

Abstract
Dysregulation of oxidative phosphorylation is associated with increased mitochondrial reactive oxygen species production and some of the most prevalent human diseases including obesity, cancer, diabetes, neurodegeneration, and heart disease. Chemical 'mitochondrial uncouplers' are lipophilic weak acids that transport protons into the mitochondrial matrix via a pathway that is independent of ATP synthase, thereby uncoupling nutrient oxidation from ATP production. Mitochondrial uncouplers also lessen the proton motive force across the mitochondrial inner membrane and thereby increase the rate of mitochondrial respiration while decreasing production of reactive oxygen species. Thus, mitochondrial uncouplers are valuable chemical tools that enable the measurement of maximal mitochondrial respiration and they have been used therapeutically to decrease mitochondrial reactive oxygen species production. However, the most widely used protonophore uncouplers such as carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) and 2,4-dinitrophenol have off-target activity at other membranes that lead to a range of undesired effects including plasma membrane depolarization, mitochondrial inhibition, and cytotoxicity. These unwanted properties interfere with the measurement of mitochondrial function and result in a narrow therapeutic index that limits their usefulness in the clinic. To identify new mitochondrial uncouplers that lack off-target activity at the plasma membrane we screened a small molecule chemical library. Herein we report the identification and validation of a novel mitochondrial protonophore uncoupler (2-fluorophenyl){6-[(2-fluorophenyl)amino](1,2,5-oxadiazolo[3,4-e]pyrazin-5-yl)}amine, named BAM15, that does not depolarize the plasma membrane. Compared to FCCP, an uncoupler of equal potency, BAM15 treatment of cultured cells stimulates a higher maximum rate of mitochondrial respiration and is less cytotoxic. Furthermore, BAM15 is bioactive in vivo and dose-dependently protects mice from acute renal ischemic-reperfusion injury. From a technical standpoint, BAM15 represents an effective new tool that allows the study of mitochondrial function in the absence of off-target effects that can confound data interpretation. From a therapeutic perspective, BAM15-mediated protection from ischemia-reperfusion injury and its reduced toxicity will hopefully reignite interest in pharmacological uncoupling for the treatment of the myriad of diseases that are associated with altered mitochondrial function.
Abbreviations: ANT, adenine nucleotide translocase; ECAR, extracellular acidification rate; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; OCR, oxygen consumption rate; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride; TMRM, tetramethylrhodamine
Keywords: Mitochondria, Bioenergetics, FCCP, CCCP, DNP, Ischemia
1. Introduction
Oxidative phosphorylation involves the coupling of nutrient oxidation to ATP production through a proton cycle across the mitochondrial inner membrane. Any pathway that enables proton re-entry into the matrix independent of ATP synthase thereby ‘uncouples’ nutrient oxidation from ATP production. Physiological uncoupling is mediated by protein complexes embedded in the mitochondrial inner membrane, most notably the uncoupling proteins (UCPs) [1]. Pharmacological uncouplers are small molecules that mimic the actions of UCPs, enabling protons to enter the mitochondrial matrix along their concentration and electrochemical gradient. Pharmacological uncouplers generally belong to one of two general classes – protonophore uncouplers and the non-protonophores that activate proton leak through protein complexes such as the adenine nucleotide translocase (ANT).
Mitochondrial uncouplers also have beneficial antioxidant effects that are a consequence of reduced proton motive force and shortened occupancy time of single electrons at electron carriers within the electron transport chain [2]. In contrast, a high proton motive force leads to a more reduced state of the electron transport chain resulting in an increased rate of mitochondrial superoxide production [2–4]. Genetic and pharmacologic uncoupling have favorable effects on disorders that are linked to mitochondrial oxidative stress including ischemic-reperfusion injury [5–8], Parkinson's disease [9], insulin resistance [10,11], aging [12], and heart failure [13], and also on disorders that may benefit from increased energy expenditure such as obesity [14].
The most widely used protonophore uncoupler for mitochondrial bioenergetics studies, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), was discovered more than 50 years ago. FCCP is often used for analysis of mitochondrial function in living cells, tissues, and isolated mitochondrial preparations due to its relatively high potency compared to other uncouplers. However, the protonophore activity of mitochondrial uncouplers such as FCCP is not restricted to mitochondria, and their use for mitochondrial bioenergetics studies is limited by plasma membrane depolarization [15–18]. Based on the known non-mitochondrial effects of currently available pharmacological uncouplers, we developed a cell-based small molecule phenotypic screen to identify new mitochondrial uncouplers that have a broad effective range and do not affect plasma membrane electrophysiology. Herein we report the identification and validation of compound BAM15, (2-fluorophenyl){6-[(2-fluorophenyl)amino](1,2,5-oxadiazolo[3,4-e]pyrazin-5-yl)}amine, as a new chemotype mitochondrial uncoupler that satisfies these criteria.
2. Materials and methods
2.1. Materials
BAM15 solid compound and the ApexScreen 5040 library was purchased from TimTec (Newark, DE). 96-well Becton Dickinson (BD) Oxygen Biosensor (OBS) microplates were obtained from BD Biosciences (Bedford, MA). FCCP, antimycin A, and succinate were purchased from Sigma-Aldrich. Rotenone was purchased from MP Biomedicals (Santa Ana, CA). Pyruvic acid was purchased from Acros organics (Geel, Belgium).
2.2. Non-quantitative oxygen consumption assay
L6 myoblasts were grown to confluence, washed with PBS, trypsinized and then seeded into a 96-well plate in cell culture media wherein an O2-sensitive fluorophore tris 1,7-diphenyl-1,10 phenanthroline ruthenium (II) chloride is embedded in silicone at the base of each well of a 96-well plate (BD-OBS microplate, BD Biosensor). The reporter dye is quenched by O2, thus fluorescence intensity increases as O2 is consumed from the media. Cells were incubated with 0.5% (v/v) library compound or vehicle control (DMSO) and fluorescence intensity recorded over 45–90 min (1 read/min) at 37 °C by a SpectraMax M5 dual-monochromator microplate reader (Molecular Devices, CA) using a bottom-read configuration and with the excitation and emission filters set at 485 nm and 630 nm, respectively. The mitochondrial uncoupling agent FCCP was used as positive control increase oxygen consumption. Fluorescence data were recorded on SoftMax Pro (version 4.8) software. Fluorescence tracers were evaluated manually and the top 25 ‘hits’ that increased oxygen consumption over DMSO control were selected for secondary screening. More precise analysis of oxygen consumption rates were determined by Seahorse XF24 analysis as described in Section 2.4.
2.3. ROS production assay
L6 myoblasts were seeded into black-walled clear-bottom 96-well microplates in L6 growth media and grown to confluence. Cells were then washed twice with PBS and co-incubated with 7.5 μM CM-H2DCFDA (Molecular Probes, Invitrogen, Carlsbad, CA) and 0.5 ng/μL of each hit compound or vehicle control (DMSO) in Krebs-Ringer phosphate buffer (136 mM NaCl, 4.7 mM KCl, 10 mM NaPO4, 0.9 mM MgSO4, 0.9 mM CaCl2, pH 7.4) supplemented with 25 mM d-glucose at 37 °C in 5% CO2/95% air for 1 h. 100 nM H2O2 was used as a positive control for ROS production. Following incubation, cells were washed three times with PBS to remove excess probe. Cells were then covered with 100 μL/well PBS and fluorescence intensity measured by a Tecan Infinite® M200 microplate reader (Tecan Group Ltd., Switzerland) using a top-read configuration and with the excitation and emission filters set at 495±9 nm and 530±20 nm, respectively. Fluorescence data were recorded on Magellan (version 6.4) software and exported to Microsoft Excel for subsequent analysis. Having subtracted the background fluorescence (that emitted from a well which did not receive the CM-H2DCFDA probe) from each well, ROS production was expressed in terms of percentage fluorescence of the vehicle control for each condition. Compounds which increased ROS levels by greater than 20% were eliminated.
2.4. Measurements of oxygen consumption and extracellular acidification in whole cells
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XF-24 Flux Analyzer (Seahorse Biosciences, North Billerica, MA). NMuLi, C2C12, and L6 cells were seeded in a Seahorse 24-well tissue culture plate at a density of 3.5×104 cells/well, isolated cardiomyocytes at a density of 4×104 cells/well, and human primary fibroblasts at a density of 1.1×104 cells/well. The cells were then allowed to adhere for 24 h. Prior to the assay, the media was changed to unbuffered DMEM containing pyruvate and glutamine (Gibco #12800-017, pH=7.4 at 37 °C) and the cells were equilibrated for 30 mins at 37 °C. Compounds were injected during the assay and OCR and ECAR were measured using 2 min measurement periods.
2.5. Mitochondria isolation
Mitochondria were isolated from the livers of male C57BL/6 mice. Mice were euthanized by CO2 inhalation and cervical dislocation. Livers were removed, minced with scissors, and immediately placed in 1 mL ice-cold isolation medium (250 mM sucrose, 10 mM Tris–HCl, 1 mM EGTA, 1% fatty acid free BSA, pH 7.4). The tissue was homogenized using four strokes of a custom automated Potter-Elvehjem tissue homogenizer. After adding 4 mL of isolation medium the homogenate was centrifuged at 800×g for 10 min at 4 °C. The supernatant was then divided into four 2 mL Eppendorf tubes and centrifuged at 12,000×g for 10 min at 4 °C. The supernatant was removed and any white debris was aspirated from the brown mitochondria pellet. The pellets were then combined in 1 mL isolation medium and centrifuged at 10,000×g for 10 min. The supernatant and any white debris were removed and the mitochondria were resuspended in 1 mL mitochondrial assay solution (MAS, 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% fatty acid free BSA, pH 7.2).
2.6. Electron flow assay
Electron flow assays were performed using the methods described in Rogers et al. [19]. Briefly, 5 μg of mitochondrial protein in MAS was loaded into a Seahorse 24-well tissue culture plate and centrifuged at 2000×g for 15 min at 4 °C. Prior to the assay, mitochondria were incubated at 37 °C for 10 mins in MAS containing 10 mM pyruvate, 2 mM malate, and 5 μM BAM15 or FCCP. Rotenone (2 μM), succinate (10 mM), antimycin A (4 μM), and N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD, 100 μM) plus ascorbate (10 mM) were added sequentially as indicated in the figure. N=3 wells/plate of a representative of 3 plates.
2.7. Mitochondrial titration assays
Mouse liver mitochondria were isolated and respiration was measured according to Rogers et al. [19]. Oxygen consumption was measured using a Seahorse XF96 Flux Analyzer on mitochondria respiring on pyruvate (10 mM) and malate (2 mM) or succinate (10 mM) and rotenone (2 μM).
2.8. Mitochondrial membrane potential in whole cells
L6 cells were incubated with the fluorescent indicator of mitochondrial membrane potential tetramethylrhodamine (TMRM, 125 nM) or DMSO (1%) control for 30 min. The cells were then centrifuged for 5 min at 700×g and resuspended in unbuffered DMEM at a concentration of 1×105 cells/mL. The cells were then treated with BAM15/FCCP or DMSO (0.1%) for 10 min prior to flow cytometric analysis.
2.9. ANT-independence assay
C2C12 cells (ATCC) were plated in XF96 tissue culture plates (15,000 cells/well) and cultured overnight as recommended by the supplier. Just before initiation of the XF assay (XF96, Seahorse Bioscience), cells were washed twice with 200 μl/well MAS buffer, permeabilized with 1.0 nM XF PMP (Seahorse Bioscience), and simultaneously provided 4 mM ADP, 10 mM succinate, and 10 μM cyclosporin A (Sigma-Aldrich) to obtain State 3 respiration as described [20]. The plate was immediately placed into the XF96 instrument and assay initiated. Respiration (State 3) was recorded, followed by sequential injections of 3 μg/mL oligomycin or carboxyatractyloside (CAT, Sigma) and then 1.0 μM BAM15 or FCCP with measurements taken after each injection.
2.10. Mitochondrial swelling assay
Mitochondrial swelling was calculated as previously described by Shchepinova et al. [21]. Briefly, isolated liver mitochondria from female WT FVB mice were added to 1 mL isotonic acetate buffer (145 mM potassium acetate, 5 mM Tris–HCl, 0.5 mM EDTA, 3 μM valinomycin, and 1 μM rotenone, pH 7.4) in a cuvette at a final concentration of 0.25 mg/mL mitochondrial protein. Absorbance at 600 nm of the mitochondrial suspension was measured at a rate of 30 measurements/min using a SpectraMax M5 dual-monochromator microplate reader (Molecular Devices, CA) using a cuvette-read configuration for 60 s before the addition of 10 μM uncouplers. Data was recorded on SoftMax Pro (version 4.8) software.
2.11. Mitochondrial membrane potential in isolated mitochondria
Isolated liver mitochondria from male WT FVB mice incubated with 200 nM TMRM in MASSRO buffer (MAS buffer supplemented with 10 mM succinate, 1 μM rotenone, and 1 μM oligomycin) for 20 min at 25 °C. The mitochondria were then centrifuged for 5 min at 3000×g and resuspended MASSRO. Mitochondria were then added to MASSRO containing the indicated concentrations of uncouplers and incubated at room temperature for 20 min. The mitochondria were then centrifuged for 5 min at 3000×g. The supernatant was then removed and placed into a black clear bottom 96-well plate (100 μL/well). TMRM fluorescence was determined using an excitation emission of 545ex/580em. Fluorescence data were recorded on Magellan (version 6.4) software and exported to Microsoft Excel for subsequent analysis. Having subtracted the background fluorescence (that emitted from a well which did not receive the TMRM probe) from each well, TMRM fluorescence was expressed in terms of percentage fluorescence of the vehicle control for each condition.
2.12. Plasma membrane electrophysiology
In preparation for recording, L6 cells were plated onto poly-l-lysine-coated glass coverslips and returned to the incubator to adhere for at least 1 h prior to use. Cells were used within 1 day of plating. Whole cell recordings were performed at room temperature with 3–5 MΩ Sylgard-coated borosilicate glass patch pipettes and an Axopatch 200B amplifier (Molecular Devices). The internal solution contained 120 mM KCH3SO3, 4 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 10 mM HEPES, 10 mM EGTA, 3 mM ATP-Mg and 0.3 mM GTP-Tris (pH 7.2). The bath solution was composed of 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES and 10 mM glucose (pH 7.3) and was flowed over the cells at approximately 2 mL/min. For voltage clamp experiments, cells were held at −70 mV and a 750 ms ramp from −150 mV to +80 mV was applied at 10 s intervals using pCLAMP software and a Digidata 1322A digitizer (Molecular Devices). Conductance measurements were taken between −130 mV and −60 mV. For current clamp experiments, cells were recorded at the resting membrane potential.
2.13. Cytotoxicity assays
Cells were seeded into 96 well plates at a density of 5000 cells/well for NMuLi, L6 and C2C12 cells and 10,000 cells/well for primary rat left ventricular cardiomyocytes. Cells were incubated overnight at 37 °C prior to drug treatment. Drugs were diluted in cell culture medium (10% fetal calf serum in Dulbecco's Modified Eagle Medium) (Gibco Life Technologies, Grand Island, NY, USA) and added to each well at the indicated concentrations. Cell viability was measured 48 h later using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution (MTT) (Amresco, Solon, Ohio, USA) or crystal violet staining (0.5% w/v in 50% methanol). Absorbance was measured using a SpectraMax M5 dual-monochromator microplate reader (Molecular Devices, CA). Cell viability of drug-treated cells is displayed as a percentage of control cells i.e. cells with equivalent concentrations of the vehicle, dimethylsulfoxide (DMSO). The final concentration of DMSO exposed to the cells was no more than 0.1% (v/v) for the duration of the experiment.
2.14. Renal ischemic reperfusion injury
All animals were handled and procedures were performed in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the University of Virginia Institutional Animal Care and Use Committee. Male mice (8-week old, C57BL/6, from the National Cancer Institute, Frederick, MD) were anesthetized with a mixture (i.p.) of ketamine (120 mg/kg), xylazine (12 mg/kg), and atropine (0.324 mg/kg) and were subjected to bilateral ischemic reperfusion injury (26 min ischemia, then 24 h or 48 h reperfusion) as previously described [22]. During the surgery, mouse core temperature was maintained at 34–36 °C with a heating pad; during the recovery and reperfusion period, mice were housed in a warming incubator with ambient temperature at 30–32 °C. Control, sham-operated mice underwent a similar procedure, but the renal pedicles were not clamped. Mice were i.p. injected with BAM15 at 1 or 5 mg/kg, 1 h before kidney IR. Vehicle mice were also injected with the same solution BAM15 was prepared with (3% DMSO in 50% PEG400).
2.15. Assessment of kidney function and histology
Plasma creatinine, as a measure of kidney function, was determined using a colorimetric assay according to the manufacturer's protocol (Sigma-Aldrich). For histology, kidneys were fixed overnight in 0.2% sodium periodate/1.4% dl-lysine/4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) and embedded in paraffin. Kidneys were prepared for H&E staining and viewed by light microscopy (Zeiss Axioskop). Photographs were taken and brightness/contrast adjustment was made with a SPOT RT camera (software version 3.3; Diagnostic Instruments). Acute tubular necrosis was assessed as previously described [23]. Stained kidney sections were scored in a blinded manner. The score was based on the percentage of outer medulla tubules with pink casts on the inside, which is a marker of tubular necrosis. The scoring system was as follows: 1 (<10%), 2 (10–25%), 3 (25–75%), and 4 (>75%).
2.16. Kidney FACS analysis
Flow cytometry was used to analyze kidney leukocyte content. In brief, kidneys were extracted, minced, digested, and passed through a filter and a cotton column as described [24]. After blocking non-specific Fc binding with anti-mouse CD16/32 (2.4G2), fresh kidney suspensions were incubated with fluorophore-tagged anti-mouse CD45 (30-F11) to determine total leukocyte cell numbers. CD45-labeled samples were further used for labeling with different combinations of anti-mouse F4/80 (BM8), GR-1 (Ly6G), CD11b, CD11c, IA (MHCII). 7-Aminoactinomycin D (7-AAD; BD Biosciences) was added 15 min before analyzing the sample to separate live from dead cells. Flow cytometry data acquisition was performed on a FACSCalibur (Becton Dickinson). Data were analyzed by FlowJo software 9.0 (Tree Star).
3. Results
3.1. A small molecule library screen identifies compound BAM15 as an energy expenditure agonist with a broad maximally effective range
Enhanced O2 consumption is a consequence of mitochondrial uncoupling; therefore, we measured cellular O2 consumption as a primary screen to identify novel uncouplers using a non-quantitative assay. Rat L6 myoblasts were chosen for this screen because they are fast growing, have abundant mitochondria, and have spare mitochondrial reserve capacity. L6 cell number was optimized such that 500,000 cells per well produced a reliable 2-fold increase in fluorescence over background across a time course of up to 50 min (Suppl. Figure 1A). Using FCCP as a positive control for increased O2 consumption (Suppl. Figure 1B) and DMSO as a vehicle control, we screened a small molecule diversity library of 5040 compounds at a concentration of 5 μg/mL (TimTec Apex Screen). Hits were identified as those that increased O2 consumption over DMSO control (Suppl. Figure 1C). Hits were then subjected to a secondary screen to identify, and eliminate, compounds that increased the production of reactive oxygen species (ROS). Compounds that did not increase ROS production and were structurally unrelated to known uncouplers were then tested across a concentration range from 10 nM to 10 μM on a Seahorse XF Analyzer to identify those with a broad activity range. This algorithm identified BAM15 as a compound that increased O2 consumption across a broad dosing range (Figure 1) without increasing ROS (Suppl. Figure 2). With respect to O2 consumption, BAM15 had equal potency to FCCP; therefore, we compared BAM15 to FCCP throughout this study. BAM15 and FCCP are structurally unrelated (Figure 1A) and we observed that low doses of BAM15 from 100 nM to 1 μM increased cellular O2 consumption rate (OCR) to a similar degree as FCCP, but higher concentrations from 1 μM to 50 μM revealed that BAM15 was able to maintain uncoupled respiration at a high rate in a range of cell lines including rat L6 myoblasts, primary rat neonatal ventricular cardiomyocytes, mouse C2C12 myoblasts, normal murine liver cells, and human primary fibroblasts (Figure 1B–F). Similarly, we found that the cellular extracellular acidification rate (ECAR), a measure of both glycolysis (proton co-transport with lactate out of the cell) and nutrient oxidation (the hydration of CO2 to form HCO3− and H+), was maintained at a higher level in BAM15-treated cells (Suppl. Figure 3).
Figure 1.
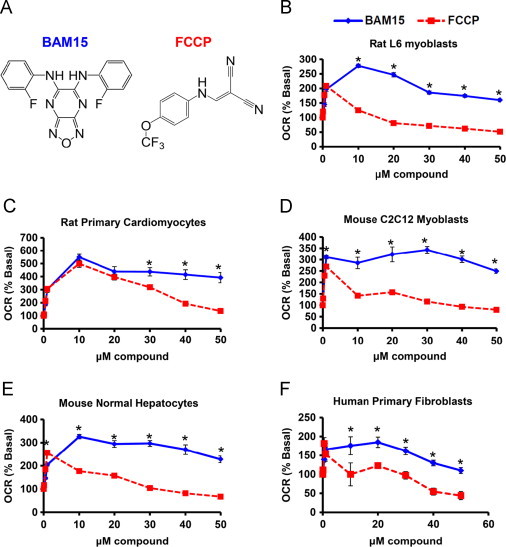
(A) BAM15 and FCCP are structurally unrelated. (B–F) FCCP- and BAM15-stimulated oxygen consumption rate (OCR) in L6 myoblasts, rat primary cardiomyocytes, mouse C2C12 myoblasts, mouse normal hepatocytes, and human primary fibroblasts at the indicated concentrations. Error bars indicate SEM. For (B–F), * indicates p<0.05 by two-way ANOVA with Bonferroni's posttest and N=6–8 wells per condition from three separate experiments.
Figure 2.
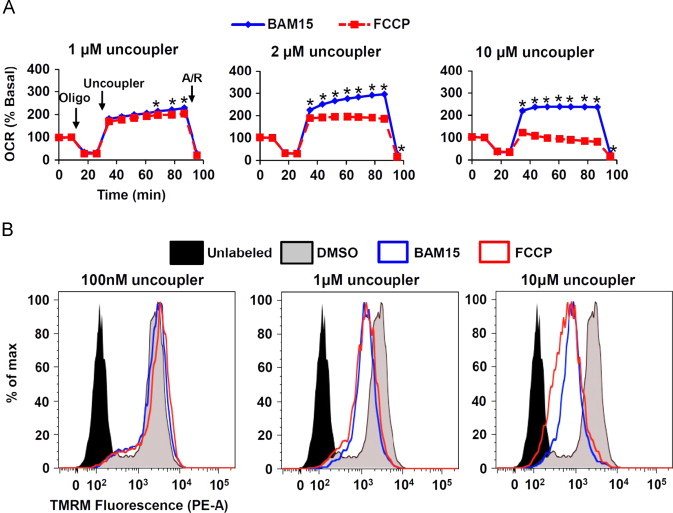
BAM15 causes mitochondrial uncoupling in cells. (A) L6 cells were sequentially treated with oligomycin (Oligo, 1 µM), the indicated concentration of BAM15 or FCCP (Uncoupler), and antimycin A (10 µM) plus rotenone (1 µM) (A/R) as indicated by arrows. (B) TMRM-loaded L6 cells were treated with the indicated concentrations of BAM15 or FCCP for 10 min prior to FACS analysis in the phycoerythrin (PE) channel. Uncoupler-treated cells are left-shifted, indicating loss of mitochondrial membrane potential. Error bars indicate SEM. For (A) * indicates p<0.05 by two-way ANOVA with Bonferroni's posttest. N=5 wells per condition over one experiment. For (B), N=one representative from three separate experiments.
3.2. BAM15 is a mitochondrial protonophore uncoupler
We next sought to determine whether BAM15 was a bona-fide mitochondrial uncoupler. The ATP synthase is the major pathway whereby protons enter the mitochondrial matrix; therefore, we investigated whether BAM15 could stimulate O2 consumption in the presence of the ATP synthase inhibitor oligomycin. Using FCCP as an equipotent positive control, we found that BAM15 was fully capable of increasing mitochondrial respiration in the presence of oligomycin and did so across a broader concentration range than FCCP in both myoblasts and hepatocytes (Figure 2A and Suppl. Figure 4). Increasing proton leak into mitochondria is expected to induce mitochondrial depolarization, and we observed that BAM15 and FCCP had similar effects on mitochondrial depolarization in L6 myoblasts treated with concentrations of each uncoupler at 1 μM and 10 μM as measured by tetramethylrhodamine (TMRM) fluorescence (Figure 2B).
To determine whether BAM15 was acting directly on mitochondria, we isolated rat liver mitochondria and treated them with increasing concentrations (1–64 μM) of BAM15 and FCCP. As shown in Figure 3A, we found that BAM15 and FCCP had comparable effects on the stimulation of oxygen consumption on mitochondria respiring on pyruvate and malate (complex I substrates), or succinate (a complex II substrate) in the presence of the complex I inhibitor rotenone. Treatment with BAM15 and FCCP also depolarized isolated mitochondria respiring on succinate (Figure 3B) and succinate in the presence of oligomycin (Suppl. Figure 5).
Figure 3.
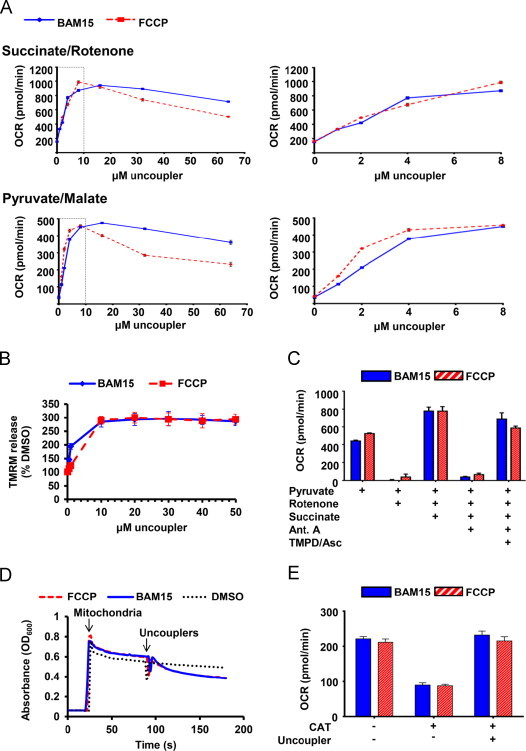
BAM15 is a mitochondrial protonophore. (A) Oxygen consumption rate (OCR) of isolated mouse liver mitochondria respiring on succinate in the presence of rotenone and pyruvate/malate was measured following treatment with increasing concentrations of BAM15 or FCCP. Right graphs expand the area that is indicated by the dotted box in the left graph. (B) BAM15 depolarizes isolated mitochondria. TMRM release in isolated mitochondria respiring on succinate (10 mM) in the presence of rotenone (1 µM) was measured following treatment with increasing concentrations of BAM15 and FCCP. Data is expressed as a percentage of fluorescence from DMSO (0.1%)-treated mitochondria. (C) Isolated mouse liver mitochondria respiring on pyruvate and malate in the presence of FCCP (5 µM) or BAM15 (5 µM) were treated sequentially with rotenone (4 µM), succinate (10 mM), antimycin A (4 µM), and the electron donors TMPD (100 µM) and ascorbate (10 mM). (D) BAM15 induces proton-dependent mitochondrial swelling. Absorbance at 600 nm was read over time following the addition of isolated mouse liver mitochondria respiring on succinate (10 mM) in the presence of rotenone (1 µM) and valinomycin. Mitochondria and 10 µM uncouplers were added at the indicated time points. (E) BAM15 increases oxygen consumption independent of the adenine nucleotide translocase. Oxygen consumption rate (OCR) was measured on permeabilized C2C12 cells respiring on succinate (10 mM) and treated with rotenone (2 µM) and ADP (4 mM). Mitochondria were then treated with carboxyatractyloside (CAT, 3 µg/mL), followed by FCCP or BAM15 (1 µM). Error bars indicate SEM. For (A and E) N=6–9 wells per condition over three separate experiments. For (B) N=3 experiments. For (C) N=3 wells/condition. For (D) N=one representative of three separate experiments.
To determine whether BAM15 was altering mitochondrial electron flow we compared FCCP and BAM15 (5 µM) in mouse liver mitochondria respiring on pyruvate and malate, and sequentially treated with rotenone, succinate, antimycin A (a complex III inhibitor), and the complex IV electron donor system N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) and ascorbate as described [19]. As shown in Figure 3C, rotenone inhibited respiration in uncoupled mitochondria respiring on the complex I-linked respiratory substrates pyruvate and malate. These data demonstrate that neither FCCP nor BAM15 donate electrons to the ETC. Succinate was then added to enable complex II-dependent respiration, then antimycin A was added to block respiration. Since both BAM15 and FCCP-mediated respiration was inhibited by antimycin A, these data demonstrate that neither molecule can donate electrons from succinate to cytochrome c or complex IV. The addition of the electron donor system of TMPD and ascorbate was able to fully rescue respiration at complex IV in the presence of both BAM15 and FCCP.
To determine whether BAM15 was transporting protons across the mitochondrial inner membrane, we measured proton-dependent mitochondrial swelling [21]. Mitochondria were incubated in isotonic potassium acetate buffer supplemented with the potassium ionophore valinomycin. Acetate is used as a solute in this assay because the dissociated acetate anion is membrane impermeable and causes the mitochondria to swell when trapped inside the matrix. Under these conditions, only the protonated form of acetate can enter the mitochondria, but the accumulation of protons inside the matrix limits further acetate entry. In this system the addition of a protonophore enables proton efflux from the matrix, which relieves the proton gradient and allows more acetate accumulation and swelling [25]. As shown in Figure 3D, both FCCP and BAM15 induced mitochondrial swelling, demonstrating that BAM15 is a protonophore.
Finally, some non-protonophore uncouplers increase proton transport into the matrix via interaction with the mitochondrial inner membrane adenine nucleotide translocase (ANT) [26]. To determine whether the ANT was necessary for BAM15-mediated respiration we treated permeabilized cells with the ANT inhibitor carboxyatractyloside prior to BAM15 or FCCP treatment. These data demonstrated that both BAM15 and FCCP did not require the ANT to increase mitochondrial respiration (Figure 3E).
3.3. BAM15 does not depolarize the plasma membrane and has less cytotoxicity than the equipotent protonophore uncoupler FCCP
Since FCCP is known to have off-target effects on depolarization of the plasma membrane, we measured the effects of BAM15 versus FCCP on plasma membrane electrophysiology using whole cell voltage and current clamp recordings. As expected, under voltage clamp conditions at a holding potential of −70 mV, FCCP induced an inward current that was dose-dependent and associated with an increase in conductance (Figure 4A–F). In contrast, BAM15 elicited no appreciable change in current in the same cells. Under current clamp conditions, FCCP caused reversible and repeatable plasma membrane depolarization, whereas BAM15 had no effect (Figure 4G and H). The differential effects of BAM15 and FCCP on plasma membrane properties were independent of the order of uncoupler application (not shown). Furthermore, BAM15-treated cells were more viable than FCCP-treated cells when administered across a broad dosing range up to 50 μM (Suppl. Figure 6). These data indicate that BAM15 does not share the adverse plasma membrane effects that may contribute to cytotoxicity.
Figure 4.
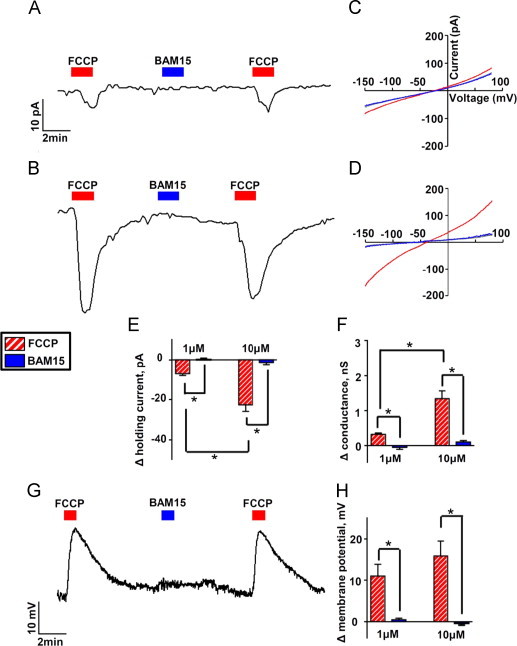
BAM15 does not alter plasma membrane electrophysiology. (A) Representative whole cell voltage clamp recording from a L6 cell showing the holding current (at −70 mV) during exposure to FCCP and BAM15 (both 1 µM). (B) Voltage clamp with 10 µM FCCP and BAM15. (C and D) Currents were elicited with a voltage ramp from −150 mV to +80 mV using 1 µM uncouplers in (C) and 10 µM uncouplers in (D); I–V relationships are plotted under control conditions and in the presence of the uncouplers. (E) Average data comparing the change in holding current caused by FCCP and BAM15 at 1 µM and 10 µM. (F) Average data comparing the change in conductance generated by either drug in the range of −130 mV to −60 mV. (G) Representative whole cell current clamp recording at concentrations of 10 µM for FCCP and BAM15. (H) Average data comparing the change in membrane potential by both drugs. Error bars indicate SEM. For (E)–(H), * indicates p<0.05 by two-way ANOVA with Bonferroni's posttest, N=7–9 cells per condition.
3.4. BAM15 protects from kidney ischemia-reperfusion injury
Mitochondrial uncoupling, both pharmacological and endogenous, is protective in preclinical models of ischemia-reperfusion injury [5,8,27]. To determine whether BAM15 treatment could protect mice from ischemic reperfusion injury we administered BAM15 as a single intraperitoneal bolus at 1 or 5 mg/kg 1 h prior to 26 min of bilateral renal ischemia. At 24 and 48 h following reperfusion, plasma creatinine levels were measured and the mice were euthanized at 48 h for analysis of renal pathology. Compared to vehicle-treated mice, animals that received BAM15 were protected from kidney injury as indicated by lower plasma creatinine levels at 24 and 48 h post-ischemia, reduced tubular necrosis, less depletion of brush border villi, less obstruction of proximal tubules, and less immune cell infiltration (Figure 5 and Suppl. Figure 7).
Figure 5.
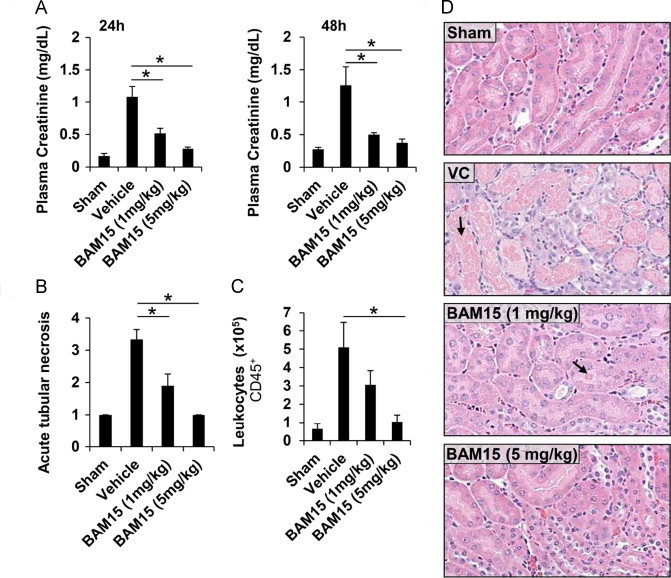
BAM15 protects against kidney ischemic-reperfusion injury. Male mice (8-week old, C57BL/6) were treated with vehicle control (VC) or BAM15 at 1 or 5 mg/kg 1 h prior to bilateral ischemia for 26 min followed by 48 h of reperfusion. Sham-operated mice underwent an identical surgical procedure, but the renal pedicles were not clamped. (A) BAM15 pretreatment dose-dependently protected from kidney damage, as indicated by the decreased plasma creatinine levels at 24 and 48 h following reperfusion, compared to VC. (B-D) BAM15 pretreatment decreased acute proximal tubular necrosis and leukocyte invasion of the kidney medulla 48 h following reperfusion. Arrows indicate sites of tubular cell death. Error bars indicate SEM. * indicates p < 0.05 compared to vehicle control by one-way ANOVA with Dunnett's posttest. N = 3-6 mice per group.
4. Discussion and conclusions
Synthetic mitochondrial uncouplers are invaluable tools for the analysis of mitochondrial function and they represent possible therapeutics for diabetes, obesity, neurodegeneration, cancer, ischemia-reperfusion injury, and other disorders linked to mitochondrial dysfunction. However, a possible factor limiting the use of available protonophore uncouplers is their unwanted activity at other membranes such as the plasma membrane. The overarching goal of this study was to identify new mitochondrial uncouplers that lack activity at the plasma membrane. To achieve this, we performed a cell-based phenotypic screen that involved a series of filters including assays for O2 consumption, ROS production, dynamic range, oligomycin- and carboxyatractyloside-independence, and plasma membrane electrophysiology. This screen identified BAM15 as a bona-fide mitochondrial uncoupler that does not affect plasma membrane conductance and has a broad effective range. The mechanism by which BAM15 has a preference for protonophore activity at mitochondria but not the plasma membrane is unclear, but may be due to a pKa of its ionizable protons, such that BAM15 is only able to donate protons in the alkaline environment of the mitochondrial matrix. However, other alterative explanations exist and include the possibility that BAM15 may have a structural preference for the unique lipid composition of the mitochondrial inner membrane, or that either the protonated or deprotonated form of BAM15 is membrane impermeable. Future organic chemistry and structure–activity studies will be required to identify the precise pharmacophore mechanisms of BAM15.
Our initial characterization of BAM15 has answered several important questions concerning the effects of pharmacological mitochondrial uncoupling on cellular function. For example, since some metabolic substrates utilize the pH gradient to enter mitochondria, it is thought that mitochondrial depolarization is a limiting factor for mitochondrial respiration [28]. Indeed, FCCP-induced mitochondrial failure coincides with the loss of mitochondrial membrane potential; however, we observed that BAM15 was fully capable of stimulating maximal mitochondrial respiration at concentrations above 10 μM where the mitochondria were fully depolarized (Figures 1A and 2B). These data suggest that FCCP-induced mitochondrial failure is not due to mitochondrial depolarization and instead may be caused by off-target effects, such as plasma membrane depolarization. Furthermore, when BAM15 and FCCP were used at high concentrations (20–60 μM) in isolated mitochondria, BAM15 was able to continuously drive a greater rate of respiration than FCCP (Figure 3A). The FCCP-induced decline in mitochondrial respiration was less pronounced in isolated mitochondria than the decline observed in whole cells, which indicates that the deleterious effects of FCCP are most likely due to its extra-mitochondrial actions. Consistent with this notion, we observed that BAM15 was significantly less cytotoxic than FCCP (Suppl. Figure 5). Finally, we observed that BAM15 treatment stimulated ECAR to a greater rate than FCCP. Since ECAR is a correlative measure of both glycolysis (proton co-transport with lactate out of the cell) and nutrient oxidation (the hydration of CO2 to form HCO3− and H+), these data demonstrate that FCCP is not able to maximally drive the metabolic capacity of the cell. Although FCCP has been used for more than 50 years to assay maximal mitochondrial function, these data demonstrate that FCCP underestimates maximal mitochondrial and cellular metabolic capacity. Therefore, BAM15 is a more reliable chemical tool for the study of mitochondrial function, particularly in living cells.
Therapeutically, mitochondrial uncoupling is an attractive drug target due to its ability to lower mitochondrial ROS production [29]. Low concentrations of uncouplers are demonstrated to have beneficial effects in cell culture and animal models of insulin resistance and obesity [11,12], neurodegeneration and ischemic reperfusion injury [7,9,30–32] and cardiovascular disease [13,33]. Our data demonstrated that BAM15 treatment dose-dependently protected from renal ischemic-reperfusion injury, indicating that BAM15 is bioactive in vivo. These data are promising; however, future pharmacokinetic analysis will be required to determine if BAM15 is suitable for long-term treatment of pathologies that require constant exposure such as progressive metabolic disorders.
In summary, we report the identification of BAM15 as a new chemotype mitochondrial uncoupler. BAM15 is highly potent and demonstrates a greater maximally effective dynamic range than the gold-standard uncoupler FCCP. One of the most important advantages of BAM15 is that it depolarizes mitochondria without affecting plasma membrane potential. This property enables a sustained maximal rate of mitochondrial respiration with low cytotoxicity. We also identified that BAM15 is bioactive in vivo and protects from acute renal ischemic reperfusion injury in mice. Collectively, BAM15 represents a valuable new tool for the study of mitochondrial function and its low cytotoxicity and lack of off-target membrane depolarization provides renewed optimism that mitochondrial uncouplers may again be useful for medical intervention in the myriad disorders linked to mitochondrial dysfunction.
Conflict of interest
BMK and KLH are co-inventors on a patent application involving BAM15.
Acknowledgments
This work was funded through start-up funds provided by the Department of Pharmacology at the University of Virginia (KLH), the American Diabetes Association (1-11-JF-17, KLH), and the University of Virginia Launchpad Biomedical Innovation in Diabetes Fund (KLH). We thank Drs. Anne Murphy and David Ferrick for their helpful consultation. We thank Dr. Shayn Peirce-Cottler and Mr Stephen Cronk for the gift of primary human fibroblasts.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.11.005.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Rousset S., Alves-Guerra M.C., Mozo J., Miroux B., Cassard-Doulcier A.M., Bouillaud F. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53(Suppl 1):S130–S135. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- 2.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 3.Turrens J.F. Superoxide production by the mitochondrial respiratory chain. Bioscience Reports. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 4.Lambert A.J., Brand M.D. Reactive oxygen species production by mitochondria. Methods in Molecular Biology. 2009;554:165–181. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 5.Sack M.N. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovascular Research. 2006;72:210–219. doi: 10.1016/j.cardiores.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 6.McLeod C.J., Aziz A., Hoyt R.F., Jr., McCoy J.P., Jr., Sack M.N. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. Journal of Biological Chemistry. 2005;280:33470–33476. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- 7.Korde A.S., Pettigrew L.C., Craddock S.D., Maragos W.F. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. Journal of Neurochemistry. 2005;94:1676–1684. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- 8.Modriansky M., Gabrielova E. Uncouple my heart: the benefits of inefficiency. Journal of Bioenergetics and Biomembranes. 2009;41:133–136. doi: 10.1007/s10863-009-9212-z. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y.N., Munhall A.C., Johnson S.W. Mitochondrial uncoupling agents antagonize rotenone actions in rat substantia nigra dopamine neurons. Brain Research. 2011;1395:86–93. doi: 10.1016/j.brainres.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 10.Anderson E.J., Lustig M.E., Boyle K.E., Woodlief T.L., Kane D.A., Lin C.T. Mitochondrial h2o2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoehn K.L., Salmon A.B., Hohnen-Behrens C., Turner N., Hoy A.J., Maghzal G.J. Insulin resistance is a cellular antioxidant defense mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldeira da Silva C.C., Cerqueira F.M., Barbosa L.F., Medeiros M.H., Kowaltowski A.J. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–560. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- 13.Brennan J.P., Southworth R., Medina R.A., Davidson S.M., Duchen M.R., Shattock M.J. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovascular Research. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Tseng Y.H., Cypess A.M., Kahn C.R. Cellular bioenergetics as a target for obesity therapy. Nature Reviews Drug Discovery. 2010;9:465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park K.S., Jo I., Pak K., Bae S.W., Rhim H., Suh S.H. FCCP depolarizes plasma membrane potential by activating proton and Na+ currents in bovine aortic endothelial cells. Pflügers Archiv. 2002;443:344–352. doi: 10.1007/s004240100703. [DOI] [PubMed] [Google Scholar]
- 16.Juthberg S.K., Brismar T. Effect of metabolic inhibitors on membrane potential and ion conductance of rat astrocytes. Cellular and Molecular Neurobiology. 1997;17:367–377. doi: 10.1023/A:1026331226241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brismar T., Collins V.P. Effect of external cation concentration and metabolic inhibitors on membrane potential of human glial cells. Journal of Physiology. 1993;460:365–383. doi: 10.1113/jphysiol.1993.sp019476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckler K.J., Vaughan-Jones R.D. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. Journal of Physiology. 1998;513(Pt 3):819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Divakaruni A.S., Wiley S.E., Rogers G.W., Andreyev A.Y., Petrosyan S., Loviscach M. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shchepinova M.M., Denisov S.S., Kotova E.A., Khailova L.S., Knorre D.A., Korshunova G.A. Dodecyl and octyl esters of fluorescein as protonophores and uncouplers of oxidative phosphorylation in mitochondria at submicromolar concentrations. Biochimica et Biophysica Acta. 2013;1837:149–158. doi: 10.1016/j.bbabio.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 22.Li L., Huang L., Ye H., Song S.P., Bajwa A., Lee S.J. Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. Journal of Clinical Investigation. 2012;122:3931–3942. doi: 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bajwa A., Jo S.K., Ye H., Huang L., Dondeti K.R., Rosin D.L. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. Journal of the American Society of Nephrology. 2010;21:955–965. doi: 10.1681/ASN.2009060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L., Huang L., Sung S.S., Lobo P.I., Brown M.G., Gregg R.K. Nkt cell activation mediates neutrophil ifn-gamma production and renal ischemia-reperfusion injury. Journal of Immunology. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 25.Nicholls D.G., Ferguson S.J. Bioenergetics. 2002;3:297. (xviii, 297 p.) [Google Scholar]
- 26.Lou P.H., Hansen B.S., Olsen P.H., Tullin S., Murphy M.P., Brand M.D. Mitochondrial uncouplers with an extraordinary dynamic range. Biochemical Journal. 2007;407:129–140. doi: 10.1042/BJ20070606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minners J., van den Bos E.J., Yellon D.M., Schwalb H., Opie L.H., Sack M.N. Dinitrophenol, cyclosporin a, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovascular Research. 2000;47:68–73. doi: 10.1016/s0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- 28.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochemical Journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandya J.D., Pauly J.R., Sullivan P.G. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Experimental Neurology. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 31.Stout A.K., Raphael H.M., Kanterewicz B.I., Klann E., Reynolds I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nature Neuroscience. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 32.Maragos W.F., Rockich K.T., Dean J.J., Young K.L. Pre- or post-treatment with the mitochondrial uncoupler 2,4-dinitrophenol attenuates striatal quinolinate lesions. Brain Research. 2003;966:312–316. doi: 10.1016/s0006-8993(02)04225-7. [DOI] [PubMed] [Google Scholar]
- 33.Brennan J.P., Berry R.G., Baghai M., Duchen M.R., Shattock M.J. FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovascular Research. 2006;72:322–330. doi: 10.1016/j.cardiores.2006.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material