

ABSTRACT
Centriole biogenesis depends on the polo-like kinase (PLK4) and a small group of structural proteins. The spatiotemporal regulation of these proteins at pre-existing centrioles is essential to ensure that centriole duplication occurs once per cell cycle. Here, we report that phosphatidylinositol 4-phosphate 5-kinase type-1 gamma (PIP5K1C, hereafter referred to as PIPKIγ) plays an important role in centriole fidelity. PIPKIγ localized in a ring-like pattern in the intermediate pericentriolar materials around the proximal end of the centriole in G1, S and G2 phases, but not in M phase. This localization was dependent upon an association with centrosomal protein of 152 KDa (CEP152). Without detaining cells in S or M phase, the depletion of PIPKIγ led to centriole amplification in a manner that was dependent upon PLK4 and spindle assembly abnormal protein 6 homolog (SAS6). The expression of exogenous PIPKIγ reduced centriole amplification that occurred as a result of endogenous PIPKIγ depletion, hydroxyurea treatment or PLK4 overexpression, suggesting that PIPKIγ is likely to function at the PLK4 level to restrain centriole duplication. Importantly, we found that PIPKIγ bound to the cryptic polo-box domain of PLK4 and that this binding reduced the kinase activity of PLK4. Together, our findings suggest that PIPKIγ is a novel negative regulator of centriole duplication, which acts by modulating the homeostasis of PLK4 activity.
KEY WORDS: Centriole duplication, PLK4, CEP152, CEP192, phosphatidylinositol 4-phosphate 5-kinase type-1 gamma
INTRODUCTION
The centrosome, comprising one pair of centrioles and the surrounding pericentriolar material (PCM), is the major microtubule organization center and plays a crucial role in mitotic spindle assembly, primary ciliogenesis and cell morphogenesis (Debec et al., 2010; Nigg and Raff, 2009). In cycling cells, the centrosome duplicates once in S phase so that each daughter cell can inherit one centrosome after mitosis. The possession of an abnormal number of centrosomes results in mis-assembled spindles, and these frequently give rise to chromosome mis-segregation, genomic instability and aneuploid daughter cells (Ganem et al., 2009), linking centrosomal anomalies to many human diseases, including a variety of different aneuploid tumors (Ghadimi et al., 2000; Lingle et al., 2002; Nigg, 2006; Pihan et al., 1998; Yamamoto et al., 2004) and microcephaly syndromes (Megraw et al., 2011; Nigg and Raff, 2009). The procentriole is assembled by a small group of highly conserved proteins and is strictly limited to one per pre-existing centriole, with assembly occuring once per cell cycle. The availability and activity of these proteins at the centrosome have to be carefully controlled to maintain the fidelity of centriole and centrosome duplication (Bettencourt-Dias et al., 2005; Eckerdt et al., 2011; Kleylein-Sohn et al., 2007; Nigg, 2007; Nigg and Stearns, 2011).
It has been demonstrated that the polo-like kinase PLK4 is a key regulator of centriole formation and that excessive PLK4 activity leads to centriole amplification (Bettencourt-Dias et al., 2005; Eckerdt et al., 2011; Kleylein-Sohn et al., 2007; Nigg, 2007; Nigg and Stearns, 2011). PLK4 is recruited to the centrosome by CEP152 and controls the onset of centriole assembly together with this protein, which also provides a platform for centromere protein J (CENPJ or CPAP) to bind to the centrosome (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010). PLK4 can carry out both autophosphorylation and the phosphorylation of other substrates, thereby priming either itself or a substrate (such as FBXW5) for ubiquitinylation and destruction. In this way, PLK4 can control its own availability (Brownlee and Rogers, 2012; Cunha-Ferreira et al., 2009; Guderian et al., 2010; Holland et al., 2010; Rogers et al., 2009) or that of other components of the centriole duplication machinery (such as SAS6) (Puklowski et al., 2011), to restrict centriole number. By contrast, protein phosphatase 2 (PP2A) was reported to promote centriole formation by facilitating SAS6 accumulation at the nascent centriole (Brownlee et al., 2011; Kitagawa et al., 2011; Song et al., 2011). These results support the existence of an intrinsic important balance between kinase and phosphatase activities during centriole duplication and highlight the importance of PLK4 to the centriole assembly machinery. In spite of the importance of controlling PLK4 activity accurately, the underlying mechanism is far from fully understood.
Phosphatidylinositol 4-phosphate 5-kinase type-1 gamma (PIP5K1C, hereafter referred to as PIPKIγ) is a lipid kinase that phosphorylates phosphatidylinositol 4-phosphate to generate phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2]. To date, six alternatively spliced isoforms of PIPKIγ have been reported (Giudici et al., 2004; Giudici et al., 2006; Ishihara et al., 1998; Schill and Anderson, 2009; Xia et al., 2011). PIPKIγ isoforms are targeted to distinct subcellular sites. By regulating the dynamics of regional PI(4,5)P2 pools, they are implicated in distinct cellular processes such as vesicular trafficking (Bairstow et al., 2006; Ling et al., 2007; Sun et al., 2013; Thapa et al., 2012; Thieman et al., 2009; Xiong et al., 2012), calcium signaling (Wang et al., 2004) or cell adhesion and migration (Di Paolo et al., 2002; El Sayegh et al., 2007; Ling et al., 2007a; Ling et al., 2002; Sun et al., 2007; Thapa et al., 2012; Thieman et al., 2009; Wang et al., 2004; Xiong et al., 2012). Moreover, PIPKIγ can regulate some proteins, such as E-cadherin (Ling et al., 2007), by directly interacting with them. Here, we report that PIPKIγ isoform 3 (PIPKIγ_i3) is targeted to the centrosome and negatively regulates centriole duplication. This lipid kinase associated with the proximal ends of parental centrioles in a cell-cycle-dependent manner, just like many other centrosomal proteins. Similar to PLK4 and CPAP, the residence of PIPKIγ at the centrosome depended upon CEP152. We show that loss of PIPKIγ results in centriole amplification without blocking the cells in S or G2/M phase, indicating that PIPKIγ is a negative regulator of centriole duplication. Indeed, further investigation revealed that PIPKIγ directly interacts with PLK4 and inhibits its activity. These results suggest the existence of an additional level of regulation of PLK4 activity and centriole biogenesis, acting in combination with phosphorylation-dependent protein ubiquitinylation and degradation.
RESULTS
PIPKIγ localizes at the centrosome
Using previously characterized rabbit polyclonal antibodies that recognize all of the PIPKIγ isoforms, we observed that PIPKIγ localizes to multiple subcellular sites including focal adhesions, adherens junctions and recycling endosomes (Ling et al., 2007; Ling et al., 2002), as well as the centrosome. To investigate the unexpected centrosomal localization of PIPKIγ, we generated new rabbit polyclonal and monoclonal antibodies to ensure that this localization pattern was not an artifact from the previous antibody. Both the purified polyclonal antibodies (supplementary material Fig. S1B) and a monoclonal antibody against PIPKIγ (Fig. 1A, left panel) stained the centrosome, which is highlighted by the centrosomal marker centrin 2 in HeLa cells. By contrast, purified rabbit polyclonal antibodies specifically recognizing PIPKIα or PIPKIβ (supplementary material Fig. S1A) did not show staining around the centrosome (Fig. 1A). The centrosomal binding of anti-PIPKIγ antibody was completely abolished after treatment with a PIPKIγ siRNA that targeted all PIPKIγ isoforms (Fig. 1B), or in the presence of excess PIPKIγ but not PIPKIα protein (supplementary material Fig. S1B), supporting the authenticity of this PIPKIγ localization. In addition, we isolated centrosomes from HeLa cells (Fig. 1C) or mouse kidney (supplementary material Fig. S1C) using sucrose gradient centrifugation and analyzed the fractions by western blot. Similar to other centrosomal proteins that were identified using this method (Kaplan et al., 2004), PIPKIγ co-fractionated with the centrosome components human SAS6 (hereafter referred to HsSAS-6), CPAP and γ-tubulin – a result that further supported the hypothesis that PIPKIγ associates with centrosomes as suggested by immunofluorescence microscopy. Moreover, treatment with nocodazole, an inhibitor of microtubule polymerization, had no effect on PIPKIγ signal at the centrosome, although it efficiently disrupted microtubule integrity (supplementary material Fig. S1D), indicating that the association between PIPKIγ and the centrosome is independent of the microtubule cytoskeleton. These results demonstrated that PIPKIγ was a bona fide component of the centrosome. In addition to its presence in HeLa cells, we also observed PIPKIγ at the centrosome in human MDA-MB-231 and U-2 OS cells and in murine IMCD3 and NIH3T3 cells (supplementary material Fig. S1E and data not shown), suggesting a pervasive, potentially functional role of PIPKIγ at the centrosome.
Fig. 1.

PIPKIγ associates with the centrosome. (A) PIPKIγ, but not PIPKIα or PIPKIβ, is targeted to the centrosome. HeLa cells were fixed and stained with the indicated antibodies to visualize PIPKIγ, PIPKIα or PIPKIβ (green) along with centrin 2 (red). (B) siRNA-mediated depletion of PIPKIγ abolishes the centrosome signal recognized by anti-PIPKIγ antibody. HeLa cells were treated with nonspecific control (siNC) or PIPKIγ-specific (siPIPKIγ) siRNA and then stained with the indicated antibodies. Anti-γ-tubulin was used to label centrosomes. (C) PIPKIγ co-sedi-ments with centrosomes in 40–70% sucrose gradients. Centrosome fractions were isolated from HeLa cells and immunoblotted using the indicated antibodies. (D) HeLa cells were transfected with the HA-tagged full-length (HA–PIPKIγ), N-terminal fragment (HA–PIPKIγ 1-445) and C-terminal fragment (HA–PIPKIγ 445-End) of PIPKIγ_i3. Cells were then stained with anti-HA (green) and anti-centrin 2 (red) antibodies. (E) Schematic diagram comparing the sequence and centrosome targeting of known PIPKIγ isoforms. NT, N-terminus; KD, kinase domain; CT, C-terminus. PIPKIγ_i3 is the only isoform localized to centrosomes. The white shading represents the polypeptide translated from exon 16b, the hatched shading represents the polypeptide translated from exon 16c of the pip5k1c gene. Scale bars: 5 µM (A,B,D). Enlarged centrosome images were shown as inserts. DNA was stained with DAPI.
To determine which PIPKIγ isoform was targeted to centrosomes, we fused an HA tag to the N-terminus of all six known PIPKIγ splice variants. As shown in Fig. 1D and supplementary material Fig. S1F, only PIPKIγ_i3 displayed a centrosome-associated signal. Other isoforms showed vesicle-like cytoplasmic targeting, or localized to focal adhesions (PIPKIγ_i2), as reported previously (Di Paolo et al., 2002; Ling et al., 2002). PIPKIγ_i3 contains a unique 26-amino-acid insertion (amino acids 641-666) at the C-terminus, between the sequence representing the end of PIPKIγ_i1 and the beginning of the PIPKIγ_i2 C-terminal extension (Fig. 1E). However, the C-terminal region of PIPKIγ_i3 (amino acids 445-End) is predominantly cytoplasmic (Fig. 1D, right panel). The N-terminal fragment of PIPKIγ_i3 (amino acids 1-445), which is common to all PIPKIγ variants, localized to the centrosome (Fig. 1D, middle panel). These results suggest that the centrosome-targeting motif is likely to be located within the N-terminal 445 amino acids. Because PIPKIγ_i3 is the only isoform that showed centrosome targeting, it is possible that the unique C-terminal insertion of PIPKIγ_i3 might provoke a conformational adjustment that acts to expose the centrosome-targeting motif embedded in the first 445 amino acids.
PIPKIγ is targeted to the proximal end of centrioles in a cell cycle-dependent manner
To examine the fine structural details of the association of PIPKIγ with the centrosome, we co-labeled HeLa cells with antibodies against PIPKIγ and various centrosome components. The localizations of these proteins around the centrosome were analyzed using three dimensional structured illumination microscopy (3D-SIM). Co-staining for ninein (a subdistal appendage marker on the mother centriole) (Nakagawa et al., 2001) and centrin 2 (which marks the distal end of the centriole cylinder) (Paoletti et al., 1996) showed that PIPKIγ was positioned below these two proteins (Fig. 2A). Further examination indicated that PIPKIγ localized between ninein and C-Nap1, a linker protein between centrioles (Mayor et al., 2000) (Fig. 2B), as well as between C-Nap1 and outer dense fiber protein 2 (ODF2), which marks the distal appendage of the mature mother centriole (Piel et al., 2000) (Fig. 2C). In these images, PIPKIγ seemed to be distributed along the side of the centriole wall in the pericentriolar matrix and appeared likely to form a ring around the proximal end of centrioles with a diameter smaller than that of ninein.
Fig. 2.
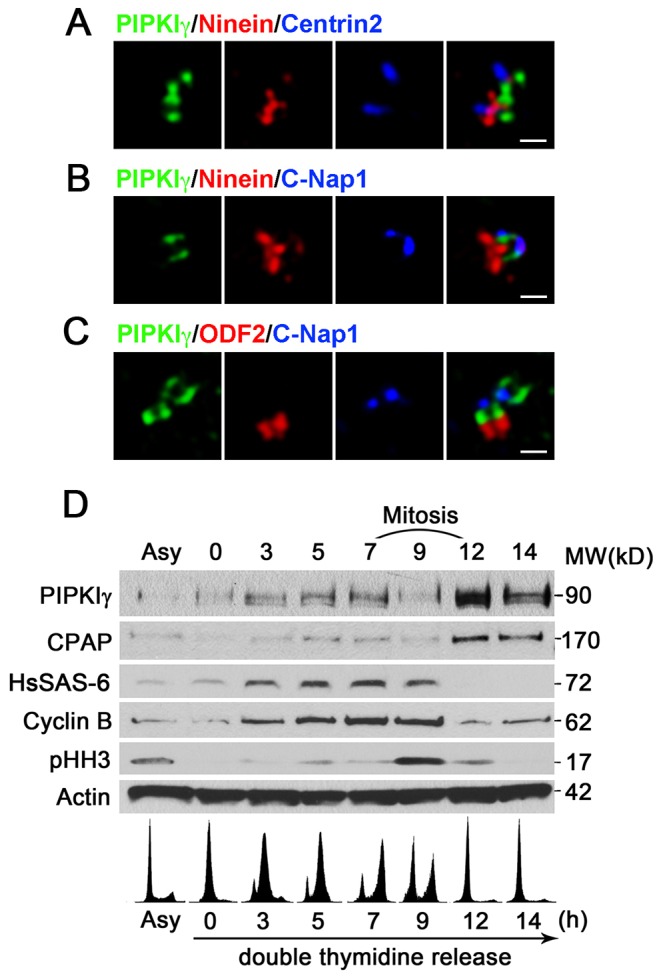
PIPKIγ localizes around the proximal end of centrioles and its protein level is regulated by the cell cycle. (A-C) HeLa cells were subjected to indirect immunofluorescence microscopy to visualize PIPKIγ along with ninein and centrin 2 (A), ninein/C-Nap-1 (B) and ODF2/C-Nap-1 (C) using 3D-SIM. SIM images of representative centrosomes show the positions of PIPKIγ and the indicated centrosome markers. These images indicate that PIPKIγ localizes below the distal and subdistal appendages and close to the proximal end of centrioles. Scale bars: 0.5 µm. (D) HeLa cells released from a double thymidine block were collected at the indicated time-points and were analyzed by immunoblotting using appropriate antibodies. The lower panel shows flow cytometry analyses of propidium iodide-stained cells at each time-point, confirming successful synchronization. Asy, asynchronous cells.
To address whether the centrosomal localization of PIPKIγ is cell-cycle dependent, as is the case for many other centrosome proteins, we subjected HeLa cells to immunofluorescence microscopy to visualize PIPKIγ together with centrin 2 and DAPI. Based on the staining of centrin 2 and DAPI, cells were categorized as being in G1 phase (two centrin 2 foci and intact nuclear membrane), S phase (four clustered centrin 2 foci and intact nuclear membrane), G2 phase (two separated pairs of centrin 2 foci and intact nuclear membrane) and various stages of M phase (broken nuclear membrane and condensed chromosome DNA in varying arrangements). PIPKIγ staining around the centrosome was observed in cells at G1, S and G2 phase (supplementary material Fig. S1G). At prophase, the PIPKIγ signal diminished and it remained absent from centrosomes until late telophase, when PIPKIγ was recruited back to both centrosomes that would soon be inherited by each daughter cell (supplementary material Fig. S1G). Consistent with its centrosome targeting, the protein level of PIPKIγ decreased significantly as cells progressed through mitosis and then recovered at mitotic exit in synchronized HeLa cells (Fig. 2D). This followed the same trend as CPAP (Tang et al., 2009) (Fig. 2D), a CEP152-interacting protein that functions early in centriole duplication; however, this trend is different from that of HsSAS-6 (Fig. 2D), which is degraded during late M phase and is absent in G1 phase (Strnad et al., 2007), suggesting that PIPKIγ might have a specific function at the centrosome.
PIPKIγ targeting to the centrosome is dependent on an association with CEP152
CEP152 is a scaffold protein localizing around the proximal end of centrioles and is required for the centrosomal localization of PLK4 and CPAP (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010). Because PIPKIγ also localizes around the proximal end of centrioles, we examined whether CEP152 was responsible for the targeting of PIPKIγ to the centrosome. The physical association between PIPKIγ and CEP152 was first determined using a co-immunoprecipitation assay. Indeed, HA-tagged PIPKIγ_i3 was pulled down with overexpressed GFP–CEP152 in HEK293T cells (Fig. 3A), suggesting that these two proteins have the potential to bind to each other. The CEP152 antibody recognized multiple bands (Fig. 3B), which could be all knocked down by CEP152-specific siRNA (Fig. 3D), suggesting that endogenous CEP152 exists in variant forms. Endogenous PIPKIγ co-immunoprecipitated with the endogenous CEP152 protein that migrated faster on SDS-PAGE gels (Fig. 3B). Although it is not known whether this PIPKIγ-associated CEP152 is an unmodified, truncated or alternatively spliced form of CEP152, our results support the existence of an in vivo physical connection between these two proteins. Because the recombinant full-length CEP152 expressed in Escherichia coli was highly degraded or insoluble no matter whether it was fused to a GST, His or maltose-binding protein (MBP) tag, we constructed and purified MBP-tagged CEP152 fragments and tested their interaction with His–PIPKIγ. Both the N-terminal 748 residues (CEP1521-748) and the C-terminal 906 residues (CEP152749-1654) pulled down PIPKIγ (supplementary material Fig. S2A). However, the N-terminal 217 residues of CEP152 (CEP1521-217), to which PLK4 directly binds (Hatch et al., 2010), did not interact with PIPKIγ (supplementary material Fig. S2A). These data suggest that PIPKIγ might directly bind to the C-terminus of CEP152, but further investigation is needed to obtain a definitive conclusion because of the heavy degradation of our recombinant CEP152 polypeptides.
Fig. 3.
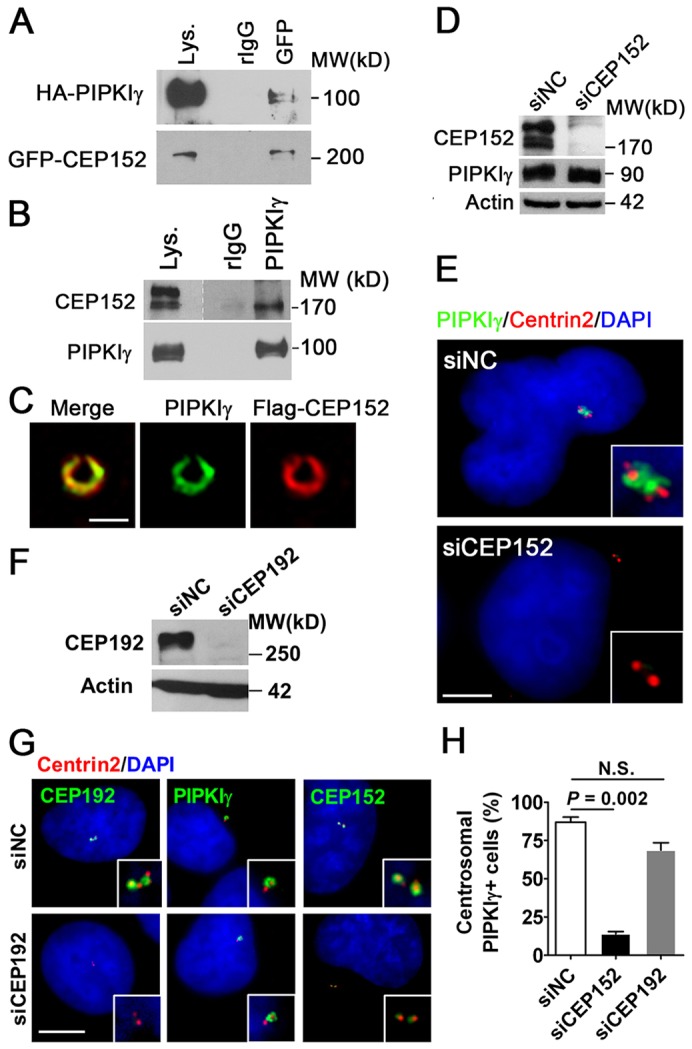
CEP152 associates with PIPKIγ and regulates PIPKIγ targeting to the centrosome. (A,B) CEP152 and PIPKIγ potentially associate with one another. (A) HEK293T cells co-transfected with vector encoding HA–PIPKIγ and GFP–CEP152 were subjected to immunoprecipitation using normal rabbit IgG (rIgG) or rabbit anti-GFP (GFP) antibodies. Lys., cell lysate. (B) Co-precipitation of endogenous PIPKIγ and endogenous CEP152 from HeLa cells. The precipitates from A and B were analyzed by immunoblotting with the indicated antibodies. (C) PIPKIγ and CEP152 colocalize at the centrosome. HeLa cells expressing FLAG–CEP152 were stained with anti-PIPKIγ and anti-FLAG antibodies and then analyzed by 3D-SIM. (D) CEP152-specific siRNA (siCEP152) efficiently depleted endogenous CEP152 from HeLa cells. siNC, non-specific control siRNA. (E) Loss of CEP152 eliminates endogenous PIPKIγ localization at centrosomes. HeLa cells were treated with siNC or siCEP152 and then stained with the indicated antibodies. Representative images of control or CEP152-depleted cells are shown. (F) CEP192-specific siRNA (siCEP192) efficiently depleted endogenous CEP192. (G) PIPKIγ localization at centrosomes was not significantly affected in CEP192-depleted cells, although CEP192 was completely abolished from the centrosome and the centrosomal signal of CEP152 was strongly decreased in these cells. (H) Quantification of the number of cells with robust centrosomal PIPKIγ staining in each group described in E and G indicates that the loss of CEP152, but not CEP192, significantly eliminates PIPKIγ targeting to the centrosome. Results from at least three independent experiments were analyzed, with >200 cells examined in each experiment. Error bars indicate s.d.; N.S., no significant difference. (E,G) Enlarged centrosome images are shown as inserts. Scale bars: 0.5 µM (C) 5 µM, (E,G). DNA was stained with DAPI.
To understand the biophysical significance of the association between PIPKIγ and CEP152, we first tested whether they colocalized at the centrosome. It has been reported recently by multiple groups that centrosomal proteins, including CEP152, form a ring-like structure around the centriole (Fu and Glover, 2012; Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). Consistently, our 3D-SIM images supported a ring-like colocalization between PIPKIγ and FLAG–CEP152 (Fig. 3C), suggesting that PIPKIγ localizes to the intermediate PCM around the proximal end of centrioles in a manner similar to CEP152 (Fu and Glover, 2012; Lawo et al., 2012; Mennella et al., 2012; Sonnen et al., 2012). More importantly, the colocalization between CEP152 and PIPKIγ reinforced the evidence for a physical interaction and suggested a functional association between these two proteins. Indeed, siRNA-mediated depletion of CEP152 (Fig. 3D) led to a loss of PIPKIγ signal from the centrosome (Fig. 3E,H), indicating that CEP152 is necessary for the recruitment of PIPKIγ to the centrosome. Moreover, depletion of CEP152 also eliminated the centrosome targeting of exogenous PIPKIγ_i31-445 (supplementary material Fig. S2B), further suggesting that CEP152 provides a necessary structural platform for the stable association of PIPKIγ with the centrosome.
In addition to CEP152 (Fig. 3F), CEP192 was recently shown to be important for the recruitment of centrosomal proteins (Sonnen et al., 2013). In CEP192-depleted cells, we found that the centrosomal signal of CEP152 was largely eliminated (Fig. 3G), as reported previously (Sonnen et al., 2013); however, the PIPKIγ signal around the centrosome was mostly retained (Fig. 3G,H). Combined with our previous observation that complete loss of CEP152 blocked the targeting of PIPKIγ to the centrosome (Fig. 3E,H), these results suggest that a very small amount of centrosomal CEP152 could be sufficient for the centrosomal recruitment of PIPKIγ.
PIPKIγ negatively regulates centriole duplication
The specific cell-cycle-dependent centrosomal localization of PIPKIγ led us to investigate the potential function of PIPKIγ at the centrosome. For this purpose, we knocked down PIPKIγ in HeLa cells using lentivirus-based PIPKIγ-specific short hairpin (sh)RNA (Fig. 4A). Strikingly, the loss of PIPKIγ resulted in an augmentation of centrin 2 foci (more than four per cell) in ∼20% of the cells (Fig. 4B,C). Importantly, centriole amplification caused by depletion of PIPKIγ was almost fully rescued by expression of RNAi-resistant PIPKIγ (Fig. 4D,E). A similar increase in the number of centrin 2 foci was also observed in HeLa cells treated with two distinct PIPKIγ siRNAs (siPIPKIγ-O1 and siPIPKIγ-O2) (supplementary material Fig. S3A,B) but not in cells depleted of PIPKIα or PIPKIβ (supplementary material Fig. S3F,G). This further confirmed that the phenotype was caused by PIPKIγ depletion. To determine whether the increased centrin 2 foci resulted from cell cycle arrest in S phase or G2/M phase, we examined the cell cycle distribution of PIPKIγ-depleted cells. The incorporation of BrdU (supplementary material Fig. S3C) and staining for proliferating cell nuclear antigen (PCNA) (supplementary material Fig. S3D) were used to determine the S phase population, and phospho-histone H3 (supplementary material Fig. S3E) was used to identify cells in G2/M phase. As shown in supplementary material Fig. S3C-E, no significant difference in these markers was observed between cells treated with siPIPKIγ and cells treated with control siRNA. Therefore, the excessive centrin 2 foci in PIPKIγ-depleted cells were not likely resulted from a sustained S, G2 or M phase. Furthermore, depletion of PIPKIγ, but not PIPKIα or PIPKIβ, caused an increase in centrosome number (more than two γ-tubulin loci per cell) in other cell types such as NIH3T3 (supplementary material Fig. S3H,I) and the renal collecting duct epithelial cell line IMCD3 (supplementary material Fig. S3J). Together, these results suggest that PIPKIγ likely inhibits centriole duplication.
Fig. 4.

Depletion of PIPKIγ induces excessive centrin 2 foci in cells. (A-C) HeLa cells infected for 48 h with lentivirus carrying either non-specific control (shNC) or PIPKIγ-specific (shPIPKIγ) shRNAs for were collected for immunoblotting (A) or subjected to indirect immunofluorescence microscopy using anti-centrin 2 antibody and DAPI (B). (A) shPIPKIγ efficiently knocked down endogenous PIPKIγ. (B) Loss of PIPKIγ results in increased centriole numbers. Enlarged centrosome images are shown as inserts. Scale bar: 5 µM. (C) Quantification of cells with more than four centrioles in control or PIPKIγ-depleted cells. n>200, results are derived from at least three independent experiments. (D,E) The centriole amplification caused by the depletion of PIPKIγ was rescued by expression of the RNAi-resistant form of HA-tagged wild-type (PIPKIγ-WT) or kinase-dead (PIPKIγ-KD) PIPKIγ. After being treated with siNC or siPIPKIγ, HeLa cells were transfected with empty vector (EV) or RNAi-resistant HA–PIPKIγ constructs. (D) The levels of endogenous and exogenous PIPKIγ were determined by immunoblotting with the indicated antibodies. (E) Cells in each group were stained with anti-HA, anti-centrin 2 antibodies and DAPI. The percentage of cells with more than four centrioles in the control or HA–PIPKIγ-expressing group was quantified and plotted. (F,G) HU-induced centrosome amplification in U-2 OS cells was significantly suppressed by the expression of either HA–PIPKIγ wild type (WT) or HA–PIPKIγ kinase dead (KD). U-2 OS cells were pretreated with 16 mM HU for 24 h, followed by transfection with EV or the indicated HA–PIPKIγ constructs and were re-incubated with HU for an additional 44 h before harvest. Non-treated U-2 OS cells were used as a control. Expression of endogenous and exogenous PIPKIγ was determined by immunoblotting (F). Next, cells in each group were stained with anti-HA and anti-γ-tubulin antibodies as well as DAPI, and then the samples were quantified for cells exhibiting more than two γ-tubulin foci (G). (E,G) n>100, results are derived from at least three independent experiments. N.S., no significant difference. Error bars indicate s.d.
To test this possibility, we examined whether the expression of PIPKIγ could reverse centrosome amplification caused by hydroxyurea (HU) treatment. As shown in Fig. 4F, U2 OS cells were treated with HU for 24 h before being transfected with or without wild-type or kinase-dead HA–PIPKIγ. This experimental design limited the impact of overexpressed PIPKIγ to S phase and excluded the potential influence on other phases of the cell cycle (Hemerly et al., 2009). As shown in Fig. 4G, HU-induced centrosome amplification in U-2 OS cells was significantly suppressed by the overexpression of HA–PIPKIγ, suggesting that PIPKIγ is a negative regulator of centrosome duplication. Furthermore, the expression of both wild-type and kinase-dead PIPKIγ could rescue centriole over-duplication caused by PIPKIγ depletion or HU treatment, suggesting that the production of PI(4,5)P2 might not play an important role in centriole biogenesis.
To further characterize these excessive centrin 2 foci in PIPKIγ-depleted cells, we checked the localization of several centriolar proteins, including centriolar coiled protein of 110 kDa (CP110, which localizes at the distal ends of centrioles) (Kleylein-Sohn et al., 2007), centrin 2 (localizes at the distal ends of centrioles), centrosomal protein of 135 KDa (CEP135, which localizes at the proximal end of centrioles) (Kleylein-Sohn et al., 2007) and CPAP (localizes at the proximal ends of centrioles) (Kleylein-Sohn et al., 2007). At 48 h after HeLa cells were infected with lentivirus carrying control or PIPKIγ shRNA, we observed excessive CP110 and centrin 2 foci (more than four per cell) that associated with each other in PIPKIγ-depleted cells. However, the centrosome number (represented by γ-tubulin foci) in these cells was normal (two per cell). The numbers of CEP135 and CPAP foci were also normal (two per cell) in PIPKIγ-depleted cells (Fig. 5A,C). These results suggest that the additional centrin 2 and CP110 foci in PIPKIγ-depleted cells might correspond to over-duplicated procentrioles that were still engaged with the parental centrioles. Indeed, 72 h after lentivirus infection when the disengagement of nascent centrioles occurred, excessive CEP135, CPAP and γ-tubulin foci (more than two per cell) were observed in PIPKIγ-depleted cells (Fig. 5B,C). Moreover, transmission electron microscopy analysis of serial-sectioned PIPKIγ-depleted HeLa cells showed two procentrioles (P1 and P2, Fig. 5D, left and middle panels) attached to the proximal wall of a mother centriole (identifiable by the distal appendages) (Fig. 5D, right panel), further supporting the hypothesis that the lack of PIPKIγ leads to procentriole amplification.
Fig. 5.

PIPKIγ depletion causes PLK4- and HsSAS6-dependent centriole amplification. (A,B) HeLa cells were infected with lentivirus carrying control (shNC) or PIPKIγ-specific (shPIPKIγ) shRNA for 48 h (A) or 72 h (B) and were then fixed and stained to visualize the indicated centrosomal proteins. The schematic diagrams (middle panels) illustrate the positions of the indicated centrosomal proteins at normally duplicated centrosomes. Scale bars: 1 µM. (C) Quantification of cells with more than four centrioles in samples of cells that were treated with control or PIPKIγ shRNA for 48 h or 72 h. (D) Transmission electron microscopy (TEM) images from serial-sectioned PIPKIγ-depleted cells showed two procentrioles (P1 and P2) around one mother centriole (M). The numbers of serial-sections are shown (0, +1 and +4). Scale bar: 0.2 µM. (E) Co-depletion of PLK4 or HsSAS-6 rescues the centriole duplication that results from PIPKIγ depletion. HeLa cells were transfected with negative control (siNC) or PIPKIγ siRNA (siPIPKIγ), with or without PLK4 or HsSAS-6 siRNA. Cells in each group were stained with anti-centrin 2 antibody and DAPI. The percentage of cells with more than four centrin 2 foci in each group was quantified and plotted. (F) Myc–PLK4-overexpression-induced centriole amplification in U-2 OS cells was significantly suppressed by the expression of wild-type (WT) or kinase-dead (KD) PIPKIγ. The Tet-On U-2 OS cells expressing Myc-tagged PLK4 were transfected with empty vector (EV) or HA-tagged PIPKIγ constructs before a 16 h treatment with 1 µg/ml doxycycline (+Dox) to induce PLK4 expression. Cells were then stained with anti-HA and anti-centrin 2 antibodies as well as with DAPI. Cells with more than four centrioles were quantified in each group. Cells without doxycycline treatment (−Dox) were used as a control. (E,F) n>100, results are derived from at least three independent experiments. N.S., no significant difference. Error bars indicate s.d.
It has been reported that overexpression of PLK4 (Habedanck et al., 2005) or HsSAS-6 (Leidel et al., 2005; Strnad et al., 2007) leads to biogenesis of multiple procentrioles around one pre-existing centriole. PIPKIγ depletion also yielded more than one procentriole per parental centriole, suggesting a connection with PLK4 or HsSAS-6. To test this, we designed specific siRNAs to knock down PLK4 (supplementary material Fig. S3K) or HsSAS-6 (supplementary material Fig. S3L). When PLK4 or HsSAS-6 was co-depleted along with PIPKIγ, the centriole over-duplication phenotype caused by PIPKIγ exhaustion was completely rescued (Fig. 5E), suggesting that PIPKIγ functions against PLK4 and/or HsSAS-6 at the early stage of centriole biogenesis. In agreement with this, depletion of PIPKIγ, without affecting the level of PLK4 (supplementary material Fig. S3M), synergistically enhanced the centriole amplification that resulted from PLK4 overexpression (supplementary material Fig. S3N). In addition, the centriole amplification induced in U-2 OS cells by Myc–PLK4 overexpression was significantly suppressed by expression of either wild-type or kinase-dead HA–PIPKIγ (Fig. 5F), further suggesting that PIPKIγ might counteract PLK4 function.
The cryptic polo-box domain of PLK4 directly interacts with PIPKIγ
Like PIPKIγ, PLK4 (the key regulator of centriole duplication) localizes in the PCM around the proximal end of centrioles in a CEP152-dependent manner (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010). To investigate the functional correlation between PIPKIγ and PLK4, we first tested whether they physically associate with one another. Because the endogenous level of PLK4 is undetectable by immunoblotting, we examined whether endogenous PIPKIγ associates with overexpressed GFP-tagged PLK4. GFP–PLK4 co-immunoprecipitated with PIPKIγ, indicating that they form a complex in vivo (Fig. 6A). To understand the spatial correlation between PIPKIγ and PLK4, we developed a mouse monoclonal PLK4 antibody. This antibody specifically recognized purified MBP–PLK4 but not MBP or MBP–CEP1521-748 (supplementary material Fig. S4A) and it stains PLK4 at the centrosome. This staining was abolished by two independent PLK4-specific siRNAs (supplementary material Fig. S4B). These results indicate that this new PLK4 antibody can be used to visualize endogenous PLK4 by immunofluorescence microscopy. Images obtained by 3D-SIM showed that our PLK4 antibody nicely stained the centrosome with a pattern similar to that seen for endogenous CEP152, the binding partner of PLK4 (supplementary material Fig. S4C), further endorsing the specificity of this antibody. Colocalization of endogenous PIPKIγ with endogenous PLK4 at the centrosome was observed (Fig. 6B), suggesting that the working spaces of these two proteins overlap. Both PIPKIγ and PLK4 displayed a ring-like structure with a similar shape and diameter to that of CEP152 (Fig. 3C and Fig. 6B) (Sir et al., 2011). This is in line with the notion that PLK4 and CEP152 interact and colocalize (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010) and position PIPKIγ at the inner to intermediate region of the PCM (Sonnen et al., 2012).
Fig. 6.

PIPKIγ directly interacts with the CPB domain of PLK4. (A) Endogenous PIPKIγ co-precipitates with GFP-tagged PLK4 in HeLa cells. Lys., cell lysate. (B) PIPKIγ and PLK4 colocalize at the centrosome. HeLa cells were stained with anti-PIPKIγ and anti-PLK4 antibodies and then analyzed by 3D-SIM. Scale bar: 0.5 µM. (C) PLK4 directly interacts with PIPKIγ. Purified MBP or MBP–PLK4 was incubated with His–PIPKIγ, pulled down using amylose resin and analyzed by immunoblotting. (D) PIPKIγ binds to the cryptic polo box (CPB) domain of PLK4. His–PIPKIγ was incubated with GST or GST–PLK4 polypeptides, including the N-terminus (PLK4-NT), C-terminus (PLK4-CT), cryptic polo box (PLK4-CPB) or polo box (PLK4-PB). (E) The kinase domain (KD) of PIPKIγ binds to PLK4. GST or GST–PLK-CT was incubated with His-tagged PIPKIγ N-terminus (PIPKIγ-NT), C-terminus (PIPKIγ-CT) or N-terminus truncated (PIPKIγ-ΔN) and analyzed by immunoblotting. (C-E) The loading of MBP, MBP–PLK4, GST and GST-fused PLK4 fragments is shown by Coomassie Brilliant Blue (CBB) staining. (F) Schematic of the interaction sites between PIPKIγ and PLK4, based on the results from the in vitro GST or MBP pull-down assays shown in C-E. The identified binding regions are highlighted in red. FL, full length; NT, N-terminus; KD, kinase domain; CT, C-terminus; CPB, cryptic polo box; PB, polo box.
Next we tested the direct interaction between PIPKIγ and PLK4 with in vitro pull-down assays using purified MBP–PLK4 and His–PIPKIγ. PIPKIγ clearly shows direct binding to PLK4 (Fig. 6C). Using a series of truncated recombinant PLK4 fragments (Fig. 6F), we found that the cryptic polo-box (CPB) domain of PLK4 is necessary and sufficient for PIPKIγ interaction (Fig. 6D). Using truncated PIPKIγ proteins, we narrowed down the PLK4-binding region within PIPKIγ to the kinase domain (Fig. 6E,F). The N- or C-terminus of PIPKIγ alone could not bind to PLK4, and the PIPKIγ N-terminus is not necessary for PLK4 binding (Fig. 6E,F). These results strongly support the notion of a physical association between PIPKIγ and PLK4 and suggest a functional consequence of this interaction.
PIPKIγ impairs PLK4 activity
Because both CEP152 and PIPKIγ bind to the CPB domain of PLK4, we tested whether PIPKIγ and CEP152 compete for the interaction with PLK4. Because the full length CEP152 and the protein fragments were highly degraded when purified from E. coli, we produced the 35S-labeled CEP152 N-terminal fragment (35S–CEP1521-748) by in vitro translation. Consistent with the literature (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010), MBP-tagged full-length PLK4 pulled down 35S–CEP1521-748 well. Although PIPKIγ could still be pulled down by MBP–PLK4, the addition of increasing amounts of PIPKIγ did not affect the binding of CEP152 to PLK4 (Fig. 7A), suggesting that PIPKIγ and CEP152 do not bind to the same region of PLK4.
Fig. 7.

The binding of PIPKIγ impairs PLK4 kinase activity. (A) PIPKIγ does not affect the interaction between CEP152 and PLK4. The [35S]Met-labeled N-terminus of CEP152 (CEP1521-748) was expressed using an in vitro transcription/translation system. Its interaction with MBP–PLK4 was determined with or without the presence of the indicated amount of His–PIPKIγ, using a MBP-pull-down assay. The loading of MBP and MBP–PLK4 is shown by Ponceau S staining. (B) PLK4 phosphorylates both itself and CEP152. A FLAG-tagged CEP152 fragment (CEP1521-748) was overexpressed in HeLa cells and immunoprecipitated with anti-FLAG antibody. It was then used in a PLK4 kinase assay as a substrate for purified MBP–PLK4. Lane 1, immunoblotting (IB) showing the loading of purified MBP–PLK4. Lanes 2 and 3, IB with anti-FLAG antibody to analyze the immunoprecipitates pulled down by mouse IgG (mIgG) or anti-FLAG antibody. Lanes 4–6, autoradiographs (Autoradio.) showing the phosphorylation of PLK4 and CEP1521-748 when CEP1521-748 was incubated with or without MBP–PLK4 in the PLK4 kinase assay. (C) PLK4 could not phosphorylate PIPKIγ. The left panel shows autoradiographs of the PLK4 kinase assay when purified His–PIPKIγ was incubated with or without MBP–PLK4. The right panel shows Coomassie Brilliant Blue (CBB) staining indicating the loading of MBP–PLK4 and His–PIPKIγ. (D) PIPKIγ inhibits PLK4 activity. PLK4 autophosphorylation or PLK4-meditated phosphorylation of CEP1521-748 was determined using the PLK4 kinase assay with or without purified wild-type (WT) or kinase-dead (KD) PIPKIγ as indicated. The upper panel shows the results of autoradiography of phosphorylated PLK4 or CEP1521-748. The lower panel shows the loading of PIPKIγ proteins. (E,F) Quantification of phosphorylated PLK4 (E) or CEP1521-748 (F) from three independent experiments. A single asterisk (*) represents P<0.05; a double asterisk (**) represents P<0.01; N.S., no significant difference. (G) PIPKIγ does not affect the dimerization of PLK4. HeLa cells expressing either control construct or Myc–PLK4 construct were subjected to immunoprecipitation using an anti-FLAG antibody. A pull-down assay was used to determine the co-precipitation of the resulting immunoprecipitates with purified soluble MBP–PLK4 with or without an increasing amount of His−PIPKIγ. The final precipitates were then blotted with the indicated antibodies.
Well-controlled activity of PLK4 is essential for centriole duplication and fidelity (Bettencourt-Dias et al., 2005; Eckerdt et al., 2011; Kleylein-Sohn et al., 2007; Nigg, 2007; Nigg and Stearns, 2011). PLK4 can phosphorylate both itself (Brownlee and Rogers, 2012; Cunha-Ferreira et al., 2009; Guderian et al., 2010; Holland et al., 2010; Rogers et al., 2009) and its potential in vivo substrate CEP152 (Cizmecioglu et al., 2010; Dzhindzhev et al., 2010; Hatch et al., 2010). Therefore, we overexpressed FLAG-tagged CEP1521-748 in HeLa cells and purified this protein by immunoprecipitation. The precipitates obtained by normal mouse IgG or by anti-FLAG antibody were incubated with purified MBP–PLK4 to perform a kinase assay. Results from these experiments demonstrated that PLK4 could phosphorylate both itself and CEP152 (Fig. 7B) but not PIPKIγ (Fig. 7C), indicating that PIPKIγ would not compete with PLK4 substrates. We next tested whether the interaction with PIPKIγ influences PLK4 activity (Fig. 7D). Because PIPKIγ and PLK4 are both kinases and they might inhibit one another by competing for ATP, we compared the PLK4 activity with or without varying amounts of the wild-type (WT) or kinase-dead (KD) PIPKIγ. PIPKIγ-KD does not bind to ATP (Kunz et al., 2000) but binds to PLK4 in vitro at a comparable level to its wild-type counterpart (unpublished data). Both PIPKIγ-WT and PIPKIγ-KD inhibited PLK4 activity in a dose-dependent manner (Fig. 7D-F). When equal amounts of PIPKIγ and PLK4 were added, PIPKIγ-WT or PIPKIγ-KD resulted in a ∼20% inhibition of PLK4 autophosphorylation and CEP152 phosphorylation (Fig. 7D-F). When the amount of PIPKIγ-WT or PIPKIγ-KD was increased to threefold that of PLK4, >40% of PLK4 autophosphorylation and ∼50% of CEP1521-748 phosphorylation were inhibited (Fig. 7D-F). PIPKIγ-WT and PIPKIγ-KD consistently repressed PLK4 at a comparable level, suggesting that this inhibition was not caused by competition for ATP. These results suggest that PIPKIγ regulates PLK4 activity by binding to PLK4. To explore how PIPKIγ inhibits PLK4 activity, we determined whether the binding of PIPKIγ affects PLK4 dimerization, which is important for the self-activation of PLK4 (Guderian et al., 2010). As shown in Fig. 7G, overexpressed Myc–PLK4 were pulled down by using purified MBP–PLK4. This interaction was not interrupted by the addition of increasing amounts of recombinant PIPKIγ (Fig. 7G), indicating a more complicated mechanism that will require future exploration. Nevertheless, in the context that PLK4 activity is crucial for PLK4 to initiate procentriole biogenesis and achieve self-regulation, our results suggest that PIPKIγ could restrain centriole duplication by binding to and inhibiting PLK4 kinase activity.
DISCUSSION
How centrioles are built and how the centriole assembly is limited to one daughter per mother in a single cell cycle are associated puzzles that have not been fully resolved. In addition to the key players identified in recent years (Brito et al., 2012; Nigg and Stearns, 2011), here we report PIPKIγ as a new component of the centriole duplication machinery that restricts centriole formation by potentially limiting PLK4 activity. As a negative regulator of centriole biogenesis, the association of PIPKIγ with the centrosome is likely to be sensitive and carefully regulated, which could explain why PIPKIγ was not described in the recent centrosome proteome studies (Andersen et al., 2003; Keck et al., 2011; Müller et al., 2011; Müller et al., 2010; Nogales-Cadenas et al., 2009; Ren et al., 2010; Zellner et al., 2011). Nevertheless, our results from multiple experiments (Figs 1, 2) provide solid evidence supporting the physical association of endogenous PIPKIγ with the centrosome. The overall levels and centrosome association of PIPKIγ are regulated through the cell cycle, as is the case for many other centriolar proteins, however, this regulation operates in a rather unique way. Compared with the positive regulators of centriole biogenesis, such as PLK4, SAS6 and SCL-interrupting locus protein (STIL), the levels of which are highest in M phase and drop sharply at mitotic exit, PIPKIγ is regulated in the opposite direction: it is downregulated during mitosis but increases rapidly immediately after mitosis. In addition, the association of PIPKIγ with a centriole is first seen at the G1–S boundary when the procentriole starts to emerge from the centriole (supplementary material Fig. S1G). This timing is similar to that of centrin 2, SAS-6 and CPAP, which are recruited to centrioles in G1/S phase to support procentriole assembly (Kleylein-Sohn et al., 2007; Paoletti et al., 1996; Strnad et al., 2007; Tang et al., 2009). Although it is not fully understood why PLK4, SAS-6 and STIL accumulate in M phase when the budding of the procentriole has completed, the cell-cycle-dependent expression and centrosome association of PIPKIγ serves its function well: it counteracts the centriole assembly machinery and inhibits the formation of excessive procentrioles.
Although only PIPKIγ_i3 (and not other PIPKIγ isoforms) is targeted to the centrosome when overexpressed, the unique insert sequence in PIPKIγ_i3, which distinguishes it from other PIPKIγ isoforms, does not contain a centrosome-targeting sequence (Fig. 1E). Instead, the centrosome-targeting signal seems to be located in the kinase domain that is commonly present in other PIPKIγ splicing isoforms and is highly conserved among all type I PIPK members; however, none of the other isoforms shows centrosomal localization. This suggests that the centrosome-targeting signal in PIPKIγ is normally hidden and needs to be modified in a certain way in order to be exposed. It is plausible that dimerization of PIPKIγ (Rao et al., 1998) covers the centrosome-targeting motif in the kinase domain. The unique C-terminal insertion of PIPKIγ_i3 might alter the protein conformation and expose the centrosome-targeting motif. Because only a fraction of PIPKIγ_i3-expressing cells showed PIPKIγ_i3 at the centrosome, it is also possible that other modification mechanisms, which could have been exhausted by overexpressed PIPKIγ_i3, are necessary to achieve a more efficient exposure of the centrosome-targeting motif in PIPKIγ. Similar to other centrosomal proteins (Fu and Glover, 2012; Mennella et al., 2012; Pelletier and Yamashita, 2012; Sonnen et al., 2012), both endogenous and overexpressed PIPKIγ exhibited a toroid-shaped distribution around the centriole in most cells, suggesting that the association of PIPKIγ with the centrosome follows the same common mechanism as other centrosomal proteins. This was reinforced by the observation that the stable association between PIPKIγ and the centrosome strictly relies on the existence of CEP152 but not CEP192. This stable association is likely to be achieved through a physical interaction between PIPKIγ and CEP152. Indeed, the PIPKIγ toroid overlapped well with the CEP152 toroid, indicating that PIPKIγ, like CEP152, is an intermediate PCM protein (Sonnen et al., 2012). However, PIPKIγ is absent from the centrosome in M phase when CEP152 is still present, suggesting that CEP152 might not be sufficient to recruit PIPKIγ to the centrosome. Further investigations are required in order to decipher the signal code that governs the cell-cycle-dependent association of PIPKIγ with the centrosome.
Traditionally, PIPKIγ regulates a variety of cellular processes by targeting to specific subcellular locales and providing PI(4,5)P2 to the proximate downstream effector. This leads to the question of whether PI(4,5)P2 has a role in centriole duplication. Using GFP-tagged phospholipase C delta pleckstrin homology (PLCδ PH) domain, we failed to see clear GFP signal at the centrosome owing to the strong membrane and cytosolic background (data not shown). Antibody against PI(4,5)P2 also failed to highlight any specific subcellular locations, including the ones where PI(4,5)P2 plays important roles, such as focal adhesions and clathrin-coated pits (data not shown). However, it has been reported that there are membrane vesicles around the centrosome (Foraker et al., 2012; Westlake et al., 2011) and that phosphoinositide-binding proteins, such as clathrin (Ford et al., 2001) and FYVE-domain-containing centrosomal protein (FYVE-CENT) (Sagona et al., 2010) localize at the centrosome and regulate centrosome integrity (Foraker et al., 2012) – suggesting that phosphoinositides, including PI(4,5)P2, might occur and function at the centrosome. Nevertheless, in our hands both the wild-type and kinase-dead forms of PIPKIγ could similarly rescue the centriole or centrosome amplification caused by PIPKIγ depletion, HU treatment or PLK4 overexpression, suggesting that PI(4,5)P2 is not essential in limiting centriole duplication. We reported previously that PIPKIγ can regulate some cellular process independently of PI(4,5)P2 production. For example, PIPKIγ functions as a scaffold between E-cadherin and the clathrin-adaptor complex AP1B and thereby facilitates the transport of E-cadherin from the trans-Golgi network to the recycling endosome (Ling et al., 2007). In this process, the physical interaction of PIPKIγ with E-cadherin, but not the PI(4,5)P2 generated by PIPKIγ, is crucial. We propose that the binding of PIPKIγ to PLK4 might impair the conformation of the protein and/or the access of ATP or other PLK4 substrates such that PLK4 activity is weakened. Although this model has not been tested in vivo owing to the highly restricted expression of endogenous PLK4 (Guderian et al., 2010; Holland et al., 2010; Sillibourne et al., 2010), it is in agreement with our observation that loss of PIPKIγ causes PLK4-dependent centriole amplification without affecting cell cycle progression.
The auto-phosphorylation of PLK4 triggers both activation and degradation of PLK4 (Guderian et al., 2010; Holland et al., 2010; Sillibourne et al., 2010). It has been shown that the degradation of PLK4 requires multi-site phosphorylation, which leads to a model in which PLK4 is destroyed once its activity reaches a certain threshold level (Guderian et al., 2010; Holland et al., 2010; Sillibourne et al., 2010). Our results suggest that PIPKIγ binds to PLK4 and inhibits its activity, providing another level of regulation in addition to phosphorylation. This seems crucial to maintain the appropriate activity of the centriole duplication machinery because loss of PIPKIγ leads to the generation of multiple procentrioles and overexpression of PIPKIγ blocks centrosome overduplication. Our results suggest that CEP152, by engaging both the positive regulator PLK4 and its negative regulator PIPKIγ at the proximal ends of parental centrioles, provides a physical platform for the accurate control of centriole assembly and homeostasis. Future studies will be necessary to provide greater insight into the functional consequences of the physical interaction between PIPKIγ and PLK4. It is also important to investigate whether PLK4 is the only centrosomal protein regulated by PIPKIγ and whether PI(4,5)P2 has a functional role in other centrosome-related cellular processes.
MATERIALS AND METHODS
Cell culture and transfection
HeLa, U-2 OS, MDA-MB-231, NIH3T3 and HEK293T cells were maintained in DMEM containing 10% FBS. IMCD3 cells were cultured in DMEM:F12 containing 10% fetal bovine serum (FBS). HeLa and HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) or FuGENE 6 Transfection Reagent (Promega). U-2 OS cells were transfected using X-tremeGENE 9 (Roche) or Amaxa Cell Line Nucleofector Kit V (Lonza). siRNA duplexes were introduced into cells using Lipofectamine RNAiMAX (Invitrogen). HeLa cells were synchronized with a standard double-thymidine block [two 16 h thymidine (Sigma, 2 mM) treatments separated by a 9 h release]. U-2 OS cells were synchronized with 16 mM hydroxyurea (Sigma) for 24 h. The tetracycline-inducible cell-line expressing Myc-tagged PLK4 was kindly provided by Erich Nigg (Kleylein-Sohn et al., 2007). Myc–PLK4 expression was induced by the addition of 1 µg/ml doxycycline (Clontech).
Antibodies and constructs
Rabbit monoclonal antibody against PIPKIγ was generated at Epitomics by immunizing rabbits with recombinant His-tagged PIPKIγ. Before collecting the spleens, anti-sera were harvested and purified against an affinity column conjugated with MBP-tagged PIPKIγ C-terminus (amino acids 469-668) to obtain polyclonal PIPKIγ antibody. Rabbit polyclonal antibodies against PIPKIα and PIPKIβ were obtained by immunizing rabbits with recombinant GST–His-tagged PIPKIα or PIPKIβ. Anti-sera were purified with affinity column conjugated with the MBP-tagged C-terminus of PIPKIα (amino acids 451–562) or PIPKIβ (amino acids 397-540). Mouse monoclonal anti-PLK4 antibody was generated at Abmart, Inc. Rabbit polyclonal antibodies against the following proteins were used: phospho-histone H3 (Ser10) (Millipore), CPAP (Proteintech), β-actin, CEP135, GFP (Abcam), CEP152 (Bethyl Laboratories, A302-480A), CEP192 (a generous gift from Erich Nigg) (Schmidt et al., 2009) and CP110 (Proteintech). Mouse antibodies against the following proteins were used: centrin 2 (20H5, a generous gift from Jeffrey Salisbury), ODF2 (Abnova), C-Nap1 (BD Biosciences), His (HIS.H8, Millipore), GFP (Roche); α-tubulin (DM1A), γ-tubulin (GTU-88), FLAG (M2) and HA (HA-7) were from Sigma; Myc (9E10), HsSAS-6 (91.390.21) and cyclin B (GNS1) were from Santa Cruz. Anti-ninein (Y-16) antibody was from Santa Cruz and maltose binding protein antibody (7G4) was from Sigma. cDNAs encoding full-length or truncated human PIPKIγ (Ling et al., 2002), PLK4 and CEP152 were obtained by PCR and subcloned into pCMV–HA, pcDNA3–FLAG, pGEX–4T-1, pET-28, pET-42 or pMAL–C2X constructs. pcDNA3-Myc–PLK4 was obtained from Addgene. HA–PIPKIγ kinase dead (KD) and RNAi-resistant HA–PIPKIγ-WT or –KD, which contain mismatches to the HsPIPKIγ-O1 siRNA, were generated using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). All constructs were verified by sequencing.
Lentiviruses
Lentiviruses carrying shRNA were constructed by cloning shRNA oligonucleotides (Invitrogen) into the pLKO.1 vector (AddGene). The shRNA sequence targeting human PIPKIγ was 5′-GTCGTGGTCATGAACAACA-3′. An interfering RNA sequence targeting luciferase (5′-GTACCTGTACTTCATGCAG-3′) was used as negative control. All constructs were confirmed by sequencing. HEK293T cells were co-transfected with pLKO.1–puro carrying shRNA, pCMV–VSVG and pCMV-R8.91 (gifts from Gaoxiang Ge, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, China) at a ratio of 2∶1∶1 using FuGENE 6 (Promega). The supernatant was collected 48-72 h after transfection. Subconfluent HeLa cells were infected in the presence of 8 µg/ml polybrene (Sigma).
siRNAs
All siRNA oligonucleotides were obtained from Invitrogen. HsPIPKIγ-O1, 5′-GCGTGGTCAAGATGCACCTCAAGTT-3′; HsPIPKIγ-O2, 5′-CCTACAGGTTCATCAAGAAACTGGA-3′; mouse PIPKIγ, 5′-GCGAGAGAGAGGATGTGCAGTATGA-3′; PLK4-O1, 5′-CACTGGTTTGGAAGTTGCAATCAAA-3′; PLK4-O2, 5′-AGGAGGTGTGTGTGGAGCTTGTAAA-3′; HsSAS-6, 5′-AGAAAAGCACGTTAATCAGCTACAA-3′ (Strnad et al., 2007); CEP152-O1, 5′-CAGAACAACTGAAATGGCTCTGGAA-3′; CEP152-O2, 5′-CAGCGTTTGCTGGGTGCAACTCAA-3′; CEP192-O1, 5′-CCCAAAGGAAGACATTTTCATCTCT-3′; CEP192-O2, 5′-ATCAGACAGAGGAATCAATAATAAA-3′ (Gomez-Ferreria et al., 2007; Sonnen et al., 2013; Zhu et al., 2008); HsPIPKIα-O1, 5′-TTGAAAGGTGCCATCCAGTTAGGCA-3′; HsPIPKIα-O2, 5′-TGTTAAGAAGTTGGAGCACTCTTGG-3′ (Mellman et al., 2008); HsPIPKIβ, 5′-CAGCAAAGGGTTACCTTCCAGTTCA-3′. Stealth RNAi Negative Control siRNA (Invitrogen) was used as negative control. Similar results were obtained by using both siRNA oligonucleotides (O1, O2) for depletion of HsPIPKIγ, PLK4, CEP152, CEP192 or HsPIPKIα, respectively. Unless stated otherwise, results for oligonucleotide 1 (O1) are shown.
Immunoprecipitation and microscopy
Immunoprecipitation was performed as previously described (Ling et al., 2002) using immunoprecipitation (IP) buffer [20 mM HEPES-KOH (pH 7.2) 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 0.5% NP-40, Complete Protease Inhibitor Cocktail (Roche), PhosSTOP Phosphatase Inhibitor Cocktail (Roche) following the manufacturer's standard protocols]. For indirect immunofluorescence, cells were grown on glass coverslips, fixed with ice-cold methanol for 10 min, permeabilized and stained as described previously (Ling et al., 2002). Fluorescence images were acquired using a Nikon TE 2000-U with Metamorph (Molecular Devices). Z-Series were taken at 0.1-µm steps. Three-dimensional structured illumination microscopy (3D-SIM) was performed on a ELYRA Superresolution Microscopy system (Zeiss) equipped with an alpha ‘Plan-Apochromat’ ×100/1.46 digital interference contrast (DIC) oil immersion objective and Andor iXon 885 EMCCD camera, following standard protocol. Sections were acquired at 0.125 mm z-steps. Color channels were aligned using the alignment parameter from control measurements with 0.5 µm diameter multispectral fluorescent beads (Zeiss). Structured illumination reconstruction and image processing was performed with the ZEN software package (Zeiss). Final image processing was performed using Adobe Photoshop.
In vitro protein pull-down assay
GST or MBP-fused proteins were incubated with His-tagged proteins and glutathione-sepharose beads (GE Healthcare) or amylose resin (New England Biolabs) in binding buffer [25 mM Tris (pH 7.6), 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.5% Triton X-100, 10% glycerol] for 2 h at 4°C. The precipitates were washed five times with binding buffer before being analyzed by immunoblotting. The CEP152 N-terminus (amino acids 1-748) protein was generated using the TNT Quick Coupled Transcription/Translation System (Promega), in the presence of [35S]methionine (Perkin-Elmer), used for pull-down assay and analyzed by autoradiography.
Transmission electron microscopy
The cells were fixed for 1 h using Trump's fixative (Electron Microscopy Sciences). Embedding and serial sectioning of the cell samples was performed according to standard procedures at the Electron Microscopy Core Facility, Mayo Clinic. Specimens were observed in a JEOL 1400 transmission electron microscope (JEOL) operating at 80 kV.
PLK4 kinase assay
PLK4 activity was determined following a previously published approach (Hatch et al., 2010; Holland et al., 2010). Briefly, a FLAG-tagged CEP152 fragment (CEP1521-748) was overexpressed in HeLa cells and immunoprecipitated with anti-FLAG antibody. 0.2 µg of purified MBP–PLK4 was incubated with or without recombinant CEP152 N-terminus and/or an appropriate amount of wild-type (WT) or kinase-dead (KD) His–PIPKIγ in kinase buffer with 5 µCi [32P]ATP (Perkin Elmer) and 33 µM ATP (New England Biolabs) for 30 min at room temperature. ImageJ (NIH, Bethesda, MD) was used to quantify autoradiographs.
Centrosome isolation
Centrosomes were isolated from HeLa cells or mouse kidneys, based on a published approach (Meigs and Kaplan, 2008).
Statistical analyses
Data represent the mean and standard deviation (SD) from at least three independent experiments. Significance was calculated by Student's t-test using Excel software (Microsoft). A P-value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We sincerely thank Bing Huang and Trace Christensen (Electron Microscopy Core Facility, Mayo Clinic) for technical assistance with electron microscope studies, Gaoxiang Ge (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) for kindly sharing the lentivirus system, and Jason L. Bakeberg and Cynthia Hommerding at Mayo Clinic for technical assistance with ultra-speed centrifugation and fluorescence microscopy. We thank Gina Razidlo, Barbara Schroeder and Mark McNiven for their stimulating discussion.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
Q.X. performed most of the experiments; Y.Z. constructed and purified recombinant proteins and performed in vitro protein binding assays; X.X. constructed and purified PIPKIγ proteins, purified antibodies against PIPKIα, β and γ and performed kinase assays; Y.H. purified full-length and C-terminus of PIPKI proteins and constructed affinity columns. Q.X., J.L.S., J.H. and K.L. designed the study, analyzed the data and prepared the manuscript. All authors discussed the results and commented on the manuscript.
Funding
K.L. and co-workers were supported by research grants from the National Cancer Institute [grant number 1R01CA149039-01A1], Susan G. Komen for the Cure [grant number KG100902] and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDKD) [grant number DK90728]. J.H. and Y.Z. are funded by NIDDKD and National Institutes of Health [grant number R01-DK090038]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.141465/-/DC1
References
- Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- Bairstow S. F., Ling K., Su X., Firestone A. J., Carbonara C., Anderson R. A. (2006). Type Igamma661 phosphatidylinositol phosphate kinase directly interacts with AP2 and regulates endocytosis. J. Biol. Chem. 281, 20632–20642 10.1074/jbc.M601465200 [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M., Rodrigues-Martins A., Carpenter L., Riparbelli M., Lehmann L., Gatt M. K., Carmo N., Balloux F., Callaini G., Glover D. M. (2005). SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15, 2199–2207 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- Brito D. A., Gouveia S. M., Bettencourt-Dias M. (2012). Deconstructing the centriole: structure and number control. Curr. Opin. Cell Biol. 24, 4–13 10.1016/j.ceb.2012.01.003 [DOI] [PubMed] [Google Scholar]
- Brownlee C. W., Rogers G. C. (2012). Show me your license, please: deregulation of centriole duplication mechanisms that promote amplification. Cell. Mol. Life Sci. 70, 1021–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee C. W., Klebba J. E., Buster D. W., Rogers G. C. (2011). The Protein Phosphatase 2A regulatory subunit Twins stabilizes Plk4 to induce centriole amplification. J. Cell Biol. 195, 231–243 10.1083/jcb.201107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O., Arnold M., Bahtz R., Settele F., Ehret L., Haselmann-Weiss U., Antony C., Hoffmann I. (2010). Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J. Cell Biol. 191, 731–739 10.1083/jcb.201007107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Ferreira I., Rodrigues-Martins A., Bento I., Riparbelli M., Zhang W., Laue E., Callaini G., Glover D. M., Bettencourt-Dias M. (2009). The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr. Biol. 19, 43–49 10.1016/j.cub.2008.11.037 [DOI] [PubMed] [Google Scholar]
- Debec A., Sullivan W., Bettencourt-Dias M. (2010). Centrioles: active players or passengers during mitosis? Cell. Mol. Life Sci. 67, 2173–2194 10.1007/s00018-010-0323-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., Pellegrini L., Letinic K., Cestra G., Zoncu R., Voronov S., Chang S., Guo J., Wenk M. R., De Camilli P. (2002). Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature 420, 85–89 10.1038/nature01147 [DOI] [PubMed] [Google Scholar]
- Dzhindzhev N. S., Yu Q. D., Weiskopf K., Tzolovsky G., Cunha-Ferreira I., Riparbelli M., Rodrigues-Martins A., Bettencourt-Dias M., Callaini G., Glover D. M. (2010). Asterless is a scaffold for the onset of centriole assembly. Nature 467, 714–718 10.1038/nature09445 [DOI] [PubMed] [Google Scholar]
- Eckerdt F., Yamamoto T. M., Lewellyn A. L., Maller J. L. (2011). Identification of a polo-like kinase 4-dependent pathway for de novo centriole formation. Curr. Biol. 21, 428–432 10.1016/j.cub.2011.01.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sayegh T. Y., Arora P. D., Ling K., Laschinger C., Janmey P. A., Anderson R. A., McCulloch C. A. (2007). Phosphatidylinositol-4,5 bisphosphate produced by PIP5KIgamma regulates gelsolin, actin assembly, and adhesion strength of N-cadherin junctions. Mol. Biol. Cell 18, 3026–3038 10.1091/mbc.E06-12-1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foraker A. B., Camus S. M., Evans T. M., Majeed S. R., Chen C. Y., Taner S. B., Corrêa I. R., Jr, Doxsey S. J., Brodsky F. M. (2012). Clathrin promotes centrosome integrity in early mitosis through stabilization of centrosomal ch-TOG. J. Cell Biol. 198, 591–605 10.1083/jcb.201205116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford M. G., Pearse B. M., Higgins M. K., Vallis Y., Owen D. J., Gibson A., Hopkins C. R., Evans P. R., McMahon H. T. (2001). Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science (New York), NY 291, 1051–1055 [DOI] [PubMed] [Google Scholar]
- Fu J., Glover D. M. (2012). Structured illumination of the interface between centriole and peri-centriolar material. Open Biol. 2, 120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadimi B. M., Sackett D. L., Difilippantonio M. J., Schröck E., Neumann T., Jauho A., Auer G., Ried T. (2000). Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer 27, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudici M. L., Emson P. C., Irvine R. F. (2004). A novel neuronal-specific splice variant of Type I phosphatidylinositol 4-phosphate 5-kinase isoform gamma. Biochem. J. 379, 489–496 10.1042/BJ20031394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudici M. L., Lee K., Lim R., Irvine R. F. (2006). The intracellular localisation and mobility of Type Igamma phosphatidylinositol 4P 5-kinase splice variants. FEBS Lett. 580, 6933–6937 10.1016/j.febslet.2006.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Ferreria M. A., Rath U., Buster D. W., Chanda S. K., Caldwell J. S., Rines D. R., Sharp D. J. (2007). Human Cep192 is required for mitotic centrosome and spindle assembly. Curr. Biol. 17, 1960–1966 10.1016/j.cub.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Guderian G., Westendorf J., Uldschmid A., Nigg E. A. (2010). Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J. Cell Sci. 123, 2163–2169 10.1242/jcs.068502 [DOI] [PubMed] [Google Scholar]
- Habedanck R., Stierhof Y. D., Wilkinson C. J., Nigg E. A. (2005). The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 7, 1140–1146 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hatch E. M., Kulukian A., Holland A. J., Cleveland D. W., Stearns T. (2010). Cep152 interacts with Plk4 and is required for centriole duplication. J. Cell Biol. 191, 721–729 10.1083/jcb.201006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemerly A. S., Prasanth S. G., Siddiqui K., Stillman B. (2009). Orc1 controls centriole and centrosome copy number in human cells. Science 323, 789–793 10.1126/science.1166745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. J., Lan W., Niessen S., Hoover H., Cleveland D. W. (2010). Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188, 191–198 10.1083/jcb.200911102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H., Shibasaki Y., Kizuki N., Wada T., Yazaki Y., Asano T., Oka Y. (1998). Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J. Biol. Chem. 273, 8741–8748 10.1074/jbc.273.15.8741 [DOI] [PubMed] [Google Scholar]
- Kaplan D. D., Meigs T. E., Kelly P., Casey P. J. (2004). Identification of a role for beta-catenin in the establishment of a bipolar mitotic spindle. J. Biol. Chem. 279, 10829–10832 10.1074/jbc.C400035200 [DOI] [PubMed] [Google Scholar]
- Keck J. M., Jones M. H., Wong C. C., Binkley J., Chen D., Jaspersen S. L., Holinger E. P., Xu T., Niepel M., Rout M. P. et al. (2011). A cell cycle phosphoproteome of the yeast centrosome. Science (New York) 332, 1557–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa D., Flückiger I., Polanowska J., Keller D., Reboul J., Gönczy P. (2011). PP2A phosphatase acts upon SAS-5 to ensure centriole formation in C. elegans embryos. Dev. Cell 20, 550–562 10.1016/j.devcel.2011.02.005 [DOI] [PubMed] [Google Scholar]
- Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y. D., Nigg E. A. (2007). Plk4-induced centriole biogenesis in human cells. Dev. Cell 13, 190–202 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Kunz J., Wilson M. P., Kisseleva M., Hurley J. H., Majerus P. W., Anderson R. A. (2000). The activation loop of phosphatidylinositol phosphate kinases determines signaling specificity. Mol. Cell 5, 1–11 10.1016/S1097-2765(00)80398-6 [DOI] [PubMed] [Google Scholar]
- Lawo S., Hasegan M., Gupta G. D., Pelletier L. (2012). Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat. Cell Biol. 14, 1148–1158 10.1038/ncb2591 [DOI] [PubMed] [Google Scholar]
- Leidel S., Delattre M., Cerutti L., Baumer K., Gönczy P. (2005). SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell Biol. 7, 115–125 10.1038/ncb1220 [DOI] [PubMed] [Google Scholar]
- Ling K., Doughman R. L., Firestone A. J., Bunce M. W., Anderson R. A. (2002). Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420, 89–93 10.1038/nature01082 [DOI] [PubMed] [Google Scholar]
- Ling K., Bairstow S. F., Carbonara C., Turbin D. A., Huntsman D. G., Anderson R. A. (2007). Type I gamma phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with mu 1B adaptin. J. Cell Biol. 176, 343–353 10.1083/jcb.200606023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle W. L., Barrett S. L., Negron V. C., D'Assoro A. B., Boeneman K., Liu W., Whitehead C. M., Reynolds C., Salisbury J. L. (2002). Centrosome amplification drives chromosomal instability in breast tumor development. Proc. Natl. Acad. Sci. USA 99, 1978–1983 10.1073/pnas.032479999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor T., Stierhof Y. D., Tanaka K., Fry A. M., Nigg E. A. (2000). The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J. Cell Biol. 151, 837–846 10.1083/jcb.151.4.837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megraw T. L., Sharkey J. T., Nowakowski R. S. (2011). Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 21, 470–480 10.1016/j.tcb.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigs T. E., Kaplan D. D. (2008). Isolation of centrosomes from cultured Mammalian cells. Cold Spring Harb. Protoc. 2008, pdb prot5039 10.1101/pdb.prot5039 [DOI] [PubMed] [Google Scholar]
- Mellman D. L., Gonzales M. L., Song C., Barlow C. A., Wang P., Kendziorski C., Anderson R. A. (2008). A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451, 1013–1017 10.1038/nature06666 [DOI] [PubMed] [Google Scholar]
- Mennella V., Keszthelyi B., McDonald K. L., Chhun B., Kan F., Rogers G. C., Huang B., Agard D. A. (2012). Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat. Cell Biol. 14, 1159–1168 10.1038/ncb2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H., Schmidt D., Steinbrink S., Mirgorodskaya E., Lehmann V., Habermann K., Dreher F., Gustavsson N., Kessler T., Lehrach H. et al. (2010). Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO J. 29, 3344–3357 10.1038/emboj.2010.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H., Schmidt D., Dreher F., Herwig R., Ploubidou A., Lange B. M. (2011). Gene ontology analysis of the centrosome proteomes of Drosophila and human. Commun. Integr. Biol. 4, 308–311 10.4161/cib.4.3.14806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y., Yamane Y., Okanoue T., Tsukita S., Tsukita S. (2001). Outer dense fiber 2 is a widespread centrosome scaffold component preferentially associated with mother centrioles: its identification from isolated centrosomes. Mol. Biol. Cell 12, 1687–1697 10.1091/mbc.12.6.1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E. A. (2006). Origins and consequences of centrosome aberrations in human cancers. Int. J. Cancer 119, 2717–2723 10.1002/ijc.22245 [DOI] [PubMed] [Google Scholar]
- Nigg E. A. (2007). Centrosome duplication: of rules and licenses. Trends Cell Biol. 17, 215–221 10.1016/j.tcb.2007.03.003 [DOI] [PubMed] [Google Scholar]
- Nigg E. A., Raff J. W. (2009). Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- Nigg E. A., Stearns T. (2011). The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 13, 1154–1160 10.1038/ncb2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales-Cadenas R., Abascal F., Díez-Pérez J., Carazo J. M., Pascual-Montano A. (2009). CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res. 37, D175–D180 10.1093/nar/gkn815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti A., Moudjou M., Paintrand M., Salisbury J. L., Bornens M. (1996). Most of centrin in animal cells is not centrosome-associated and centrosomal centrin is confined to the distal lumen of centrioles. J. Cell Sci. 109, 3089–3102 [DOI] [PubMed] [Google Scholar]
- Pelletier L., Yamashita Y. M. (2012). Centrosome asymmetry and inheritance during animal development. Curr. Opin. Cell Biol. 24, 541–546 10.1016/j.ceb.2012.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel M., Meyer P., Khodjakov A., Rieder C. L., Bornens M. (2000). The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J. Cell Biol. 149, 317–330 10.1083/jcb.149.2.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan G. A., Purohit A., Wallace J., Knecht H., Woda B., Quesenberry P., Doxsey S. J. (1998). Centrosome defects and genetic instability in malignant tumors. Cancer Res. 58, 3974–3985 [PubMed] [Google Scholar]
- Puklowski A., Homsi Y., Keller D., May M., Chauhan S., Kossatz U., Grünwald V., Kubicka S., Pich A., Manns M. P. et al. (2011). The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat. Cell Biol. 13, 1004–1009 10.1038/ncb2282 [DOI] [PubMed] [Google Scholar]
- Rao V. D., Misra S., Boronenkov I. V., Anderson R. A., Hurley J. H. (1998). Structure of type IIbeta phosphatidylinositol phosphate kinase: a protein kinase fold flattened for interfacial phosphorylation. Cell 94, 829–839 10.1016/S0092-8674(00)81741-9 [DOI] [PubMed] [Google Scholar]
- Ren J., Liu Z., Gao X., Jin C., Ye M., Zou H., Wen L., Zhang Z., Xue Y., Yao X. (2010). MiCroKit 3.0: an integrated database of midbody, centrosome and kinetochore. Nucleic Acids Res. 38, D155–D160 10.1093/nar/gkp784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers G. C., Rusan N. M., Roberts D. M., Peifer M., Rogers S. L. (2009). The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184, 225–239 10.1083/jcb.200808049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagona A. P., Nezis I. P., Pedersen N. M., Liestøl K., Poulton J., Rusten T. E., Skotheim R. I., Raiborg C., Stenmark H. (2010). PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat. Cell Biol. 12, 362–371 10.1038/ncb2036 [DOI] [PubMed] [Google Scholar]
- Schill N. J., Anderson R. A. (2009). Two novel phosphatidylinositol-4-phosphate 5-kinase type Igamma splice variants expressed in human cells display distinctive cellular targeting. Biochem. J. 422, 473–482 10.1042/BJ20090638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T. I., Kleylein-Sohn J., Westendorf J., Le Clech M., Lavoie S. B., Stierhof Y. D., Nigg E. A. (2009). Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011 10.1016/j.cub.2009.05.016 [DOI] [PubMed] [Google Scholar]
- Sillibourne J. E., Tack F., Vloemans N., Boeckx A., Thambirajah S., Bonnet P., Ramaekers F. C., Bornens M., Grand-Perret T. (2010). Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 21, 547–561 10.1091/mbc.E09-06-0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir J. H., Barr A. R., Nicholas A. K., Carvalho O. P., Khurshid M., Sossick A., Reichelt S., D'Santos C., Woods C. G., Gergely F. (2011). A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet. 43, 1147–1153 10.1038/ng.971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. H., Liu Y., Anderson D. E., Jahng W. J., O'Connell K. F. (2011). Protein phosphatase 2A-SUR-6/B55 regulates centriole duplication in C. elegans by controlling the levels of centriole assembly factors. Dev. Cell 20, 563–571 10.1016/j.devcel.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen K. F., Schermelleh L., Leonhardt H., Nigg E. A. (2012). 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol. Open 1, 965–976 10.1242/bio.20122337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen K. F., Gabryjonczyk A. M., Anselm E., Stierhof Y. D., Nigg E. A. (2013). Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J. Cell Sci. 126, 3223–3233 10.1242/jcs.129502 [DOI] [PubMed] [Google Scholar]
- Strnad P., Leidel S., Vinogradova T., Euteneuer U., Khodjakov A., Gönczy P. (2007). Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 13, 203–213 10.1016/j.devcel.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Ling K., Wagoner M. P., Anderson R. A. (2007). Type I gamma phosphatidylinositol phosphate kinase is required for EGF-stimulated directional cell migration. J. Cell Biol. 178, 297–308 10.1083/jcb.200701078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Hedman A. C., Tan X., Schill N. J., Anderson R. A. (2013). Endosomal type Iγ PIP 5-kinase controls EGF receptor lysosomal sorting. Dev. Cell 25, 144–155 10.1016/j.devcel.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. J., Fu R. H., Wu K. S., Hsu W. B., Tang T. K. (2009). CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell Biol. 11, 825–831 10.1038/ncb1889 [DOI] [PubMed] [Google Scholar]
- Thapa N., Sun Y., Schramp M., Choi S., Ling K., Anderson R. A. (2012). Phosphoinositide signaling regulates the exocyst complex and polarized integrin trafficking in directionally migrating cells. Dev. Cell 22, 116–130 10.1016/j.devcel.2011.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieman J. R., Mishra S. K., Ling K., Doray B., Anderson R. A., Traub L. M. (2009). Clathrin regulates the association of PIPKIgamma661 with the AP-2 adaptor beta2 appendage. J. Biol. Chem. 284, 13924–13939 10.1074/jbc.M901017200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. J., Li W. H., Wang J., Xu K., Dong P., Luo X., Yin H. L. (2004). Critical role of PIP5KIgamma87 in InsP3-mediated Ca(2+) signaling. J. Cell Biol. 167, 1005–1010 10.1083/jcb.200408008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake C. J., Baye L. M., Nachury M. V., Wright K. J., Ervin K. E., Phu L., Chalouni C., Beck J. S., Kirkpatrick D. S., Slusarski D. C. et al. (2011). Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc. Natl. Acad. Sci. USA 108, 2759–2764 10.1073/pnas.1018823108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Irvine R. F., Giudici M. L. (2011). Phosphatidylinositol 4-phosphate 5-kinase Iγ_v6, a new splice variant found in rodents and humans. Biochem. Biophys. Res. Commun. 411, 416–420 10.1016/j.bbrc.2011.06.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Xu Q., Huang Y., Singh R. D., Anderson R., Leof E., Hu J., Ling K. (2012). An association between type Iγ PI4P 5-kinase and Exo70 directs E-cadherin clustering and epithelial polarization. Mol. Biol. Cell 23, 87–98 10.1091/mbc.E11-05-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Matsuyama H., Furuya T., Oga A., Yoshihiro S., Okuda M., Kawauchi S., Sasaki K., Naito K. (2004). Centrosome hyperamplification predicts progression and tumor recurrence in bladder cancer. Clin. Cancer Res. 10, 6449–6455 10.1158/1078-0432.CCR-04-0773 [DOI] [PubMed] [Google Scholar]
- Zellner M., Babeluk R., Jakobsen L. H., Gerner C., Umlauf E., Volf I., Roth E., Kondrup J. (2011). A proteomics study reveals a predominant change in MaoB expression in platelets of healthy volunteers after high protein meat diet: relationship to the methylation cycle. J. Neural Transm. 118, 653–662 10.1007/s00702-011-0617-6 [DOI] [PubMed] [Google Scholar]
- Zhu F., Lawo S., Bird A., Pinchev D., Ralph A., Richter C., Müller-Reichert T., Kittler R., Hyman A. A., Pelletier L. (2008). The mammalian SPD-2 ortholog Cep192 regulates centrosome biogenesis. Curr. Biol. 18, 136–141 10.1016/j.cub.2007.12.055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.