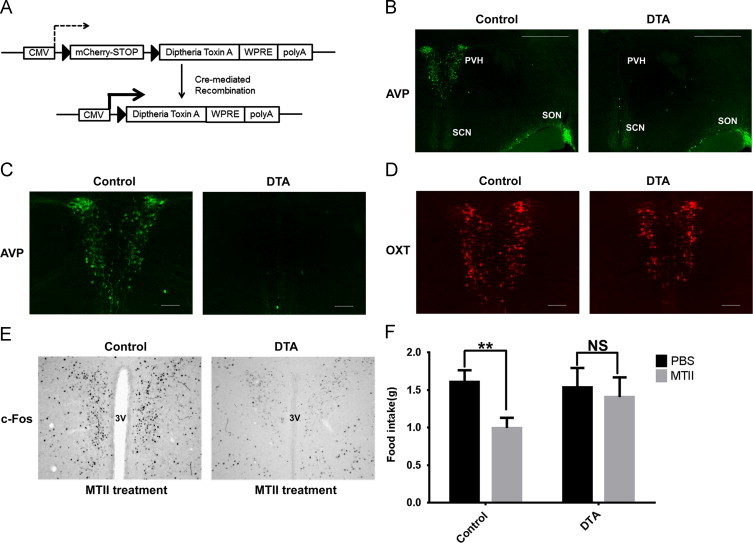
Deletion of PVH-AVP neurons blunts MTII-anorexia. (A) Schematic diagram of “toxic” lox-mCherry-lox-DTA-AAV (DTA-AAV) vector. (B) AVP neuron ablation is limited to the site (PVH) of the DTA-AAV injection. (C) Higher magnification shows complete ablation of PVH-AVP neurons by DTA injection. (D) PVH-oxytocin expression (red) is minimally affected by DTA-mediated ablation of PVH-AVP neurons. (E) c-Fos staining following treatment with MTII in control (GFP) and DTA-treated mice. (F) Dark cycle feeding responses to MTII in control and PVH-AVP ablated mice. Cumulative food intake was measured 4 h after treatment. (SEM, n=8; ⁎P<0.05, NS: not significant) Scale bar=100 µm; PVH: paraventricular nucleus of hypothalamus; SCN: suprachiasmatic nucleus; SON: supraoptic nucleus.
Since MTII induces the activation of PVH-AVP neurons, we asked whether mice lacking PVH-AVP neurons can respond normally to MTII treatment. In control mice, MTII treatment induced c-Fos immunoreactivity of PVH neurons. However, in mice lacking PVH-AVP neurons, there was little c-Fos immunoreactivity in the PVH, demonstrating that PVH-AVP neurons are required for MTII-induced c-Fos activation of the PVH (Figure 4E). To assess the physiologic effect of PVH-AVP neuron ablation, a cohort of AVP-iCre mice were stereotaxically injected with AAV-GFP control virus or DTA-AAV virus. Following recovery and allowing time for viral expression to induce cell death, the mice were treated with PBS or MTII just prior to the onset of the dark cycle, and food intake was measured over the following 4 h. As expected, MTII treatment decreased food intake in controls (62% of vehicle control), however, MTII-induced anorexia was greatly attenuated in mice lacking PVH-AVP neurons (Figure 4F). This effect was not secondary to alterations in water handling as mice receiving PVH-directed AAV-DTA virus consumed the same amount of water as controls animals (AAV-DTA mice: 7.5±0.6 mL/day (n=4) vs. controls: 7.0±0.5 mL/day (n=4); P=0.5).
4. Discussion
The melanocortin system is well known for its role in the regulation of energy balance and the PVH is a particularly important brain region for mediating these effects. Because mice lacking oxytocin neurons respond normally to the melanocortin agonist MTII [13], other non-oxytocin PVH neurons must be involved in food intake regulation. As AVP is prominently expressed in PVH and has been implicated in energy balance [14,23,24], we hypothesized that PVH-AVP neurons might participate in the regulation of feeding behaviors. We first demonstrate that the pharmacologic melanocortin agonist MTII induces c-Fos immunoreactivity in the majority of PVH-AVP neurons, whereas not in the neighboring oxytocin neurons. This suggests that PVH-AVP neurons are part of the neural substrate through which melanocortins exert some of their metabolic and homeostatic effects.
Since melanocortin agonists decrease food intake and also activate PVH-AVP neurons, we hypothesized that activation of PVH-AVP neurons would mimic melanocortin action and blunt food intake. To test this hypothesis, we used a novel Cre-driver in combination with viral vectors to spatially and temporally manipulate AVP neuron function and then assessed the effects of this manipulation on dark cycle feeding. We found that direct activation of PVH-AVP neurons acutely inhibits food intake thereby implicating PVH-AVP neurons in feeding behavior. AVP neurons project widely in the brain and spinal cord; however, it is not known if PVH-AVP neurons project to hindbrain areas involved in feeding. Other brain regions may also be the direct targets of PVH-AVP neurons and mediate their effects; one potential target may be the lateral hypothalamic area, an important regulatory region through which PVH-AVP neurons project. Given the presence of local glutamatergic interneurons within the PVH [25], it is also possible that PVH-AVP neurons are coupled to other PVH subsets and that resulting physiologic outputs from the PVH reflect an integrated summation of discrete cell type activities.
Pharmacogenetic activation of PVH-AVP neurons demonstrates the previously unrecognized ability of these neurons to alter food intake. To determine if PVH-AVP neuron activity is required for the acute anorectic response to the melanocortin agonist MTII, we used two complementary approaches to inhibit PVH-AVP neuron activity. We found that transient inhibition of PVH-AVP neurons using inhibitory DREADDs partially reverses the anorectic effects of MTII. Moreover, selective ablation of PVH-AVP neurons blunts MTII-mediated c-Fos activation within the PVH. As might be expected from this inhibition of PVH activation, mice lacking PVH-AVP neurons were relatively unresponsive to the acute anorectic effects of MTII. Thus PVH-AVP neurons are critical components for transducing the anorectic effects of melanocortin agonists.
In summary, we demonstrate that PVH-AVP neurons are activated by melanocortin agonists and that direct activation of PVH-AVP neurons suppresses food intake. Moreover, PVH-AVP neurons or neuron activity are required for the acute anorectic effects of the potent melanocortin agonist MTII thereby placing the PVH-AVP neurons within the neural circuitry affecting feeding behaviors. Future work will investigate the mechanisms and neural pathways, including intra-PVN network connectivity, through which PVH-AVP neurons regulate feeding and contribute to overall energy homeostasis.
Conflict of interest
None declared.
Acknowledgments
The authors wish to thank Dr. Martin Myers and members of the Myers' laboratory for advice and technical assistance. This work was funded by the University of Michigan Department of Pediatrics and a Pilot and Feasibility grant to DPO from the Michigan Nutrition Obesity Research Center (P30 DK089503).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.12.006.
Appendix A. Supplementary materials
Supplementary data
Supplementary data
References
- 1.Duplan S.M. Impact of Sim1 gene dosage on the development of the paraventricular and supraoptic nuclei of the hypothalamus. European Journal of Neuroscience. 2009;30(12):2239–2249. doi: 10.1111/j.1460-9568.2009.07028.x. [DOI] [PubMed] [Google Scholar]
- 2.Swanson L.W., Sawchenko P.E. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31(6):410–417. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 3.Leibowitz S.F., Hammer N.J., Chang K. Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat. Physiology & Behavior. 1981;27(6):1031–1040. doi: 10.1016/0031-9384(81)90366-8. [DOI] [PubMed] [Google Scholar]
- 4.Holder J.L., Jr., Butte N.F., Zinn A.R. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Human Molecular Genetics. 2000;9(1):101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 5.Michaud J.L. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Human Molecular Genetics. 2001;10(14):1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 6.Tolson K.P. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. Journal of Neuroscience. 2010;30(10):3803–3812. doi: 10.1523/JNEUROSCI.5444-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biag J. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. Journal of Comparative Neurology. 2012;520(1):6–33. doi: 10.1002/cne.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan W. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385(6612):165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 9.Cone R.D. Studies on the physiological functions of the melanocortin system. Endocrine Reviews. 2006;27(7):736–749. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- 10.Chen A.S. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Research. 2000;9(2):145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- 11.Pandit R. Neurobiology of overeating and obesity: the role of melanocortins and beyond. European Journal of Pharmacology. 2011;660(1):28–42. doi: 10.1016/j.ejphar.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 12.Balthasar N. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z. An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS One. 2012;7(9):e45167. doi: 10.1371/journal.pone.0045167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benarroch E.E. Oxytocin and vasopressin: social neuropeptides with complex neuromodulatory functions. Neurology. 2013;80(16):1521–1528. doi: 10.1212/WNL.0b013e31828cfb15. [DOI] [PubMed] [Google Scholar]
- 15.Meyer A.H., Langhans W., Scharrer E. Vasopressin reduces food intake in Goats. Quarterly Journal of Experimental Physiology. 1989;74(4):465–473. doi: 10.1113/expphysiol.1989.sp003294. [DOI] [PubMed] [Google Scholar]
- 16.Spruce B.A. The effect of vasopressin infusion on glucose metabolism in man. Clinical Endocrinology (Oxford) 1985;22(4):463–468. doi: 10.1111/j.1365-2265.1985.tb00145.x. [DOI] [PubMed] [Google Scholar]
- 17.Wideman C.H., Murphy H.M. Modulatory effects of vasopressin on glucose and protein metabolism during food-restriction stress. Peptides. 1993;14(2):259–261. doi: 10.1016/0196-9781(93)90039-j. [DOI] [PubMed] [Google Scholar]
- 18.Tsunematsu T. Vasopressin increases locomotion through a V1a receptor in orexin/hypocretin neurons: implications for water homeostasis. Journal of Neuroscience. 2008;28(1):228–238. doi: 10.1523/JNEUROSCI.3490-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tong Q. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nature Neuroscience. 2008;11(9):998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vong L. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71(1):142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierroz D.D. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51(5):1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- 22.Kublaoui B.M. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Molecular Endocrinology. 2006;20(10):2483–2492. doi: 10.1210/me.2005-0483. [DOI] [PubMed] [Google Scholar]
- 23.Rood B.D., Vries G.J. De. Vasopressin innervation of the mouse (Mus musculus) brain and spinal cord. Journal of Comparative Neurology. 2011;519(12):2434–2474. doi: 10.1002/cne.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hallbeck M., Blomqvist A. Spinal cord-projecting vasopressinergic neurons in the rat paraventricular hypothalamus. Journal of Comparative Neurology. 1999;411(2):201–211. [PubMed] [Google Scholar]
- 25.Boudaba C., Schrader L.A., Tasker J.G. Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. Journal of Neurophysiology. 1997;77(6):3396–3400. doi: 10.1152/jn.1997.77.6.3396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data