

ABSTRACT
MicroRNA-149 (miR-149) is located within the first intron of the glypican-1 (GPC1) gene. GPC1 is a low affinity receptor for fibroblast growth factor (FGF2) that enhances FGF2 binding to its receptor (FGFR1), subsequently promoting FGF2–FGFR1 activation and signaling. Using bioinformatic approaches, both GPC1 and FGFR1 were identified and subsequently validated as targets for miR-149 (both the mature strand, miR-149, and the passenger strand, miR-149*) in endothelial cells (ECs). As a consequence of their targeting activity towards GPC1 and FGFR1, both miR-149 and miR-149* regulated FGF2 signaling and FGF2-induced responses in ECs, namely proliferation, migration and cord formation. Moreover, lentiviral overexpression of miR-149 reduced in vivo tumor-induced neovascularization. Importantly, FGF2 transcriptionally stimulated the expression of miR-149 independently of its host gene, therefore assuring the steady state of FGF2-induced responses through the regulation of the GPC1–FGFR1 binary complex in ECs.
KEY WORDS: microRNA, miR-149, Angiogenesis, Endothelial cell, Glypican-1, FGF2
INTRODUCTION
Angiogenesis is the formation of new blood vessels from pre-existing vasculature during various physiological and pathological conditions, including embryonic development, wound repair and tumor growth (Carmeliet, 2005; Folkman, 1984; Jain, 2005). The angiogenic process involves a switch from normal quiescent vasculature to an activated state in which endothelial cells (ECs) acquire a proliferative, migratory and morphogenic phenotype (Folkman, 1984), and is a change in the balance between pro- and anti-angiogenic factors towards a pro-angiogenic function (Jain, 2005). Although there are many angiogenic inducers, vascular endothelial growth factor (VEGF, also termed VEGF-A) and basic fibroblast growth factor (bFGF, also termed FGF2), both members of the VEGF and FGF family of growth factors, are probably the most important and potent ones (Cross and Claesson-Welsh, 2001; Olsson et al., 2006; Presta et al., 2005). Their pro-angiogenic effects are mediated through VEGF receptor 2 (VEGFR2) (Cross and Claesson-Welsh, 2001; Olsson et al., 2006; Presta et al., 2005) or FGF receptor 1 (FGFR1) (Cross and Claesson-Welsh, 2001; Olsson et al., 2006; Presta et al., 2005), respectively. Interestingly, a large number of the angiogenic factors (including those mentioned above) have been found to bind to heparan sulphate proteoglycans (HSPGs) (Gengrinovitch et al., 1999; Zhang et al., 2001). HSPGs are ubiquitous components of the cell surface (syndecans and glypicans) and extracellular matrix (perlecan) (Iozzo and Sanderson, 2011). Indeed, their presence at the cell surface and in the extracellular environment is crucial to many physiological processes including angiogenesis (Iozzo and Sanderson, 2011). The aberrant expression of a number of these HSPGs has been implicated in a variety of human malignancies (Iozzo and Sanderson, 2011; Sasisekharan et al., 2002).
Glypicans (GPCs) are glycosyl-phosphatidylinositol-anchored (GPI-anchored) HSPGs (Fico et al., 2011; Filmus et al., 2008) that are expressed during normal development in a stage- and tissue-specific manner (Filmus et al., 2008). There are six members of the GPC family. GPC1 is involved in tumorigenesis and angiogenesis, and is frequently overexpressed in different human malignancies including pancreatic carcinoma, breast cancer and glioma (Aikawa et al., 2008; Kleeff et al., 1998; Kleeff et al., 1999; Matsuda et al., 2001; Su et al., 2006). GPC1 has been proposed to act as a co-receptor for FGF that enhances the binding of FGF to its receptor, subsequently promoting FGF–FGFR activation and signaling (Zhang et al., 2001). GPC1 is highly expressed in ECs from human glioma blood vessels, whereas its expression is barely detectable in normal brain ECs (Qiao et al., 2003). Interestingly, in vitro studies in mouse brain micro-vascular ECs have shown the crucial role of GPC1 in EC proliferation and cell cycle progression (Qiao et al., 2008).
It is currently accepted that small non-coding RNAs, microRNAs (miRNAs), are key regulators of several cellular processes (Bushati and Cohen, 2007), including angiogenesis (Caporali and Emanueli, 2011; Suárez and Sessa, 2009; Thum, 2012; Urbich et al., 2008; Wang and Olson, 2009). This regulatory control is carried out through repression of gene expression at the post-transcriptional level (Bartel, 2009). Interestingly, miR-149 is found within the first intron of GPC1 (Jin et al., 2011) and its expression is downregulated in different solid tumors (Jin et al., 2011; Li et al., 2011; Øster et al., 2013; Wang et al., 2013). Moreover, overexpression of miR-149 in glioma and colorectal cancer cells results in reduced proliferation and migration, thus suggesting that miR-149 might play a role as a tumor suppressor (Jin et al., 2011; Li et al., 2011). Taken together, these studies suggest that miR-149 and its host gene have different expression patterns and cellular functions.
Although the role of GPC1 in mouse brain ECs has been reported, the function of miR-149 in ECs remains to be determined. In the present study, we sought to investigate the role of miR-149 in the angiogenic activity of human ECs through targeting of FGFR1 and GPC1. In addition, we examined the mechanism that controls its independent expression from its host gene, GPC1.
RESULTS
Expression pattern of miR-149
miR-149 is encoded by intron 1 (22.9 kb) of the human GPC1 gene. The sequence of miR-149 found in human, mouse, cow and chimpanzee is identical, and the pre-miRNA is highly conserved in mammals (supplementary material Fig. S1A). In order to investigate the relationship between miR-149 and its host gene, we first analyzed the expression of GPC1, miR-149 (both mature sequences, i.e. the guide, miR-149, and the passenger, miR-149*) and the primary transcript of miR-149 (pri-miR-149) in a panel of human tissues. The expression pattern of miR-149 shows that the expression of the mature forms do not parallel that of GPC1, consistent with the idea that miR-149 is expressed independently of its host gene (Fig. 1A,C). Consistent with this, expression analysis of pri-miR-149 further revealed the lack of correlation with the expression of GPC1 (Fig. 1A,G). Interestingly, in addition to the brain, both miR-149 and miR-149* showed a high level of expression in lung and placenta (Fig. 1C,E). These tissues are highly enriched in ECs and the latter is a well-accepted angiogenic tissue (Reynolds and Redmer, 2001).
Fig. 1.
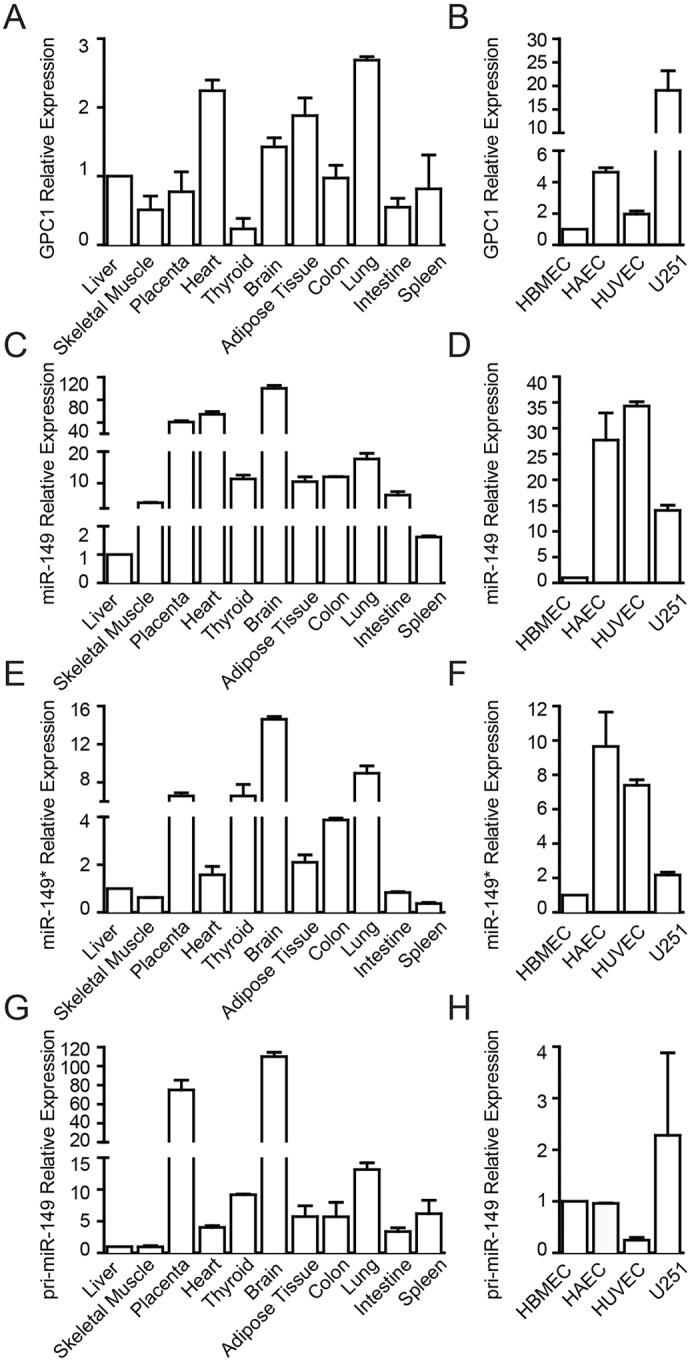
Differences in the expression of GPC1, miR-149 and miR-149* and primary transcript (pri-miR-149) in human tissues and cell lines. qRT-PCR analysis of (A) GPC1, (C) miR-149, (E) miR-149* and (G) pri-miR-149 expression in different human tissues or (B) GPC1, (D) miR-149, (F) miR-149* and (H) pri-miR-149 in cell lines. Data are expressed as relative to expression in liver (A,C,E,G) or HBMECs (B,D,F,H).
We next examined the expression pattern of GPC1, pri-miR-149 and both miR-149 and miR-149* in a panel of human primary ECs including HBMECs, HAECs and HUVECs (Fig. 1B,D,F,H). The expression level of GPC1 was higher in both HAECs and HUVECs than in HBMECs (Fig. 1B). This is consistent with a previous report that indicates that the expression of GPC1 is barely detectable in normal brain ECs (Qiao et al., 2008). The expression of GPC1 was measured in U251 glioma cells as a positive control (Fig. 1B) (Su et al., 2006). Consistent with the data observed in tissue, the expression of miR-149, miR-149* and pri-miR-149 did not correlate with the expression of the host gene (Fig. 1B,D,F,H). In particular, the expression level of GPC1 was lower in ECs than U251 cells (Fig. 1B), whereas the expression of both miRNAs was higher in ECs than U251 cells (Fig. 1D,F).
miR-149 targeting activity
To gain insight into the function of miR-149 in ECs, we first analyzed its potential gene targets using several miRNA-target-prediction algorithms (i.e. miRanda, DIANA, miRWalk, PicTar and TargetScan). This analysis revealed 6798 and 6829 predicted target genes in humans for miR-149 and miR-149*, respectively. Specifically, both strands shared predicted binding sites in 3823 genes. We next determined whether the predicted targets of miR-149 and miR-149* were preferentially connected to any specific biological process.
Using Protein Analysis Through Evolutionary Relationships (PANTHER, version 7) analysis (Mi et al., 2010), we observed an enrichment in genes implicated in the control of angiogenesis. Indeed, angiogenesis was the most significant pathway for both miR-149 and miR-149*, with P values of 5.12×10−4 and 1.71×10−3, respectively (supplementary material Fig. S2A,B). Moreover, both miRNAs shared 61 predicted target genes that are involved in the angiogenic process, most of them as positive regulators (supplementary material Table S1).
From the set of predicted targets, we selected FGFR1 and GPC1 for further analysis; the rationale for this selection was based on: (1) the well-established role of FGFR1 and its ligand FGF2 in EC angiogenesis (Gerwins et al., 2000; Presta et al., 2005), (2) the role of GPC1 as a low-affinity receptor for FGF (Zhang et al., 2001) that promotes FGF2 binding to its receptor (FGFR1), potentiating FGF signaling (Fico et al., 2011; Filmus et al., 2008; Gerwins et al., 2000; Iozzo and Sanderson, 2011; Presta et al., 2005; Qiao et al., 2003; Su et al., 2006), (3) the fact that GPC1 has been shown to modulate EC proliferation (Qiao et al., 2012; Qiao et al., 2003; Qiao et al., 2008), (4) the finding that FGFR1 regulation by miRNAs negatively controls EC angiogenic functions (Chamorro-Jorganes et al., 2011), (5) the fact that the regulation of host genes by their intronic miRNA has been shown to be biologically relevant (Nikolic et al., 2010) (Megraw et al., 2010), and (6) the finding that these genes showed a relatively high number of predicted binding sites for miR-149 and miR-149* in their 3′ untranslated regions (3′UTRs). Indeed, miRNA target algorithms found nine binding sites in the FGFR1 3′UTR (three for miR-149 and six for miR-149*) and 12 binding sites in the GPC1 3′UTR (four for miR-149 and eight for miR-149*) (supplementary material Fig. S1B).
To address whether miR-149 or miR-149* could mediate the post-transcriptional regulation of FGFR1 and GPC1 in ECs, HUVECs were transfected with miR-149, miR-149* or non-targeting control mimic, and the effect on FGFR1 and GPC1 protein levels was analyzed by western blotting (Fig. 2A) or flow cytometry (Fig. 2B), respectively. Notably, the overexpression of miR-149 or miR-149* decreased FGFR1 and GPC1 protein levels. However, the opposite results were obtained in a more physiologically relevant system where the endogenous miR-149 or miR-149* were inhibited with specific inhibitors (I-miR-149 and I-miR-149*) (Fig. 2C,D). Similar results were also observed for FGFR1 and GPC1 at the mRNA level (supplementary material Fig. S2C,D).
Fig. 2.

miR-149 and miR-149* target FGFR1 and GPC1. (A,C) Western blot analysis of FGFR1 protein expression in HUVECs transfected for 48 h with (A) control mimic (CM), miR-149 mimic (miR-149) or miR-149* mimic (miR-149*) or (C) control inhibitor (CI), inhibitor miR-149 (I-miR-149) or inhibitor miR-149* (I-miR-149*). HSP90 is used as the loading control. The data are expressed as fold activation over control mimic or CI, respectively. One representative experiment out of three independent experiments is shown. (B,D) Flow cytometry analysis of GPC1 protein expression in HUVECs transfected for 48 h with (B) control mimic (black), miR-149 (red) or miR-149* (blue), or (D) CI (black), I-miR-149 (red) or I-miR-149 (blue). Data are expressed as arbitrary fluorescence units (u.a.f.). One representative experiment out of three independent experiments is shown. (E–H) Luciferase activity in COS7 cells transfected for 24 h with control mimic, miR-149 or miR-149* and either (E) the human FGFR1 3′UTR construct, (G) the human GPC1 3′UTR, (F) the FGFR1 3′UTR or (H) GPC1 3′UTR constructs containing the indicated point mutations (PM, see supplementary material Fig. S1B) in the miR-149 or miR-149* target sites. The data are expressed as luciferase activity relative to control samples co-transfected with control mimic, and correspond to the mean±s.e.m. of three experiments performed in duplicate. *P≤0.05 compared with cells co-transfected with control mimic; and #P≤0.05 compared with cells co-transfected with miR-149 or miR-149*.
To ascertain the predicted miRNA–mRNA interactions, FGFR1 and GPC1 3′UTRs were sub-cloned into a luciferase reporter vector. The constructs were co-transfected into COS7 cells along with miR-149, miR-149* or control mimic. miR-149 significantly reduced (∼25%) the FGFR1 (Fig. 2E) and GPC1 (Fig. 3G) 3′UTR activity. Notably, the effect of miR-149* on the FGFR1 (Fig. 2F) and GPC1 (Fig. 3H) 3′UTR activity was stronger than that of miR-149 (∼30% and 50% reduction, respectively). Given the high density of predicted binding sites for miR-149 and miR-149* in both FGFR1 and GPC1 3′UTRs (supplementary material Fig. S1B), we decided to study in detail only the sites with the strongest mirSVR score provided at http://www.mircrorna.org (Bartel, 2009; Betel et al., 2010; Betel et al., 2008), which are indicated in bold in supplementary material Fig. S1B. Importantly, mutation of sites 1 and 2 for miR-149 in the FGFR1 and GPC1 3′UTRs significantly relieved the effect of miR-149 on luciferase activity (Fig. 2E,G, respectively). Mutations of sites 3, 4 and 5 for miR-149* abrogated the repression of FGFR1 and GPC1 3′UTR activity (Fig. 2F,H) by miR-149*. Taken together, these data are consistent with a direct interaction of miR-149 or miR-149* with the FGFR1 and GPC1 3′UTRs, in line with the observed effects at the protein level (Fig. 2A,B).
Fig. 3.

miR-149 and miR-149* modulate FGF2 signaling and the angiogenic phenotype in ECs. (A) Western blot analysis of the phosphorylated ERK (p-ERK) and total ERK (t-ERK) content in BAECs transfected for 48 h with control mimic, miR-149 or miR-149*, starved for 12 h and then treated with FGF2 (25 ng/ml) for the indicated times. The FGFR1 blot is shown as a control of miR-149 and miR-149* action. HSP90 is used as a loading control. Data are expressed as the fold activation over control mimic. One representative experiment out of four independent experiments is shown. (B) Cell count of HUVECs transfected for 48 h with control mimic, miR-149 or miR-149*. Cells were then harvested and counted. Data are expressed as relative to the number of cells transfected with control mimic and correspond to the mean±s.e.m. of three experiments performed in duplicate. HUVECs were transfected for 48 h with (C,E) control mimic, miR-149 or miR-149*, or transfected with (D,F) CI, I-miR-149 or I-miR-149*. (C,D) Migration in response to FGF2 (25 ng/ml) was quantified as described in the Materials and Methods section. Data are expressed as the mean number of migrated cells and correspond to the mean±s.e.m. of three experiments performed in duplicate. (E,F) Cells were counted and seeded on growth-factor-reduced Matrigel. The cumulative sprout length of capillary-like structures formed in response to 0.1% BSA (basal) or FGF2 (25 ng/ml) was quantified as described in the Materials and Methods section. Representative micrographs and quantification are shown. Scale bars: 100 µm. The data are expressed as cumulative cord length and correspond to the mean±s.e.m. of three experiments performed in duplicate. *P≤0.05 compared with cells transfected with control mimic or CI; #P≤0.05 compared with the basal condition; and §P≤0.05 compared with cells transfected with control mimic and treated with FGF2.
Role of miR-149 in the angiogenic capacity of ECs
Considering that both GPC1 and FGFR1 are necessary for the mitogenic effects of FGF in several cell types (Fico et al., 2011; Filmus et al., 2008; Gerwins et al., 2000; Iozzo and Sanderson, 2011; Presta et al., 2005; Qiao et al., 2003; Su et al., 2006), and that both miR-149 and miR-149* regulate the protein levels of GPC1 and FGFR1 in ECs, it was of interest to determine whether ERK1 and ERK2 activation was attenuated in ECs transfected with either miR-149 or miR-149*. As seen in Fig. 3A, ERK1/2 phosphorylation was reduced in response to FGF2 stimulation in ECs overexpressing miR-149 (Fig. 3A, left panel) or miR-149* (Fig. 3A, right panel). Taken together, these data suggest that these miRNAs control the angiogenic responses of ECs by regulating FGF2 signaling through GPC1 and FGFR1.
In order to address whether miR-149 and miR-149* participate in the regulation of angiogenic functions of ECs in vitro, we studied their effects on proliferation, migration and cord formation. First, we tested the effect of miR-149 or miR-149* on EC proliferation by counting the cells (Fig. 3B). Both miRNAs reduced the number of cells. This effect was not due to increased apoptosis but due to a cell cycle delay, as indicated by a diminished incorporation of BrdU (supplementary material Fig. S2E). These results are in agreement with a previous report that has shown that miR-149 overexpression reduces the proliferation of a glioblastoma cell line (Li et al., 2011) and indirectly agree with reports that indicate that GPC1 levels are important for EC proliferation and cell cycle progression (Qiao et al., 2008). We next assessed whether miR-149 or miR-149* could affect the migratory and morphogenic capacities of ECs in response to FGF2. Notably, the overexpression of both miRNAs reduced both basal and FGF2-induced migration and cord formation of ECs (Fig. 3C,E). Importantly, inhibition of the endogenous levels of both miRNAs increased migration (Fig. 3D) and cord formation (Fig. 3F) of ECs in both basal and FGF2-stimulated conditions. Taken together, these data indicate that both miRNAs regulate EC angiogenic responses, and are consistent with their targeting activity on FGFR1 and GPC1. We additionally studied the role of miR-149 in tumor-induced neovascularization. As expected, the expression of miR-149 was increased in tumors injected with miR-149-overexpressing lentiviral particles (Fig. 4C). Interestingly, and in agreement with the previously described effects of miR-149 during the angiogenic responses of ECs, a reduction of platelet endothelial cell adhesion molecule 1 (PECAM-1)-positive structures (in cross-sections; Fig. 4D) in the tumor-associated vasculature was observed, as well as an overall reduction of tumor growth (volume and weight) (Fig. 4A,B). These data suggest that the function of miR-149 in the angiogenic vessels is important for optimal tumor angiogenesis and tumor growth.
Fig. 4.
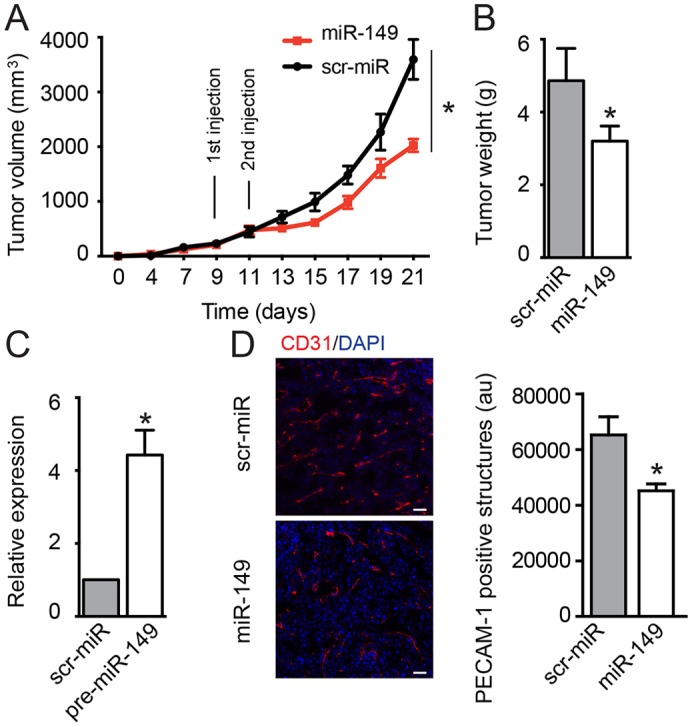
miR-149 reduces tumor-induced neovascularization. LLC cells were implanted subcutaneously into the dorsal flank of wild-type mice. On days 9 and 11, scrambled-miR (scr-miR) or miR-149 lentiviral particles were intratumorally delivered into groups of six animals and (A) tumor growth was assessed. After 21 days, animals were euthanized and tumor tissues were collected and (B) weighed. Data are expressed as mean±s.e.m. *P≤0.05. (C) qRT-PCR analysis of precursor miR-149 in tumors. The data are shown as the expression relative to scr-miR tumors and correspond to mean±s.e.m. *P≤0.05. (D) Representative PECAM-1 staining (red) of cross-sections from tumors. Tissues were counterstained with DAPI (blue). Capillary structures are expressed as PECAM-1 positive and correspond to mean±s.e.m.; *P≤0.05; a.u., arbitrary units. Scale bars: 100 µm.
Regulation of miR-149 expression in ECs
Recent reports have shown that pro-angiogenic cytokines can regulate the expression of miRNAs (Chamorro-Jorganes et al., 2011; Suárez et al., 2008; Wang and Olson, 2009). Therefore, we asked whether FGF2 could regulate miR-149 expression, thus providing negative feedback upon FGF2 stimulation by regulating the production of FGFR1 and GPC1. For this purpose, ECs were treated with FGF2 at the times indicated and the expression of pri-miR-149, its host gene (GPC1) and mature miR-149 (both guide and passenger strands) was measured (Fig. 5A,B). The genes encoding plasminogen activator inhibitor type 2 (PAI-2, also known as SERPINB2) and AXL receptor tyrosine kinase (AXL) were used as controls for FGF2 action (supplementary material Fig. S2F) because the expression of these genes in ECs has been shown to be induced or inhibited, respectively, upon FGF2 treatment (Jih et al., 2001). We found that FGF2 induced the expression of pri-miR-149 as early as 2 h after treatment, and that this expression peaked at 4 h and gradually reduced to basal levels by 16 h after treatment. By contrast, the mRNA level of the host gene, GPC1, remained unaffected for the first 6 h and then reduced at 8 and 16 h after treatment (Fig. 5A). Consistent with pri-miR-149 induction, the levels of mature miR-149 and miR-149* were also induced upon FGF2 treatment and peaked at 6 h (Fig. 5B). These results were further corroborated by northern blot analysis (Fig. 5C). To investigate whether FGF2 induced the expression of pri-miR-149, ECs were pre-treated with the transcription inhibitor actinomycin D prior to FGF2 stimulation. As shown in Fig. 5D, the level of pri-miR-149 was not increased when cells were pretreated with actinomycin D. In addition, the mRNA level of GPC1 remained unaffected (Fig. 5D). These data suggest that miR-149 is regulated at the transcriptional level by FGF2 independently of its host gene and support the idea that miR-149 has its own independent promoter within the first intron of GPC1. A previous report has shown that there is a predicted transcriptional start site (TSS) located at the 5′ end of validated expressed-sequence tags (ESTs) within 3.1 kb upstream of the miR-149 sequence. In addition, a computational algorithm has identified CpG islands located at the predicted TSS (Monteys et al., 2010). Using the ENCODE Broad Chromatin State Segmentation Track (Ernst and Kellis, 2010) to identify the potential promoter region and/or regulatory site(s) involved in the regulation of the expression of miR-149 by FGF2, we identified three regions with different chromatin states in HUVECs (supplementary material Fig. S1C). Specifically, a repressed region (indicated as region 1), a poised promoter (indicated as region 2) and a second repressed region (indicated as region 3), located 2.5 kb, 3.6 kb and 4.3 kb, respectively, upstream of the miRNA sequence were cloned in a pGL3 promoter vector. As shown in Fig. 5E, at 2 h after FGF2 stimulation only Region 3 showed an increase in luciferase activity. Interestingly, in our in vitro system Region 1 (proximal to miR-149) showed a higher basal luciferase activity than Region 3 (Fig. 5E), thereby suggesting that Region 1 maintains some degree of basal promoter activity and that Region 3 is activated upon FGF2 stimulation.
Fig. 5.

FGF modulates miR-149 and miR-149* expression in ECs. (A–C) HUVECs were starved for 12 h and then treated with FGF2 (25 ng/ml) or left untreated for the indicated times. qRT-PCR analysis of (A) GPC1 and pri-miR-149 expression or (B) miR-149 and miR-149* expression. Data are shown as expression relative to non-treated cells and correspond to mean±s.e.m. of five experiments. (C) Northern blotting of pri-miR-149, miR-149 and miR-149* expression. The inset corresponds to miR-149 expression and an overexposed blot is shown below. 5S rRNA is used as loading control. (D) HUVECs were starved for 12 h and then treated for 2 h with actinomycin D (4 ug/ml) prior to FGF2 (25 ng/ml) stimulation for the times indicated. qRT-PCR analysis of GPC1 and pri-miR-149 expression. Data are shown as expression relative to non-treated cells and correspond to mean±s.e.m. of three experiments. (E) Luciferase activity in HUVECs transfected for 5 h with different promoter regions (supplementary material Fig. S1C). Data are shown as luciferase activity relative to control samples transfected with pGL3 promoter vector in basal conditions, and correspond to the mean±s.e.m. of three experiments performed in triplicate. *P≤0.05 compared with cells transfected with the same promoter region upon FGF stimulation. (F) HUVECs were transfected for 48 h with a CI or I-miR-149 plus I-miR-149*, then cells were starved for 12 h and treated with FGF2 (25 ng/ml) as indicated. qRT-PCR analysis of GPC1 and FGFR1 expression. Data are shown as expression relative to non-treated cells and correspond to mean±s.e.m. of three experiments. *P≤0.05 compared with non-treated cells; and #P≤0.05 compared with cells transfected with CI and treated with FGF2.
Taken together, these data indicate that FGF2 stimulates the transcription of miR-149 without inducing that of its host gene. Indeed, GPC1 mRNA and protein levels decreased after 8–16 h of FGF2 stimulation (Fig. 5A and supplementary material Fig. S2G, respectively). These data suggest a possible downregulation of the host gene by FGF2-induced miR-149 and/or miR-149*, through direct targeting as shown above (Fig. 2). Interestingly, a similar effect was observed when we analyzed FGFR1 expression after FGF2 treatment (supplementary material Fig. S2H). To verify whether the downregulation of GPC1 and FGFR1 was mediated by miR-149 and miR-149* expression, we inhibited both miRNAs prior to FGF2 treatment. As shown in Fig. 5F, the mRNA levels of GPC1 and FGFR1 were decreased upon FGF2 treatment when cells were transfected with control inhibitor. By contrast, transfection with the inhibitors of miR-149 and miR-149* before FGF2 stimulation prevented the miR-149-mediated downregulation of GPC1 and FGFR1 (Fig. 5F). Taken together, these findings suggest that FGF stimulates the expression of miR-149 to fine tune the levels of FGFR1 and GPC1 and to assure the steady state level of FGF2-induced responses in ECs, thus providing a negative-feedback regulation to this pathway (Fig. 6).
Fig. 6.

A summary of the negative-feedback loop by which miR-149 and miR-149* regulate the cellular response to FGF. (A) GPC1 stabilizes the binding of FGF2 to its receptor, FGFR1, potentiating FGF signaling. (B) Upon FGF2 stimulation, the transcription of miR-149 is increased. (C) pri-miR-149 is processed to pre-miR-149 and translocates into the cytoplasm where it is further processed into duplex miR-149. (D) Mature miR-149/149* are loaded into the RISC complex and subsequently can mediate the downregulation of their target genes, FGFR1 and GPC1. (E) As a consequence, the expression of the FGFR–GPC1 complex is diminished. This negative feedback loop decreases the angiogenic endothelial cell response to FGF.
DISCUSSION
FGF2 classically transmits its signals through the high-affinity tyrosine kinase receptor FGFR1, thereby stimulating a wide range of EC functions including migration, proliferation, differentiation and survival. However, recent evidence strongly implicates other cell-surface FGFR-interacting proteins as important players in FGF signaling. In this regard, HSPGs, including GPC1, have been shown to facilitate FGF–FGFR binding and stabilization of the receptor–ligand complex in several cell types including ECs (Elfenbein et al., 2012; Qiao et al., 2003; Su et al., 2006). GPC1 has been shown to act as a co-receptor of many angiogenic factors, including FGF, and it is upregulated in different cancers, as well as in ECs isolated from human gliomas, where it enhances FGF signaling. The expression of GPC1 in both tumor cells and host ECs appears to contribute to tumor growth and angiogenesis. In this regard, ECs from gliomas express high levels of GPC1, which contributes to the angiogenic responses of ECs, including proliferation. Interestingly, miR-149 is located within the first intron of the GPC1 gene and, in contrast to its host gene, its expression is reduced in several human malignancies and it is thought to play a role as a tumor suppressor. In this work, we investigated the functional relationship between the miRNA host gene (GPC1) and putative targets of the corresponding intronic miRNA (miR-149) in the context of ECs.
There is an increasing body of evidence that suggests that the miRNA* (also known as the passenger strand) also has important regulatory activity. Therefore, we analyzed the potential targeting activity of both strands of miR-149. In this regard, recent reports suggest that, depending on the tissue, miRNA*s can have a more prominent functional role than previously assumed (Yang et al., 2011). We have found that both strands of miR-149 control a complex network of genes that are involved in the regulation of angiogenesis; genes that have roles in processes including proliferation, cell migration and adhesion, cell differentiation and morphogenesis. Interestingly, the two strands showed an overall different pattern of expression, which is in agreement with the described tissue-dependent mechanisms of strand selection (Okamura et al., 2008; Ro et al., 2007). However, we found that in addition to brain, miR-149 and miR-149* were highly co-expressed in EC-enriched tissues, such as lung and the well-accepted angiogenic tissue, placenta (Reynolds and Redmer, 2001). Even though miR-149 and miR-149* do not have the same seed sequence, they share 61 out of the 97 and 96 predicted targets involved in angiogenesis, respectively. When both strands are coexpressed in ECs, this is likely to enrich the heterogeneity and the targeting activity of miRNA-mediated repression of the angiogenic pathway, therefore promoting cooperative silencing activity. Among the predicted targets identified for miR-149 and miR-149*, of special interest was the identification of FGFR1 and GPC1 as targets for both strands of miR-149. In fact, the 3′UTR of FGFR1 and GPC1 showed high enrichment in predicted sites for both miR-149 and miR-149*. In this regard, it is well accepted that the strength of repression by a given miRNA is dependent on the density of binding sites within a 3′UTR (Bartel, 2009; Hon and Zhang, 2007). Interestingly, our data show that both miR-149 and miR-149* regulate the expression of FGFR1 and GPC1 by targeting the FGFR1 and GPC1 3′UTR. Consistent with this targeting activity, miR-149 and miR-149* negatively regulate FGF2 signaling. This involves a reduced activation of mitogen-activated protein kinase kinase (MEK), with a consequent diminished phosphorylation of extracellular signal-regulated kinases (ERK1/2) (Gerwins et al., 2000; Presta et al., 2005). In agreement with our results, other groups have demonstrated that a reduction in FGFR1 levels in ECs impairs the angiogenic responses to FGF2 (Gerwins et al., 2000; Presta et al., 2005). Additionally, GPC1 is responsible for maintaining cell surface FGF2 at levels that are sufficient to activate FGFR1, ensuring the activation of target genes. In the absence of GPC1, FGF2 levels at the cell surface are reduced and the activity of signal transduction pathways is diminished. Conversely, when GPC1 is overexpressed and FGF2 levels at the cell surface remain elevated (upon angiogenic stimulation), the activation of the FGFR1 pathway is increased. However, in this scenario the signaling activity might also be reduced if FGF2 is sequestered from FGFR1 by the increased levels of GPC1 at the cell surface (Fico et al., 2011; Qiao et al., 2003; Su et al., 2006). Interestingly, in the context of ECs, the knockdown of GPC1 inhibits cell growth, whereas its overexpression can either promote cell proliferation or disrupt cell cycle progression, depending on the expression level of GPC1 (Qiao et al., 2012; Qiao et al., 2008). Taken together, this suggests that the levels of GPC1 need to be tightly regulated in order to ensure the adequate control of cell cycle progression in ECs.
Our data suggest that both miR-149 and miR-149* participate in the regulation of EC proliferation in response to FGF by fine tuning the levels of GPC1 as well as FGFR1. In another scenario, miR-149 has been reported to inhibit proliferation and cell cycle progression by targeting ZBTB2, which is a repressor of the ARF–HDM2–p53–p21 pathway in gastric cancer cells (Wang et al., 2012). Moreover, miR-149* has been reported to negatively regulate AKT1 and E2F1 expression, thereby inducing apoptosis in human cancer cells (Lin et al., 2010). However, in the context of ECs we have not observed any effect on apoptosis.
Intronic miRNAs can be co-transcribed with their host gene [e.g. miR-208 and miR-33 are encoded in an intron of αMHC (van Rooij et al., 2007) or SREBP (Rayner et al., 2010), respectively]. This host-miRNA co-expression can have a specific functional role. In fact, an intronic miRNA can support the function of its host gene by silencing genes that are functionally antagonistic to the host, or can more generally act synergistically with the host by coordinating the expression of genes with related functions (Lutter et al., 2010). Other studies also indicate that intronic miRNAs can directly participate in the regulation of the expression of their host gene [e.g. the regulation of EGFL7 by its intronic miRNA miR-126 (Nikolic et al., 2010), and the regulation of ARPP-21 by miR-128b (Megraw et al., 2010)], providing negative feedback regulation (Lutter et al., 2010). By contrast, there are interesting examples that indicate that intronic miRNAs can also be transcribed from their own regulatory elements (e.g. miR-21 is encoded by an intron of TMEM49 gene) (Löffler et al., 2007). Indeed, independent expression from intronic promoters could be one of the molecular mechanisms that explains why the expression of the miRNA and the host gene does not always correlate (Lutter et al., 2010; Monteys et al., 2010). In our case, the low correlation in the expression profiles of miR-149 (both mature and primary transcripts) and its host gene in a panel of human tissues and cell lines was indicative of independent transcriptional regulation. Indeed, the region upstream of miR-149 contains three different chromatin states in HUVECs. Interestingly, in other cell types the chromatin state of these regions was different, which might explain the differences in miR-149 expression pattern between different cell types. We found that FGF2 stimulates the promoter activity of miR-149, thereby inducing the expression of its primary transcript without inducing the expression of its host gene. A recent report shows a melanoma-specific upregulation of both miR-149* and the host gene by p53 under endoplasmic reticulum (ER) stress (Jin et al., 2011). Although the transcriptional activation of GPC1 by p53 is experimentally supported, the authors only measure the levels of the mature miR-149*, therefore they cannot determine whether the miR-149* induction was due to ER stress-induced processing and/or stabilization of miR-149*. In fact, p53 has recently been demonstrated to interact with the Drosha miRNA processing complex, thus facilitating the processing of primary miRNAs to precursor miRNAs (Suzuki et al., 2009). In any case, our results indicate that the induction of miR-149 by FGF is additionally contributing to the overall inverse expression observed between miR-149 and GPC1. In this regard, we have previously shown that this type of miRNA-mediated regulation is important to fine tune both inflammatory (Suárez et al., 2010) and angiogenic responses of ECs (Chamorro-Jorganes et al., 2011), thereby participating in the maintenance of steady-state conditions in ECs (Inui et al., 2010).
The timely termination of the angiogenic response is as important as its initiation. A persistent or exaggerated angiogenic growth leads to detrimental effects (Yancopoulos et al., 2000). Therefore, in accordance with the complex and highly coordinated activation phase, negative regulatory processes have evolved and function at multiple levels to impose an appropriate termination. Diverse mechanisms operate in ECs to shutdown the activity of the proangiogenic signaling pathway. Several lines of evidence indicate that the regulation of miRNA levels by different stimuli might contribute to the regulation of the specific stimulus-induced responses (Inui et al., 2010).
Our present findings show that FGF2 stimulates the expression of intronic miR-149 independently of its host gene to provide a finely tuned regulation of GPC1 and FGFR1, co-receptor and receptor for FGF2, respectively, thereby assuring the steady state of FGF2-induced responses in ECs (Fig. 6). As we improve our knowledge about these events, further molecular elucidation might provide novel strategies for therapeutic intervention of vascular growth.
MATERIALS AND METHODS
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from the tissue culture core laboratory of the Vascular Biology and Therapeutics program (Yale University, New Haven, CT) and were cultured on 0.1% gelatin (Sigma, St Louis, MO) in M199 containing 20% FBS and supplemented with endothelial cell growth supplement (ECGS) (BD Biosciences, San Jose, CA). Bovine aortic endothelial cells (BAECs) were purchased from Lonza (Allendale, NJ) and were cultured in DMEM containing 10% FBS. Human aortic endothelial cells (HAECs) were purchased from Lonza and were cultured in EBM-2. Human brain microvascular endothelial cells (HBMECs) were generated and kindly provided by Ana Rodriguez (New York University, NY) and cultured in ECM (Sciencell Research Laboratories, Carlsbad, CA). U251 cells were purchased from Sigma and cultured in DMEM containing 10% FBS.
microRNA transfection
ECs were transfected with 30 nM miRNA mimics (miR-149 and miR-149*) or 60 nM miRNA inhibitor (I-miR-149 and I-miR-149*) (Dharmacon, Lafayette, CO) using Oligofectamine (Invitrogen, Carlsbad, CA) for 48 h, as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007; Suárez et al., 2008; Suárez et al., 2010). All experimental control samples were treated with an equal concentration of a non-targeting control mimic sequence or an inhibitor negative control sequence (CI).
3′UTR and promoter luciferase reporter assays
The generation of FGFR1 3′UTR luciferase reporter plasmids was reported previously (Chamorro-Jorganes et al., 2011). A similar strategy was followed for the generation of the GPC1 3′UTR luciferase reporter vector. Point mutations in the seed region of the predicted miR-149 or miR-149* target sites within the 3′UTR of FGFR1 and GPC1 were generated using Multisite-QuikChange (Stratagene, La Jolla, CA) according to the manufacturer's protocol. Briefly, COS-7 cells were co-transfected with 0.5 µg of the indicated 3′UTR luciferase reporter vectors and miR-149, miR-149* or control mimic using Lipofectamine 2000 (Invitrogen) for 24 h.
The promoter regions were cloned into a pGL3 promoter vector (Promega). The primer sequences used were: repressed region (region 1), 5′-CCCTTCCTCTTCCGCCAGGCCCATCCC-3′ and 5′-CTAAGGTACCGGAATTTGGCGTGGACAGTCAGCC-3′; poised promoter (region 2), 5′-AGGTAGAGGATGTGGAATGAC-3′ and 5′-GGAGCGGAGGATGGG-3′; and repressed region (region 3), 5′-GCCTTACAGCCAAGACAC-3′ and 5′-TCCCTCTGACATACCAAACC-3′. HUVECs were co-transfected with 0.5 µg of the indicated constructs and 0.01 µg of Renilla luciferase reporter plasmid using Lipofectamine LTX (Invitrogen). After 5 h of transfection, the cells were washed with PBS and further incubated for 8 h in M199 medium supplemented with 20% FBS and ECGS. Then cells were starved for 12 h and treated with FGF2 (25 ng/ml) for the indicated times.
Luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega, Madison, WI). Renilla luciferase activity was normalized to the corresponding firefly luciferase activity and plotted as a percentage of the control (cells co-transfected with the corresponding concentration of control mimic).
Western blot analysis
Samples were prepared as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007; Suárez et al., 2008; Suárez et al., 2010). Western blots were performed using the rabbit polyclonal antibody against FGFR1 (1∶200) (Santa Cruz Biotechnology Inc., Texas, TX) and mouse monoclonal anti-heat shock protein 90 (HSP90) (1∶2000) (BD Biosciences). Protein bands were visualized using the Odyssey Infrared Imaging System (LI-COR Biotechnology). Densitometry analysis of the gels was carried out using ImageJ software from the NIH (http://rsbweb.nih.gov/ij/).
Quantitative real-time PCR
Total RNA was isolated using miRNeasy (Qiagen, Valencia, CA) according to the manufacturer's protocol. For mRNA quantification, cDNA was synthesized using the Taqman RT kit (Applied Biosystems, Austin, TX). Quantitative real-time PCR was performed using iQTM SYBR Green Supermix (BioRad, Hercules, CA). The mRNA level was normalized using 18S rRNA as a housekeeping gene. The primer sequences used were: hsa-FGFR1, 5′-ACCGTATGCCCGTAGCTCCA-3′ and 5′-GGGTCCCACTGGAAGGGCAT-3′; hsa-GPC1, 5′-AGCGACGTGGTCCGGAAAGT-3′ and 5′-CATGGAGTCCAGGAGGTTCCTCC-3′; hsa-PAI-2, 5′-ACTGGCTTGGAGCTGCTGGA-3′ and 5′-CCCGTCCCTTGTTGAAGGCG-3′; hsa-Axl, 5′-GGACACCCCAGAGGTGCTAATGGA-3′ and 5′-CAGCTGGTGGACTGGCTGTGC-3′; and hsa-18s, 5′-GCTTAATTTGACTCAACACGGGA-3′ and 5′-AGCTATCAATCTGTCAATCCTGTC-3′.
For miRNA quantification, total RNA was reverse transcribed using the RT2 miRNA First Strand kit (SABiosciences, Frederick, MD). Primers specific for human miR-149 (SABiosciences) were used, and the value was normalized to that of human SNORD38B as a housekeeping gene, as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2010). Primers specific for human miR-149* were used as described previously (Jin et al., 2011). For pri-miRNA quantification, cDNA was synthesized using the TaqMan RT kit (Applied Biosystems). Quantitative real-time PCR was performed using TaqMan Universal Master Mix (Applied Biosystems), as described previously. When described, human RNA samples were purchased from Life Technologies (Norwalk, CT).
Northern blot analysis of miRNAs
miRNA expression was assessed by northern blot as described previously (Suárez et al., 2007; Suárez et al., 2008; Suárez et al., 2010). Locked nucleic acid (LNA) miRNA detection probes were purchased from Exiqon (Woburn, MA). Oligonucleotides for hsa-miR-149, hsa-miR-149* and 5S rRNA were 5′-GGGAGTGAAGACACGGAGCCAGA-3′, 5′-GCACAGCCCCCGTCCCTCCCT-3′ and 5′-CAGGCCCGACCCTGCTTAGCTTCCGAGATCAGACGAGAT-3′, respectively.
Cell number assessment
After 48 h of miRNA transfection, ECs were collected and the cell number was assessed using a hemocytometer as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007; Suárez et al., 2008). Viability was determined by Trypan Blue exclusion.
Migration experiments
A modified Boyden chamber was used (Costar transwell inserts; Corning), as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007). After 48 h of miRNA transfection, ECs (7×104) were suspended in a 100-µl aliquot of DMEM containing 0.1% BSA and then cells were added to the upper chamber. DMEM containing 0.1% BSA and FGF2 (25 ng/ml) (Sigma) was added to the bottom chamber of the Boyden apparatus. After 5 h of incubation, the cells on both sides of the membrane were fixed and stained with a Diff-Quik staining kit (Baxter Healthcare, Miami, FL). The mean number of cells from five randomly chosen high-power (×20) fields on the lower side of the membrane of each well was counted.
Cord formation assay
After 48 h of miRNA transfection, ECs (7×104) were cultured in one well of a 24-well plate coated with 200 µl of growth-factor-reduced Matrigel (BD Biosciences), as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007; Suárez et al., 2008). The sprout length of capillary-like structures was imaged by using microscopy (Axiovert; Carl Zeiss MicroImaging), and the cumulative tube length was measured in three fields for each replicate per experiment.
Flow cytometry analysis
The analysis of cell cycle, apoptosis (sub-G1) and DNA synthesis rate was conducted as described previously (Chamorro-Jorganes et al., 2011; Suárez et al., 2007). Briefly, at different times after transfections, cells were incubated with 100 µM 5-bromo-2′-deoxyuridine (BrdU) (Sigma) for 4 h at 37°C, then fixed in 70% cold ethanol. After 20 min of incubation at room temperature with 2 M HCl, cells were washed with PBS and incubated for 15 min at room temperature in PBS, 0.5% Tween 20 and 1% normal goat serum. Subsequently, cells were centrifuged, resuspended in 0.5% Tween 20, 1% normal goat serum containing 20 µl of FITC-labeled anti-BrdU (BD Biosciences) for 1 hour at room temperature and then stained with PI. A total of 10,000 events per sample were acquired for data analysis using CellQuest software, with selective gating to exclude doublet cells. Apoptotic cells were determined by their hypochromic subdiploid staining profiles (sub-G1 population).
Cellular protein levels of GPC1 were determined by flow cytometry (Suárez et al., 2007). Briefly, cells were harvested and fixed with 2% paraformaldehyde for 15 min at room temperature. Then cells were washed in PBS and incubated with 0.1% of Triton X-100 for 7 min at 4°C. Non-specific binding was blocked by incubation with PBS containing 3% BSA for 20 min. The cells were stained with a rabbit polyclonal human glypican 1 biotinylated antibody (1∶100) (R&D Systems, Minneapolis, MN) for 1 h, washed with cold PBS and incubated with streptavidin–phycoerythrin (R&D Systems). Immunostained cells were then washed twice and analyzed on a FACSort using CellQuest software.
Tumor-induced neovascularization
Lewis lung carcinoma (LLC) cells (106) were injected subcutaneously into the dorsal flank of 8-week-old male mice as described (Suárez et al., 2008). When tumors became palpable, scrambled-miR (scr-pre-miR) or pre-miR-149 lentiviral particles (1.5×108) were delivered intratumorally. Tumor growth was assessed by measuring the length and width of the tumor using a caliper, and tumor volume was determined by the following formula: volume = 0.52×(width)2×(length). After 21 days, the animals were euthanized and tumor tissues were collected and frozen in optimal cutting temperature (OCT) compound and were subsequently immunostained with monoclonal anti-mouse PECAM-1 antibody (1∶200) (BD Biosciences). For each tumor, five images from three different sections were quantified. All animal experiments were approved by the Institutional Animal Care Committee of New York University Medical Center.
Statistical analysis
Data are expressed as mean±s.e.m. Statistical differences were measured by either the Student t-test or two-way ANOVA with Bonferroni correction for multiple comparisons when appropriate. A value of P≤0.05 was considered statistically significant. Data analysis was performed using the Prism program (Statistical Graphics).
Supplementary Material
Acknowledgments
The authors thank Maria del Valle Guijarro (New York University) for assistance with in vivo studies, Carlos Fernandez-Hernando (Yale University) for helpful comments, Leigh Goedeke (Yale University) for the editing work on this manuscript and Ana Rodriguez (New York University) for providing human brain microvascular endothelial cells.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
A.C.-J., E.A., N.R. and D.C.-S. performed experiments. A.C.-J., E.A. and Y.S. analyzed and interpreted the data. Y.S. conceived and designed the experiments. A.C.-J., E.A. and Y.S. wrote the manuscript.
Funding
This study was supported by a grant from the National Institutes of Health [grant number R01HL105945 to Y.S.]. E.A. is a Howard Hughes Medical Institute International Student Research Fellow. N.R. is supported by a postdoctoral fellowship from the Spanish Ministry (Programa Nacional de Movilidad de Recursos Humanos del Plan Nacional de I+D+i, 2008-2011). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.130518/-/DC1
References
- Aikawa T., Whipple C. A., Lopez M. E., Gunn J., Young A., Lander A. D., Korc M. (2008). Glypican-1 modulates the angiogenic and metastatic potential of human and mouse cancer cells. J. Clin. Invest. 118, 89–99 10.1172/JCI32412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., Koppal A., Agius P., Sander C., Leslie C. (2010). Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 10.1186/gb-2010-11-8-r90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., Wilson M., Gabow A., Marks D. S., Sander C. (2008). The microRNA.org resource: targets and expression. Nucleic Acids Res. 36, D149–D53 10.1093/nar/gkm995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N., Cohen S. M. (2007). microRNA functions. Annu. Rev. Cell Dev. Biol. 23, 175–205 10.1146/annurev.cellbio.23.090506.123406 [DOI] [PubMed] [Google Scholar]
- Caporali A., Emanueli C. (2011). MicroRNA regulation in angiogenesis. Vascul. Pharmacol. 55, 79–86 10.1016/j.vph.2011.06.006 [DOI] [PubMed] [Google Scholar]
- Carmeliet P. (2005). Angiogenesis in life, disease and medicine. Nature 438, 932–936 10.1038/nature04478 [DOI] [PubMed] [Google Scholar]
- Chamorro-Jorganes A., Araldi E., Penalva L. O., Sandhu D., Fernández-Hernando C., Suárez Y. (2011). MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler. Thromb. Vasc. Biol. 31, 2595–2606 10.1161/ATVBAHA.111.236521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross M. J., Claesson-Welsh L. (2001). FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 22, 201–207 10.1016/S0165-6147(00)01676-X [DOI] [PubMed] [Google Scholar]
- Elfenbein A., Lanahan A., Zhou T. X., Yamasaki A., Tkachenko E., Matsuda M., Simons M. (2012). Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci. Signal. 5, ra36 10.1126/scisignal.2002495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2010). Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 10.1038/nbt.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fico A., Maina F., Dono R. (2011). Fine-tuning of cell signaling by glypicans. Cell. Mol. Life Sci. 68, 923–929 10.1007/s00018-007-7471-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J., Capurro M., Rast J. (2008). Glypicans. Genome Biol. 9, 224 10.1186/gb-2008-9-5-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. (1984). What is the role of endothelial cells in angiogenesis? Lab. Invest. 51, 601–604 [PubMed] [Google Scholar]
- Gengrinovitch S., Berman B., David G., Witte L., Neufeld G., Ron D. (1999). Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J. Biol. Chem. 274, 10816–10822 10.1074/jbc.274.16.10816 [DOI] [PubMed] [Google Scholar]
- Gerwins P., Sköldenberg E., Claesson-Welsh L. (2000). Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Crit. Rev. Oncol. Hematol. 34, 185–194 10.1016/S1040-8428(00)00062-7 [DOI] [PubMed] [Google Scholar]
- Hon L. S., Zhang Z. (2007). The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol. 8, R166 10.1186/gb-2007-8-8-r166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui M., Martello G., Piccolo S. (2010). MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 11, 252–263 10.1038/nrm2868 [DOI] [PubMed] [Google Scholar]
- Iozzo R. V., Sanderson R. D. (2011). Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 15, 1013–1031 10.1111/j.1582-4934.2010.01236.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62 10.1126/science.1104819 [DOI] [PubMed] [Google Scholar]
- Jih Y. J., Lien W. H., Tsai W. C., Yang G. W., Li C., Wu L. W. (2001). Distinct regulation of genes by bFGF and VEGF-A in endothelial cells. Angiogenesis 4, 313–321 10.1023/A:1016080321956 [DOI] [PubMed] [Google Scholar]
- Jin L., Hu W. L., Jiang C. C., Wang J. X., Han C. C., Chu P., Zhang L. J., Thorne R. F., Wilmott J., Scolyer R. A. et al. (2011). MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc. Natl. Acad. Sci. USA 108, 15840–15845 10.1073/pnas.1019312108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J., Ishiwata T., Kumbasar A., Friess H., Büchler M. W., Lander A. D., Korc M. (1998). The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Invest. 102, 1662–1673 10.1172/JCI4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J., Wildi S., Kumbasar A., Friess H., Lander A. D., Korc M. (1999). Stable transfection of a glypican-1 antisense construct decreases tumorigenicity in PANC-1 pancreatic carcinoma cells. Pancreas 19, 281–288 10.1097/00006676-199910000-00009 [DOI] [PubMed] [Google Scholar]
- Li D., Chen P., Li X. Y., Zhang L. Y., Xiong W., Zhou M., Xiao L., Zeng F., Li X. L., Wu M. H. et al. (2011). Grade-specific expression profiles of miRNAs/mRNAs and docking study in human grade I-III astrocytomas. OMICS 15, 673–682 10.1089/omi.2011.0064 [DOI] [PubMed] [Google Scholar]
- Lin R. J., Lin Y. C., Yu A. L. (2010). miR-149* induces apoptosis by inhibiting Akt1 and E2F1 in human cancer cells. Mol. Carcinog. 49, 719–727 [DOI] [PubMed] [Google Scholar]
- Löffler D., Brocke-Heidrich K., Pfeifer G., Stocsits C., Hackermüller J., Kretzschmar A. K., Burger R., Gramatzki M., Blumert C., Bauer K. et al. (2007). Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110, 1330–1333 10.1182/blood-2007-03-081133 [DOI] [PubMed] [Google Scholar]
- Lutter D., Marr C., Krumsiek J., Lang E. W., Theis F. J. (2010). Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genomics 11, 224 10.1186/1471-2164-11-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K., Maruyama H., Guo F., Kleeff J., Itakura J., Matsumoto Y., Lander A. D., Korc M. (2001). Glypican-1 is overexpressed in human breast cancer and modulates the mitogenic effects of multiple heparin-binding growth factors in breast cancer cells. Cancer Res. 61, 5562–5569 [PubMed] [Google Scholar]
- Megraw M., Sethupathy P., Gumireddy K., Jensen S. T., Huang Q. H., Hatzigeorgiou A. G. (2010). Isoform specific gene auto-regulation via miRNAs: a case study on miR-128b and ARPP-21. Theor. Chem. Acc. 125, 593–598 10.1007/s00214-009-0647-4 [DOI] [Google Scholar]
- Mi H., Dong Q., Muruganujan A., Gaudet P., Lewis S., Thomas P. D. (2010). PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–D210 10.1093/nar/gkp1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteys A. M., Spengler R. M., Wan J., Tecedor L., Lennox K. A., Xing Y., Davidson B. L. (2010). Structure and activity of putative intronic miRNA promoters. RNA 16, 495–505 10.1261/rna.1731910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic I., Plate K. H., Schmidt M. H. (2010). EGFL7 meets miRNA-126: an angiogenesis alliance. J. Angiogenes Res. 2, 9 10.1186/2040-2384-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K., Phillips M. D., Tyler D. M., Duan H., Chou Y. T., Lai E. C. (2008). The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat. Struct. Mol. Biol. 15, 354–363 10.1038/nsmb.1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006). VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- Øster B., Linnet L., Christensen L. L., Thorsen K., Ongen H., Dermitzakis E. T., Sandoval J., Moran S., Esteller M., Hansen T. F. et al. (2013). Non-CpG island promoter hypomethylation and miR-149 regulate the expression of SRPX2 in colorectal cancer. Int. J. Cancer 132, 2303–2315 [DOI] [PubMed] [Google Scholar]
- Presta M., Dell'Era P., Mitola S., Moroni E., Ronca R., Rusnati M. (2005). Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 16, 159–178 10.1016/j.cytogfr.2005.01.004 [DOI] [PubMed] [Google Scholar]
- Qiao D., Meyer K., Mundhenke C., Drew S. A., Friedl A. (2003). Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J. Biol. Chem. 278, 16045–16053 10.1074/jbc.M211259200 [DOI] [PubMed] [Google Scholar]
- Qiao D., Yang X., Meyer K., Friedl A. (2008). Glypican-1 regulates anaphase promoting complex/cyclosome substrates and cell cycle progression in endothelial cells. Mol. Biol. Cell 19, 2789–2801 10.1091/mbc.E07-10-1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao D., Meyer K., Friedl A. (2012). Glypican-1 stimulates Skp2 autoinduction loop and G1/S transition in endothelial cells. J. Biol. Chem. 287, 5898–5909 10.1074/jbc.M111.325282 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rayner K. J., Suárez Y., Dávalos A., Parathath S., Fitzgerald M. L., Tamehiro N., Fisher E. A., Moore K. J., Fernández-Hernando C. (2010). MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573 10.1126/science.1189862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds L. P., Redmer D. A. (2001). Angiogenesis in the placenta. Biol. Reprod. 64, 1033–1040 10.1095/biolreprod64.4.1033 [DOI] [PubMed] [Google Scholar]
- Ro S., Park C., Young D., Sanders K. M., Yan W. (2007). Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 35, 5944–5953 10.1093/nar/gkm641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasisekharan R., Shriver Z., Venkataraman G., Narayanasami U. (2002). Roles of heparan-sulphate glycosaminoglycans in cancer. Nat. Rev. Cancer 2, 521–528 10.1038/nrc842 [DOI] [PubMed] [Google Scholar]
- Su G., Meyer K., Nandini C. D., Qiao D., Salamat S., Friedl A. (2006). Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am. J. Pathol. 168, 2014–2026 10.2353/ajpath.2006.050800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez Y., Sessa W. C. (2009). MicroRNAs as novel regulators of angiogenesis. Circ. Res. 104, 442–454 10.1161/CIRCRESAHA.108.191270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez Y., Fernández-Hernando C., Pober J. S., Sessa W. C. (2007). Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res. 100, 1164–1173 10.1161/01.RES.0000265065.26744.17 [DOI] [PubMed] [Google Scholar]
- Suárez Y., Fernández-Hernando C., Yu J., Gerber S. A., Harrison K. D., Pober J. S., Iruela-Arispe M. L., Merkenschlager M., Sessa W. C. (2008). Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl. Acad. Sci. USA 105, 14082–14087 10.1073/pnas.0804597105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez Y., Wang C., Manes T. D., Pober J. S. (2010). Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J. Immunol. 184, 21–25 10.4049/jimmunol.0902369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H. I., Yamagata K., Sugimoto K., Iwamoto T., Kato S., Miyazono K. (2009). Modulation of microRNA processing by p53. Nature 460, 529–533 10.1038/nature08199 [DOI] [PubMed] [Google Scholar]
- Thum T. (2012). MicroRNA therapeutics in cardiovascular medicine. EMBO Mol. Med. 4, 3–14 10.1002/emmm.201100191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C., Kuehbacher A., Dimmeler S. (2008). Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 79, 581–588 10.1093/cvr/cvn156 [DOI] [PubMed] [Google Scholar]
- van Rooij E., Sutherland L. B., Qi X., Richardson J. A., Hill J., Olson E. N. (2007). Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316, 575–579 10.1126/science.1139089 [DOI] [PubMed] [Google Scholar]
- Wang S., Olson E. N. (2009). AngiomiRs – key regulators of angiogenesis. Curr. Opin. Genet. Dev. 19, 205–211 10.1016/j.gde.2009.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zheng X., Zhang Z., Zhou J., Zhao G., Yang J., Xia L., Wang R., Cai X., Hu H. et al. (2012). MicroRNA-149 inhibits proliferation and cell cycle progression through the targeting of ZBTB2 in human gastric cancer. PLoS ONE 7, e41693 10.1371/journal.pone.0041693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Ma Y. L., Zhang P., Shen T. Y., Shi C. Z., Yang Y. Z., Moyer M. P., Zhang H. Z., Chen H. Q., Liang Y. et al. (2013). SP1 mediates the link between methylation of the tumour suppressor miR-149 and outcome in colorectal cancer. J. Pathol. 229, 12–24 10.1002/path.4078 [DOI] [PubMed] [Google Scholar]
- Yancopoulos G. D., Davis S., Gale N. W., Rudge J. S., Wiegand S. J., Holash J. (2000). Vascular-specific growth factors and blood vessel formation. Nature 407, 242–248 10.1038/35025215 [DOI] [PubMed] [Google Scholar]
- Yang J. S., Phillips M. D., Betel D., Mu P., Ventura A., Siepel A. C., Chen K. C., Lai E. C. (2011). Widespread regulatory activity of vertebrate microRNA* species. RNA 17, 312–326 10.1261/rna.2537911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Coomans C., David G. (2001). Membrane heparan sulfate proteoglycan-supported FGF2-FGFR1 signaling: evidence in support of the “cooperative end structures” model. J. Biol. Chem. 276, 41921–41929 10.1074/jbc.M106608200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.