

Abstract
Constitutive JAK2 activation in hematopoietic cells by the JAK2V617F mutation recapitulates myeloproliferative neoplasm (MPN) phenotypes in mice, establishing JAK2 inhibition as a potential therapeutic strategy. Although most polycythemia vera patients carry the JAK2V617F mutation, half of those with essential thrombocythemia or primary myelofibrosis do not, suggesting alternative mechanisms for constitutive JAK-STAT signaling in MPNs. Most patients with primary myelofibrosis have elevated levels of JAK-dependent proinflammatory cytokines (eg, interleukin-6) consistent with our observation of JAK1 hyperactivation. Accordingly, we evaluated the effectiveness of selective JAK1/2 inhibition in experimental models relevant to MPNs and report on the effects of INCB018424, the first potent, selective, oral JAK1/JAK2 inhibitor to enter the clinic. INCB018424 inhibited interleukin-6 signaling (50% inhibitory concentration [IC50] = 281nM), and proliferation of JAK2V617F+ Ba/F3 cells (IC50 = 127nM). In primary cultures, INCB018424 preferentially suppressed erythroid progenitor colony formation from JAK2V617F+ polycythemia vera patients (IC50 = 67nM) versus healthy donors (IC50 > 400nM). In a mouse model of JAK2V617F+ MPN, oral INCB018424 markedly reduced splenomegaly and circulating levels of inflammatory cytokines, and preferentially eliminated neoplastic cells, resulting in significantly prolonged survival without myelosuppressive or immunosuppressive effects. Preliminary clinical results support these preclinical data and establish INCB018424 as a promising oral agent for the treatment of MPNs.
Introduction
The myeloproliferative neoplasms (MPNs) are a group of related clonal diseases probably arising from hematopoietic progenitor or stem cells.1,2 Patients with MPNs have an increased risk of thrombotic and bleeding complications and disease progression to acute myeloid leukemia. Currently, the treatment of MPNs involves the palliative use of cytoreductive agents. Until recently, the nebulous pathogenesis of MPNs has hampered development of molecularly targeted agents capable of changing the natural history of MPNs, as has been the case for ABL1 tyrosine kinase inhibitors (TKIs) in the treatment of chronic myeloid leukemia. The discovery of a recurrent gain-of-function mutation in MPN patients at nucleotide 1849 of the Janus kinase 2 (JAK2) gene has given hope for new targeted agents.3–7 This mutation substitutes phenylalanine for valine at amino acid 617 (V617F) of the autoinhibitory pseudokinase (JH2) domain, resulting in its constitutive activation.8 V617F is present in 50% to 60% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF), and in 95% of those with polycythemia vera (PV).3–7 Importantly, expression of JAK2V617F confers growth factor independence to cells and ectopic expression in mice results MPN-like phenotypes.4,9–11 These findings support a fundamental role for this mutation and, more broadly, JAK2 activation in the etiology of human MPNs.
JAK2 is a member of the JAK family of cytoplasmic tyrosine kinases, which also include JAK1, JAK3, and TYK2. The JAK enzymes are required for signaling by cytokine and growth factor receptors that lack intrinsic kinase activity.12,13 Although there may be some overlapping role for the different JAKs, each has a primary role in mediating signaling by a subset of factors.12 JAK1 plays a major role in the signaling of a number of proinflammatory cytokines,12,13 often in association with other JAK family members. JAK2 is used primarily by receptors for hematopoietic growth factors, such as erythropoietin and thrombopoietin (TPO). JAK3 appears to have a primary role in mediating immune function, whereas Tyk2 functions in association with JAK2 or JAK3 to transduce signaling of cytokines, such as interleukin-12 (IL-12).12,13
Although JAK2 mutations may account for the majority of deregulated oncogenic signaling in MPN patients, the complex nature of the BCR-ABL1− MPNs and of JAK signaling suggests that patients may benefit from inhibition of both JAK2 and the closely related JAK1. As alluded to earlier in the “Introduction,” JAK1 and JAK2 may interact, resulting in their transactivation.14,15 Interestingly, cytokines capable of signaling through JAK1/2 have recently been shown to convey resistance to inhibition of JAK2V617F with siRNA or TKIs, suggesting a cell autonomous benefit of JAK1/2 inhibition.16 Moreover, it has been documented that patients with primary myelofibrosis (MF) have extremely high levels of circulating inflammatory cytokines, such as IL-6 and tumor necrosis factor-α (TNF-α),17–20 and these cytokines are probably responsible for the hypercatabolic state and constitutional symptoms, such as weight loss and fatigue, frequently seen in patients with MF.21 Recognizing that many of these proinflammatory cytokines use JAK1, and to some extent also JAK2, we hypothesize that selective inhibition of both kinases may provide greater clinical benefit.
In this report, we describe the preclinical characterization of INCB018424, a potent, selective, and orally bioavailable inhibitor of JAK1 and JAK2. Further, we show that JAK1 is hyperactivated in the peripheral blood of patients with MF, implying that combined inhibition of JAK1 and JAK2 may provide superior clinical benefit. INCB018424 is currently undergoing clinical evaluation in MPNs, including MF, PV, and ET.
Methods
Full-length hJAK2 was cloned with a hemagglutinin epitope into pMSCV-puro. The JAK2V617F mutation was generated by site-directed mutagenesis and confirmed by sequencing. BaF/3 cells (DMSZ) were cultured in RPMI with 10% fetal bovine serum and 1 ng/mL IL-3 or IL-6, respectively. The Ba/F3 cell models were generated by nucleoporation of pMSCV-neo-hEPOR and antibiotic selection. Ba/F3-EpoR-JAK2 cells were generated by nucleoporation of Ba/F3-EpoR cells with pMSCV-puro-JAK2 followed by secondary selection. BaF/3-EpoR-JAK2V617F cells were similarly generated with an added selection for IL-3–independent growth. Clones used in these studies were confirmed to have exogenous JAK2 expression by Western analysis. TF-1-BCR-Abl cells were created in similar fashion from parental cells (ATCC) using pMSCV-puro. HEL92.1.7 cells were acquired from the ATCC and cultured in RPMI 1640 with 10% fetal bovine serum.
DNA sequencing and estimation of percentage JAK2V617F allele
DNA sequencing was performed using Big Dye terminator reagent and an ABI3100 instrument. To determine the frequency of JAK2V617F allele in patient samples, the Mutation Surveyor program (SoftGenetics) was used to estimate comparative areas under the curve for the g (wild-type) and the t (mutant) alleles based on a standard curve generated from an admixture of wild-type and Hel92.6 cell (homozygous mutant) DNA. This limit of detection for the mutant allele was 15% using this technique.
Quantification of JAK2V617F cell burden
A quantitative polymerase chain reaction (PCR) method was used to determine the abundance of the Ba/F3-JAK2V617F cells from tissue samples from mice. Two quantitative PCR primer and probe sets were designed against the backbone of the pMSCV-puro-JAK2V617F expression plasmid. These values were normalized to expression levels of the murine CCR2 transcript. JAK2V717F was detected in DNA samples extracted from peripheral blood specimens obtained from patients with MF by PCR and quantified by pyrosequencing. Briefly, exon 12 of JAK2 was amplified by PCR from genomic DNA by primers JAK200F 5′-GCAGAGAGAATTTTCTGAACTAT and JAK200Rbio 5′biotin-CTCTGAGAAAGGCATTAGAAAG and JAK115Fbio 5′biotin-GCAGCAAGTATGATGAGCA and JAK115R 5′CTCTGAGAAAGGCATTAGAAAG for the antisense assay. The captured biotinylated strand was annealed with the sequencing primers JAK200S 5′-GGTTTTAAATTATGGAGTATGT and JAK115S 5′-TCTCGTCTCCACAGA for the antisense strand. The sense and antisense strands were assayed separately by pyrosequencing using PSQ HS 96 Gold SNP reagents and the PSQ HS 96 pyrosequencing machine (Biotage). The sensitivity of the assay is 5%.
Biochemical assays
The kinase domains of human JAK1 (837-1142), JAK2 (828-1132), JAK3 (781-1124), and Tyk2 (873-1187) were cloned by PCR with N-terminal epitope tags. Recombinant proteins were expressed using Sf21 cells and baculovirus vectors and purified with affinity chromatography. JAK kinase assays used a homogeneous time-resolved fluorescence assay with the peptide substrate (-EQEDEPEGDYFEWLE). Each enzyme reaction was carried out with test compound or control, JAK enzyme, 500nM peptide, adenosine triphosphate (ATP; 1mM), and 2.0% dimethyl sulfoxide (DMSO) for 1 hour. The 50% inhibitory concentration (IC50) was calculated as the compound concentration required for inhibition of 50% of the fluorescent signal. Biochemical assays for CHK2 and c-MET enzymes were performed using standard conditions (Michaelis constant [Km] ATP) with recombinantly expressed catalytic domains from each protein and synthetic peptide substrates.
An additional panel of kinase assays (Abl, Akt1, AurA, AurB, CDC2, CDK2, CDK4, CHK2, c-kit, c-Met, EGFR, EphB4, ERK1, ERK2, FLT-1, HER2, IGF1R, IKKα, IKKβ, JAK2, JAK3, JNK1, Lck, MEK1, p38α, p70S6K, PKA, PKCα, Src, and ZAP70) was performed using standard conditions (CEREP; www.cerep.com) using 200nM INCB018424. Significant inhibition was defined as more than or equal to 30% (average of duplicate assays) compared with control values.
Cell proliferation assay
Cells were seeded at 2000/well of white bottom 96-well plates, treated with compounds from DMSO stocks (0.2% final DMSO concentration), and incubated for 48 hours at 37°C with 5% CO2. Viability was measured by cellular ATP determination using the Cell-Titer Glo (Promega) luciferase reagent or viable cell counting. Values were transformed to percent inhibition relative to vehicle control, and IC50 curves were fitted according to nonlinear regression analysis of the data using PRISM GraphPad.
Immunoblotting
Cells were treated for 2.5 hours with inhibitor and total lysates collected. Primary antibodies used for immunoblotting included: phospho-JAK2 (pY1007/1008), phospho-ERK1/2, phospho-Akt (Ser473), and JAK2 from Cell Signaling Technologies, phospho-STAT5 (Y694; Upstate Biotechnology), anti-HA epitope (Sigma-Aldrich), and STAT5a/b (Lab Vision). Antibodies were detected using appropriate horseradish peroxidase-linked secondary antibodies (Pierce Chemical) and developed with chemiluminescent detection reagents and visualized with film or a Kodak Imaging station 440cf.
Detection of active JAK1 in primary patient samples
Peripheral blood mononuclear cells from MF patients were collected with patient consent and Institutional Review Board approval. After lysis and protein quantification, 2 mg of protein from each sample was immunoprecipitated using a polyclonal JAK1 antibody (Cell Signaling) and protein A/G agarose overnight. After washing (3 times), samples were eluted with 2 times loading buffer, and Western blots were performed as described above. Membranes were probed with antiphosphotyrosine antibody (Santa Cruz Biotechnology) and subsequently stripped and reprobed with rabbit anti-JAK1.
Apoptosis
Annexin V staining.
Cells were treated for 20 to 24 hours and stained with annexin V and propidium iodide for analysis of early apoptotic and dead cells, respectively (BD Biosciences). Analysis was performed using a FACSCaliber flow cytometer.
Mitochondrial membrane potential.
Cells were treated for 24 hours and then incubated with 2μM of the dye JC-1 (Invitrogen). Analysis was performed by flow cytometry using 488-nm excitation and 530-nm and 585-nm emission filters. JC-1 exhibits potential-dependent accumulation in the mitochondria where its emission is in the red spectrum (590nM). A fluorescence shift from red (590nM) to green (530nM) indicates redistribution of the dye to the cytoplasm resulting from loss of mitochondrial membrane potential, an early marker for apoptosis.
Colony-forming assay
Mononuclear cells were isolated from peripheral blood from patients with PV or normal control persons by centrifugation through Ficoll (Allcells). A total of 2 × 105 cells from control or patients with PV were plated onto methocult H88434 (StemCell Technologies) supplemented with recombinant cytokines (50 ng/mL stem cell factor, 10 ng/mL granulocyte-macrophage colony-stimulating factor, 10 ng/mL granulocyte colony-stimulating factor, 10 ng/mL IL-3, and 3 U/mL erythropoietin) and with indicated concentrations of INCB018424 or DMSO vehicle. For evaluation of endogenous erythroid colony growth, 3 to 4 × 105 cells from PV patients were plated onto minimal methocult medium (H4531, StemCell Technologies) with INCB018424 or vehicle. Each condition was performed in triplicate. Colonies derived from erythroid (burst-forming units [BFU] and colony-forming units [CFU]-E) and myeloid (CFU-granulocyte macrophage) progenitor cells were counted after 14 days.
In vivo treatment with INCB018424 in a myeloproliferative neoplasm mouse model
All of the procedures were conducted in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals. Mice were fed standard rodent chow and provided with water ad libitum. Ba/F3-JAK2V617F cells (105 per mouse) were inoculated intravenously into 6- to 8-week-old female BALB/c mice (Charles River). Survival was monitored daily, and moribund mice were humanely killed and considered deceased at time of death. Treatment with vehicle (5% dimethyl acetamide, 0.5% methocellulose) or INCB018424 began within 24 hours of cell inoculation, twice daily by oral gavage. Hematologic parameters were measured using a Bayer Advia120 analyzed, and statistical significance was determined using Dunnett testing.
Histology and morphometric analysis
Tissue samples of spleen were fixed in 10% neutral buffered formalin and processed through graded alcohols and a clearing agent, infiltrated and embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. To quantify the effects of INCB018424 on white pulp, a simple morphometric method using point-counts was devised. Images of spleen at 2 times magnification were overlaid with a standardized grid. Point counts were by made tabulating the grid intersects that overlaid total spleen and white pulp. Point counts for white pulps were summed for each group, that is, naive (N = 3), vehicle-treated (N = 6), and INCB018424-treated (N = 6), and mean values calculated. An approximate mean mass of white pulp was calculated using the mean weights of spleens for each group and the relative mean point counts for total spleen and white pulp. Photographic images were acquired with a Nikon Eclipse E800 microscope equipped with a Nikon 20×/0.75 Plan Apo objective, and a Nikon DXM1200 digital camera. Images were processed on a Dell computer with Nikon ACT/1 software and Adobe Photoshop 7.0.
Cytokine analysis
Terminal blood samples were collected by intracardiac puncture with a 1-mL syringe and a 25-G needle and transferred into a 0.5-mL ethylenediaminetetraacetic acid containing tube. Plasma was centrifuged at 10 000g for 10 minutes and then stored at −80°C until the time of analysis. Plasma levels of murine IL-6 and TNF-α were measured according to the manufacturer's directions in commercially available kits from R&D Systems (M6000B and MTA00, respectively). Analysis of cytokine levels in cell culture experiments was performed after culturing cells for multiple days in a restricted volume of culture media.
Results
Identification and biochemical characterization of INCB018424
In an effort to evaluate the therapeutic potential of JAK inhibition in MPNs, we identified INCB018424,22 a potent and selective inhibitor of JAK1 and JAK2 with IC50 values of 3.3nM and 2.8nM, respectively (Table 1). INCB018424 demonstrated modest selectivity against Tyk2 (∼ 6-fold) and marked selectivity (≥ 130-fold) against JAK3. Additional testing revealed remarkable selectivity against Chk2 and c-MET when assessed at their respective Km ATP concentrations (Table 1). Moreover, no significant inhibition against a commercial panel of 26 additional kinases was observed when INCB018424 was tested at a concentration approximately 100-fold the IC50 of JAK1/2 (data not shown).
Table 1.
Enzymatic and functional potency of INCB018424
| IC50, nM (mean ± SD) | N | |
|---|---|---|
| Enzyme assays | ||
| JAK1 | 3.3 ± 1.2 | 7 |
| JAK2 | 2.8 ± 1.2 | 8 |
| JAK3 | 428 ± 243 | 5 |
| Tyk2 | 19 ± 3.2 | 8 |
| CHK2 | > 1000* | 7 |
| cMET | > 10 000* | 1 |
| Whole blood assays | ||
| IL-6 stimulation | 282 ± 54 | 6 |
| TPO stimulation | 281 ± 62 | 4 |
Highest concentration evaluated.
SD indicates standard deviation; IL, interleukin; and TPO, thrombopoietin.
To further characterize the activity of INCB018424 in a clinically relevant setting, we used cytokine-stimulated whole blood assays. After a 10-minute preincubation with increasing concentrations of INCB018424, heparinized whole blood was stimulated for 30 minutes with either IL-6 or TPO. Signaling from these cytokines was previously demonstrated to be specifically sensitive to selective inhibitors of JAK1 and JAK2, respectively (P.A.S. and M.C., unpublished results, February 2008). Therefore, although additional kinases may be activated on cytokine stimulation (eg, JAK2 by IL-6), we interpret inhibition of IL-6–mediated STAT3 phosphorylation as inhibition of JAK1 and antagonism of TPO-mediated STAT3 phosphorylation to reflect selective JAK2 inhibition in this model system. The ability of INCB018424 to inhibit activation of the JAK/STAT pathway was assessed using enzyme-linked immunosorbent assays specific for phosphorylated STAT3. Equipotent inhibition of STAT3 phosphorylation was observed for both cytokines (IC50 ∼ 280nM, Table 1), confirming the ability of INCB018424 to potently inhibit JAK1 and JAK2 in the blood. Whole blood IC50 values from these assays were used as guides for dose selection in the in vivo mouse model studies.
Selective inhibition of JAK2V617F signaling, proliferation, and survival in cells
The results shown in Table 1 demonstrated the potent in vitro activity of INCB018424 on wild-type JAK1 and JAK2. Although there is no biochemical basis for differential activity of ATP-competitive kinase inhibitors on the mutant JAK2V617F, the effect of INCB018424 on cells driven by JAK2V617F is not known. Therefore, we used an engineered cell system (Ba/F3 cells) and HEL cells to evaluate the effects of INCB018424 on viability and JAK-mediated signaling. Cytokine-dependent Ba/F3 cells were transformed to growth factor independence by ectopic expression of JAK2V617F and a requisite type I cytokine receptor (EpoR),23 referred to as Ba/F3-EpoR-JAK2V617F cells. This resulted in constitutive phosphorylation of JAK2 as well as downstream targets, such as STAT5 and ERK.24 Using these cells, we examined the effects of increasing concentrations of INCB018424 on JAK2 signaling. Equal amounts of protein were analyzed by Western blot using antibodies specific for the phosphorylated forms of JAK2, STAT5, and ERK1/2 (Figure 1A). Antibodies detecting total JAK2, STAT5, and ERK1/2 were used for comparison. At concentrations more than or equal to 51nM, we observed a dose-dependent reduction in the phosphorylated forms of each protein with maximal effects at approximately 300nM. There was no change in total levels of the respective proteins, suggesting that the effects were specific for the phosphorylated forms. Similar effects on STAT3 and STAT5 phosphorylation were obtained in the human HEL cell line, which endogenously expresses JAK2V617F (Figure 1B). We also attempted to analyze the effects of INCB018424 on Akt but found the signal to be modest. Nonetheless, when amplified with exogenous cytokine, Akt phosphorylation was observed and was inhibited by INCB018424 with similar potency as JAK, STAT, and ERK (data not shown).
Figure 1.
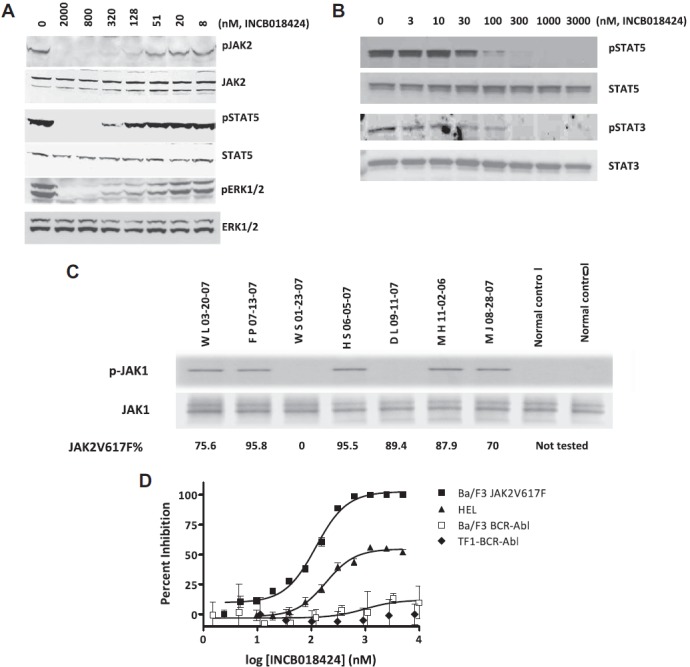
INCB018424 potently and selectively inhibits JAK2V617F-mediated signaling and proliferation. Ba/F3-EpoR-JAK2V617F (A) or HEL (B) cells were treated with increasing concentrations of INCB018424 for 2.5 hours, and extracts from these cells were subjected to immunoblot analysis for phosphorylated (p) or total forms of proteins associated with the JAK/STAT signaling pathway. JAK1 phosphorylation was assessed in primary MF patient samples (C) by immunobloting after immunoprecipitation of total JAK1. The numbers below describe the mutant JAK2V617F allele burden in patient samples. The effect of INCB018424 on viable cell number (D) was assessed after a 48-hour treatment period in cells expressing JAK2V617F (Ba/F3-EpoR-JAK2V617F and HEL) or, to confirm the selective nature of INCB018424, BCR-ABL (TF-1-BCR-ABL and Ba/F3-BCR-ABL).
Attempts to detect activated JAK1 in Ba/F3-EpoR-JAK2V617F cells were unsuccessful and probably reflect the use of EpoR as a scaffolding protein, which signals through JAK2 alone. Therefore, we analyzed the activation state of JAK1 in primary MF patient peripheral blood mononuclear cells. In 5 of 7 MF patients, we detected constitutively phosphorylated JAK1, although not detecting any signal in the healthy donor samples (Figure 1C). These results indicate that, in the majority of MF patients, JAK1 pathway is abnormally active, although at this time it is not known whether this is cell intrinsic or not. This is the first evidence of hyperactivated JAK1 in MF patients and may be a consequence of high levels of circulating inflammatory cytokines, such as IL-6. Additional studies are required to understand the biologic implications of this observation.
To better understand the consequences of JAK1/2 inhibition in cells expressing mutated JAK2, Ba/F3-EpoR-JAK2V617F cells were treated with INCB018424 and a viable cell number was assessed after 48 hours. A dose-dependent reduction in viability was observed with an IC50 of 126nM (Figure 1D square). Growth of HEL cells was also affected by INCB018424 with a 50% effective concentration (EC50) of 186nM. Unlike the BaF/3 cells, HEL cell proliferation was not completely inhibited by INCB018424 (Figure 1B), even though pSTAT3 and pSTAT5 were completely absent at concentrations more than 100nM (Figure 1D triangle) suggesting that the proliferation in these cells is not entirely JAK2V617F dependent. Importantly, the sensitivity of these cell lines to the effects of INCB018424 was in stark contrast to the lack of effect on TF-1 or BaF/3 cells transformed with the BCR-ABL oncoprotein. In these cells, no significant effect on viability was observed, consistent with the selective nature of INCB018424 (Figure 1D, diamond and open square). Likewise, 2 cell lines expressing activating mutations in c-KIT (HMC1.1 and HMC1.2) were also resistant (data not shown). INCB018424 also had no effect on BCR-ABL signaling, as demonstrated by an inability to reduce ABL-mediated pSTAT5, in contrast to imatinib (supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article).
To distinguish between cytostatic and cytotoxic effects of INCB018424 on Ba/F3-EpoR-JAK2V617F cells, we analyzed the percentage of cells in the early stages of apoptosis by fluorescence-activated cell sorter analysis, gating on those cells staining positive for annexin V and negative for propidium iodide. Treatment with INCB018424 markedly increased apoptosis compared with DMSO, with a 4.3-, 7.2-, and 13.2-fold increase at concentrations of 150, 400, and 1000nM, respectively. Similar trends were observed looking at cells that lost their plasma membrane integrity and stained positively for propidium iodide with or without annexin (dead or late apoptotic cells; Figure 2A upper quadrants). To confirm these results, we assessed the loss of mitochondrial membrane potential in Ba/F3-EpoR-JAK2V617F cells treated with similar concentrations of INCB018424. Depolarization of the mitochondria membrane potential is a hallmark of apoptosis and suggests involvement of the intrinsic apoptotic pathway. Treatment with INCB018424 (64nM) resulted in a doubling of cells with depolarized mitochondria, an effect that was dose-dependent (Figure 2B). These results indicate that induction of apoptosis is an important basis of the observed cellular effect of INCB018424 in Ba/F3-EpoR-JAK2V617F cell system.
Figure 2.
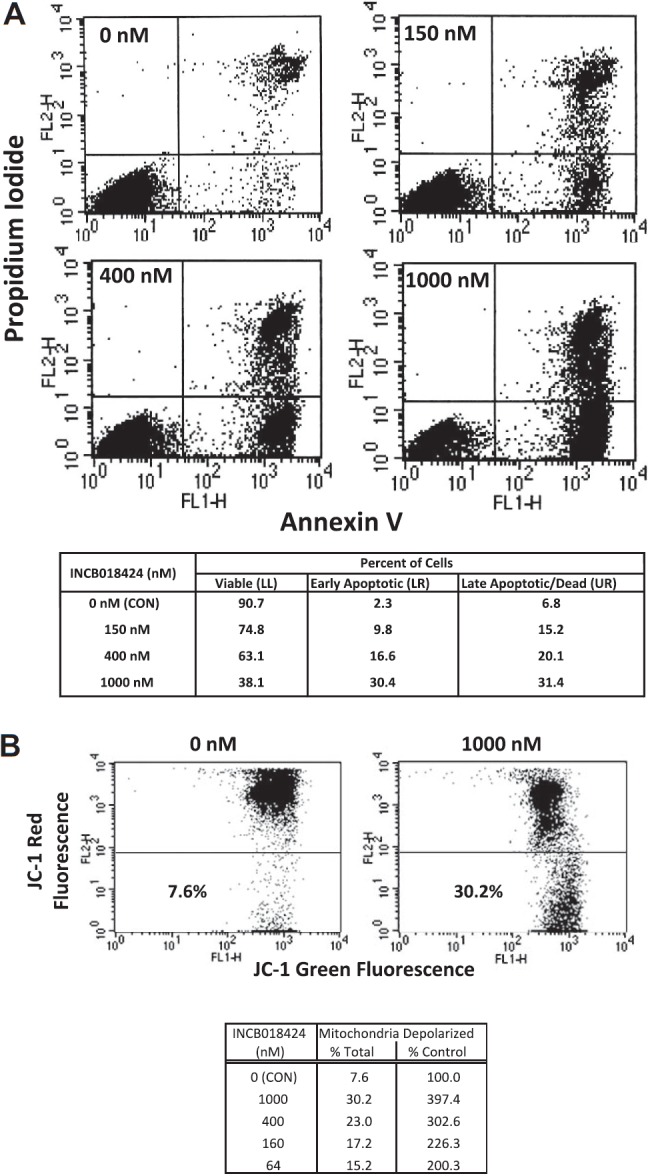
Cells expressing JAK2V617F undergo apoptosis when treated with a selective JAK inhibitor. Ba/F3-EpoR-JAK2V617F cells were treated with various concentrations of INCB018424 for approximately 24 hours and analyzed for hallmarks of apoptosis by annexin V/propidium iodide staining (A) or mitochondrial membrane depolarization (B). Cells treated with INCB018424 were stained with a combination of fluorescein isothiocyanate-annexin V and propidium iodide and analyzed using flow cytometry to determine the percentages of viable (bottom left), early apoptotic (bottom right), and late apoptotic or dead (top right) cells. The effects of JAK inhibition on mitochondrial membrane potential were determined by flow cytometry using JC-1 as a molecular probe (B). Total and relative percentages of cells with depolarized mitochondria are shown.
INCB018424 inhibits hematopoietic progenitor cell proliferation in primary MPN patient samples
To evaluate the effects of INCB018424 on primary JAK2V617F+ cells in MPN patients, mononuclear cells from 3 healthy controls and 3 patients with PV expressing the JAK2V617F mutant allele at frequencies more than 90% (not shown) were obtained. Growth of clonogenic progenitors of erythroid (BFU-E) and myeloid origin (CFU-M) was assessed in colony-forming assays in the presence of increasing concentrations of INCB018424. Dose-dependent inhibition of the growth of erythroid and myeloid progenitors was observed with INCB018424. Sufficient numbers of erythroid progenitors (range, 31-95 per plate for normal donors and 21-34 per plate for PV donors on DMSO control plates) were present to allow reliable quantitation. The mean IC50 for INCB018424 against erythroid progenitors was 407nM for normal donors and 223nM for PV donors (Table 2). A similar effect was observed on myeloid progenitors (CFU-M), with IC50 values of 511nM and 444nM for control and PV samples, respectively; however, the latter should be viewed as estimates because of the low numbers of myeloid progenitors (< 15 colonies per plate, Table 2).
Table 2.
Inhibition of hematopoietic progenitor colony formation from healthy volunteers and polycythemia patients
| Healthy controls (n = 3) | PV (n = 3) | PV without cytokine (n = 3) | |
|---|---|---|---|
| BFU-E IC50, nM | 407 | 223 | 67 |
| CFU-M IC50, nM | 551 | 444 | ND |
BFU-E indicates burst forming unit-erythroid; CFU-M, colony forming unit-myeloid; and PV, polycythemia vera.
JAK2V617F+ human MPNs are characterized by the presence of Epo-independent erythroid colony formation. To assess the effect of INCB018424 on these progenitors, we performed experiments in which PV samples were cultured in the absence of recombinant cytokines. Under these conditions, INCB018424 demonstrated remarkable potency against erythroid colony formation (IC50 = 67nM; Table 2). Moreover, phenotypic assessment of colonies showed that erythroid (BFU-E) colonies were smaller and more dispersed compared with BFU-E colonies arising in complete medium. In agreement with these data, the effects of INCB018424 on proliferation were investigated in erythroid progenitor cells from 3 additional patients with PV after ex vivo expansion, resulting in an IC50 value of 60nM (T.M. and S.V., unpublished results, December 2008). Together, the data suggest that INCB018424 potently inhibits hematopoietic progenitor cell colony formation and that cells derived from patients with PV with mutated JAK2 are more sensitive to JAK1/2 inhibition than normal donors, particularly in the absence of hematopoietic growth factor support.
INCB018424 is efficacious in an in vivo model of JAK2V617F-driven malignancy
We next investigated the in vivo activity of INCB018424 using a modified version of a JAK2V617F-driven malignancy model described previously.25 Briefly, 1 × 105 Ba/F3-EpoR-JAK2V617F cells were injected into the tail vein of Balb/c mice and mice were monitored for splenomegaly and survival over a 3-week period. Those animals receiving no treatment have progressive splenomegaly, a classic feature of myeloproliferative disorder. Further, proliferation and massive accumulation of injected cells resulted in moribund or dead mice in 2 to 3 weeks. We first determined the impact of INCB018424 on the course of disease by randomizing mice to receive vehicle or INCB018424 after inoculation of the Ba/F3-EpoR-JAK2V617F cells (t = day 0). Beginning on day 15, mice from the vehicle group died of disease. By day 22, greater than 90% of the vehicle group had died, whereas, in contrast, greater than 90% of mice treated with INCB018424 survived (Figure 3A). A cohort of mice (n = 6/group) was killed on the first day mortality was noted (day 15), and the effects of INCB018424 on splenomegaly and malignant cell burden were assessed. Vehicle-treated animals had remarkable splenomegaly with a mean weight of 471 mg (∼ 5 times normal), whereas spleen weights of mice treated with INCB018424 were 110 mg, a value comparable with historical naive controls (Figure 3B). Moreover, genomic PCR analysis of spleen samples to detect JAK2V617F cells were significantly decreased by treatment with INCB018424 (33%, P < .01). The dose of INCB018424 used in these studies was demonstrated to normalize uncontrolled JAK/STAT signaling in spleens from mice inoculated with Ba/F3-EpoR-JAK2V617F cells (Figure 3D) and significantly inhibit (> 50%) wild-type JAK2 signaling for at least 16 hours each day, based on documented pharmacodynamic activity in a TPO-stimulated pSTAT3 whole blood assay (supplemental Figure 2). Although inhibiting JAK1/2 continuously and completely may have resulted in a greater reduction in malignant cell burden, it is unlikely compatible with life because of the central roles these kinases play in normal physiology.
Figure 3.

INCB018424 treatment improves viability and splenomegaly in a JAK2V617F-driven model of malignant disease. (A) Kaplan-Meier analysis of mice inoculated intravenously with Ba/F3-EpoR-JAK2V617F cells and treated with INCB018424 or vehicle (orally, twice a day). (B) Survival was significantly improved with INCB018424 treatment (n = 14 mice/group; P < .001, log-rank test). Effects of JAK inhibition on spleen weights were assessed in a separate cohort of animals treated for 2 weeks with vehicle or INCB018424 (n = 6 /group; P < .001; 2-sided t test). Group means are represented by horizontal bars. (C) Quantitative genomic PCR analysis demonstrated a preferential diminution of cells expressing JAK2V617F after treatment with INCB018424 for 2 weeks (P < .01, 2-sided t test). (D) Phospho-STAT3 assay was performed on spleen lysates collected 4 hours after dosing using the Milliplex MAP system (Millipore), normalized to total protein, and read on a Luminex 200 instrument (Luminex).
To confirm that the observed decreases in spleen size and JAK2V617F DNA levels represented a selective decrease in neoplastic cell burden, we performed a histologic analysis of treated spleens. In mice given vehicle, spleens contained coalescing sheets of enlarged, irregular, basophilic cells with large, vesicular, pleomorphic nuclei, occasionally with multiple nucleoli (Figure 4). Large pleomorphic multinucleated cells were scattered throughout as were frequent and unusual mitotic figures. In addition, the sheets of tumor tissue filled the splenic parenchyma and compressed the normal lymphoid components into small, centrally located islands. Conversely, spleens from INCB018424-treated mice demonstrated significantly fewer of these neoplastic features and retained normal lymphoid components (Figure 4B). Quantitative analysis showed that the mass of white pulp in INCB018424-treated mice (0.034 g) was similar to that of naive (0.04 g) and vehicle-treated (0.042) mice (“Histology and morphometric analysis”). Therefore, lower spleen weights in INCB018424-treated mice appear to be a result of reduction in transformed tissue mass, without loss of normal lymphoid tissue, consistent with the PCR analysis.
Figure 4.
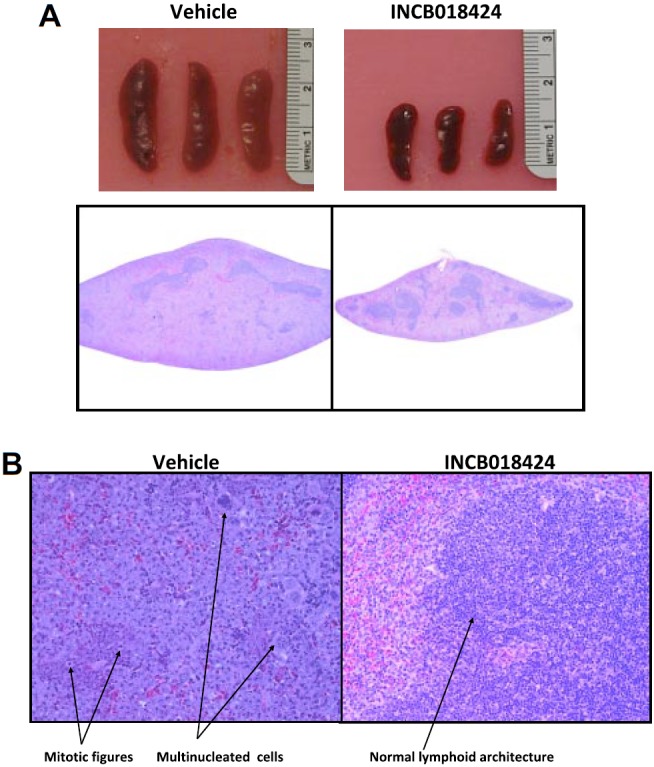
Macroscopic and microscopic effects of INCB018424 on spleens from mice inoculated with Ba/F3-EpoR-JAK2V617F cells. Massive splenomegaly is observed in vehicle-treated mice as assessed by gross appearance (A top) or hematoxylin and eosin-stained transverse sections collected at the midline (A bottom), although markedly reduced with INCB018424 treatment. (B) Microscopic examination (original magnification ×20) of tissue sections shows the massive accumulation of malignant cells in the vehicle group with an abundance of mitotic figures and multinucleated cells present. Treatment with INCB018424 dramatically reduces the malignant cell burden and restores normal lymphoid architecture.
Effects of INB018424 on peripheral blood cell counts in a JAK2V617F-driven malignancy mouse model
We evaluated the effect of INCB018424 dosed chronically for 28 days on peripheral blood cell counts (Figure 5A). No significant alteration in hemoglobin, total red blood cells count, or reticulocyte cell count was observed in mice treated with INCB018424 (n = 5). In contrast, treatment of mice with cyclophosphamide, a known myelosuppressive agent, significantly reduced hemoglobin (26%, P < .01), red blood cells (24%, P < .01), and reticulocytes (70%, P < .01; Figure 5B).
Figure 5.
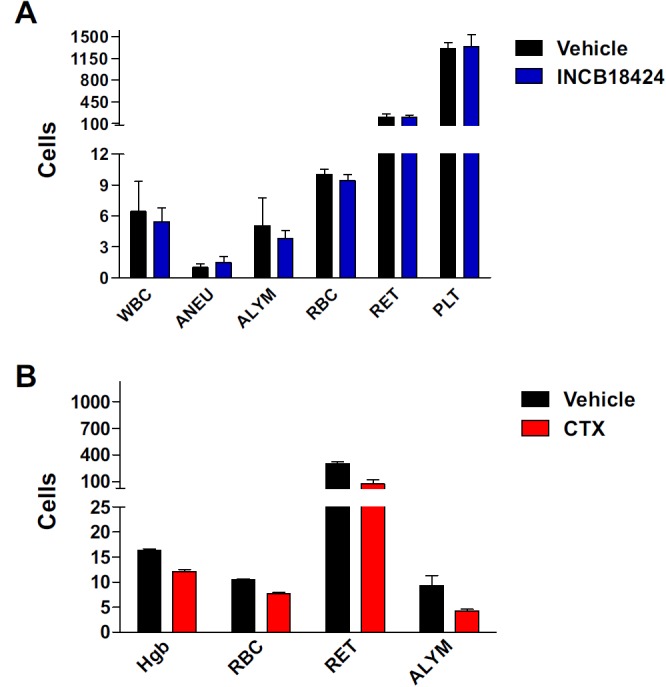
INCB018424 does not affect normal hematologic parameters. Mice (n = 5/group) were treated with INCB018424 or vehicle for 4 weeks at a total daily dose of 180 mg/kg (A). Data are reported for total white blood cell counts (WBC, 103/μL), absolute neutrophil counts (ANEU, 103/μL), absolute lymphocyte counts (ALYM, 103/μL), red blood cells (RBC, 106/μL), reticulocytes (RET, 103/μL), and platelets (PLT, 103/μL). No significant differences were found between treatment groups. For comparison, a subset of parameters was measured in mice treated with cyclophosphamide for 2 weeks (CTX, B). All parameters were significantly decreased by CTX (P < .01). Statistical significance was determined using Dunnett test.
It has been shown that mutations in JAK3, in both humans and mice, are associated with severe combined immunodeficiency disease.26,27 In agreement with the biochemical selectivity of INCB018424 against JAK3 (Table 1), whereas cyclophosphamide significantly decreased the absolute lymphocyte counts compared with vehicle-treated animals after 2 weeks of treatment (Figure 5A, 32%, P < .01), daily administration of INCB018424 for a period of 4 weeks had no significant effect on circulating lymphocyte numbers (P > .05; Figure 5B). Similarly, INCB018424 had minimal effects on thymus weights (data not shown) compared with the 10- to 100-fold reduction reported in the JAK3 knockout mice.26 Taken together, these data demonstrate that the relative selective inhibition of JAK1/2 by INCB018424 reduces the tumor burden of mice inoculated with JAK2V617F-expressing cells without causing anemia or lymphopenia.
Effects of INB018424 on the inflammatory cytokines in a JAK2V617F-driven malignancy in vivo
Next, we evaluated the impact of INCB018424 on cytokine production in the Ba/F3-EpoR-JAK2V617F model. We measured the levels of IL-6 and TNF-α by enzyme-linked immunosorbent assay in plasma samples from inoculated animals treated with vehicle or INCB018424 for 12 days. The levels of both cytokines were markedly increased in vehicle-treated animals compared with those treated with INCB018424 or naive mice (no tumor cell inoculation or treatment, Table 3). INCB018424 resulted in significant suppression of elevated IL-6 levels and reduction of TNF-α levels to normal (Table 3). These data agree with those recently reported in patients with PMF treated with INCB018424.28 In an attempt to determine the source of these elevated cytokines, we analyzed the supernatants of cultured Ba/F3-EpoR-JAK2V617F cells for IL-6 and TNF-α but were unable to detect any (data not shown). These data suggest that the excessive generation of inflammatory cytokines may be part of a reactive process triggered in response to JAK2V617F-expressing cells.
Table 3.
Relative levels of circulating inflammatory cytokines in naive mice compared with those bearing JAK2V617F-driven malignancy
| Naive | Vehicle | INCB018424 | |
|---|---|---|---|
| IL-6 | 1 | 13 | 6.7 |
| TNF-α | 1 | 2.4 | 1 |
IL indicates interleukin; and TNF, tumor necrosis factor.
Discussion
Treatment options for patients with MPNs have been largely empiric and result in limited clinical activity. Despite the use of cytoreductive agents, the median survival of patients with PMF is approximately 5 years and their quality of life is greatly diminished because of progressively worsening marrow hematopoietic function resulting in anemia and thrombocytopenia, the development of increasingly severe splenomegaly, and profound cytokine-mediated constitutional symptoms. By contrast, the clinical outcome of patients with BCR-ABL1+ chronic myeloid leukemia, another MPN, has dramatically improved with the introduction of TKIs (eg, imatinib) that inhibit the activity of the BCR-ABL1 kinase, which drives the leukemic clone. The discovery of the JAK2V617F mutation not only provides a common molecular link that unifies the pathogenesis of these MPNs but also renders these disorders amenable to targeted therapeutic interventions. The JAK2V617F mutation is thought to lead to constitutive JAK2 kinase activity by eliminating a negative feedback signal normally mediated by the pseudokinase domain. JAK2V617F expression transforms a variety of hematopoietic cell lines,4,6 the efficiency of which is increased in the presence of erythropoietin, TPO, or granulocyte colony-stimulating factor receptors.23 The ability of JAK2V617F-expressing hematopoietic cells to recapitulate a polycythemic phenotype, including evolution to myelofibrosis, in murine models supports the notion that constitutive JAK2 signaling caused by the V617F mutation plays a central role in the pathogenesis of MPNs. Supporting the therapeutic potential of JAK2 inhibitors for the treatment of MPNs is the fact that they dramatically decrease the in vitro proliferation of JAK2V617F-carrying cell lines and primary cells obtained from patients with JAK2V617F+ MPNs.6,29 Several small-molecule TKIs with potent activity against JAK2 V617F have been developed, and some are already being evaluated in clinical trials for patients with MPNs and other disorders associated with elevated JAK activity.
Here, we report on the preclinical activity of INCB018424, a potent and selective JAK1/JAK2 TKI. We demonstrate that, at nanomolar concentrations, INCB018424 inhibits JAK2V617F, STAT5, and ERK1/2 phosphorylation resulting in reduced cellular proliferation and the induction of apoptosis by JAK2V617F+ Ba/F3 cells. INCB018424 potently inhibited the proliferation of ex vivo expanded erythroid progenitors obtained from patients with JAK2V617F+ PV. In a murine model of JAK2V617F-driven malignancy, therapy with INCB018424 significantly reduced splenomegaly and increased survival. In addition, INCB018424 decreased circulating levels of proinflammatory cytokines, IL-6 and TNF-α, which have been implicated in the pathogenesis of debilitating constitutional symptoms seen in advanced MPNs. Although animals were not cured of their disease, our results are consistent with those observed in BCR-ABL1–driven mouse models.30 The in vivo efficacy of INCB018424 is unlikely to be impacted by selection for resistance mutations in JAK2 because in vitro studies in Ba/F3-EpoR-JAK2V617F cells suggest that resistant clones are extremely infrequent compared with those observed in BCR-ABL1–expressing cells treated with imatinib (T.B. and P.L., unpublished results, August 2006 to December 2007). Moreover, the mutants that do arise have only a modestly (< 10-fold) decreased sensitivity and no detectable kinase domain mutations. It should, however, be noted that these were not exhaustive studies and may have identified only rare putative resistance alleles. Further studies will be required to ascertain the basis of response and resistance to JAK inhibitors.
INCB018424 is somewhat unique among the JAK kinase inhibitors in clinical development as it potently inhibits JAK1 in addition to JAK2. There may be both cell autonomous and nonautonomous benefits of inhibiting JAK1 as well as JAK2, although these remain hypothetical at this time. For instance, Jedidi et al have recently demonstrated that cytokines capable of signaling through JAK1 can convey resistance to JAK2-targeted therapies.16 Moreover, analogous to certain examples of cytokine signaling, these 2 JAKs can interact and may activate one another.14,15 Interestingly, here we show, for the first time, that JAK1 is hyperactivated in the peripheral blood of MF patients, although the mechanism is currently an area of ongoing investigation. Systemically, inhibition of JAK1 may help alleviate many of the constitutional symptoms patients endure because of their remarkably high levels of JAK-activating inflammatory cytokines (eg, IL-6) and their resulting state of chronic inflammation. For instance, infusion of recombinant IL-6 was recently shown to result in increased JAK/STAT signaling in skeletal muscle, atrophy, and reduced strength, all of which were reversed with a selective JAK1/2 inhibitor.31 Perhaps if selective JAK1 inhibitors are developed in the future, these therapeutic hypotheses may be tested. Alternatively, experiments in genetically engineered mouse models of JAK2-driven MPNs performed in mice lacking JAK1 may provide some insights into the contribution of JAK1 in these diseases.
JAK3 kinase activity is critical for lymphopoiesis.26,32 INCB018424 was designed to spare JAK3 to avoid impairing lymphopoiesis with chronic administration. The potency of INCB018424 against JAK3 kinase (IC50 = 322nM) is 2 logs higher than that required to inhibit the kinase activity of the JAK2 protein (IC50 = 4.5nM). This implies that malignant JAK2V617F+ clones harbored by patients with MPNs can be inhibited at a dose that will not cause JAK3 inhibition. In this regard, it is worth noting that none of the mice treated herein with INCB018424 for a month developed lymphopenia.
The preclinical data herein presented indicate that INCB018424, a potent inhibitor of JAK1 and JAK2, has the potential to be an efficacious therapy for patients with JAK2V617F+ MPNs. To date, INCB018424 has been given to more than 100 patients with PMF, post-PV, or post-ET MF in an ongoing phase 1/2 study. In this setting, INCB018424 administration has resulted in swift and dramatic reduction of splenomegaly. Interestingly, INCB018424 has proven equally effective in JAK2V617F+ and JAK2V617F− patients. In addition, most patients have experienced a marked improvement in constitutional symptoms, nutritional status, cachexia, and quality of life. These improvements are paralleled by a dramatic and rapid reduction in MF-associated inflammatory cytokine levels, closely mirroring the data predicted by our in vivo model of JAK2V617F-driven malignancy.
Supplementary Material
Acknowledgments
The authors thank Roberto Nussenzveig for his contribution of data in expanded cultures of erythroid progenitors, and various members of Incyte Corporation for their technical assistance and review of this manuscript.
This work was supported in part by the Chambers Medical Foundation, the Joe W. and Dorothy Dorsett Brown Foundation, and the Marshall Heritage Foundation (all S.V.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.Q.-C., H.K., and S.V. interpreted results from experiments and contributed to the authorship of the manuscript; J.S.F. and K.V. designed experiments, interpreted results, and contributed to the authorship of the manuscript; P.L., J.L., P.A.S., E.C., X.W., Y. Li, M.C., T.B., P.W., Y. Lo, T.M., J.K., M.R., and P.H. conducted various experiments and/or helped analyze their results; and S.S. and J.D.R. optimized a series of compounds to identify and synthesize INCB018424.
Conflict-of-interest disclosure: All authors listing Incyte Corporation as their affiliated institution are or have been full time employees of Incyte Corporation. As part of this employment, authors may own shares in Incyte and/or hold stock options for shares in Incyte. The remaining authors declare no competing financial interests.
Correspondence: Srdan Verstovsek, University of Texas M. D. Anderson Cancer Center, Department of Leukemia, Unit 428, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: sverstov@mdanderson.org; and Jordan S. Fridman, Incyte Corporation, Experimental Station, E400, Wilmington, DE 19880; e-mail: jfridman@incyte.com.
References
- 1.Prchal JF, Axelrad AA. Bone-marrow responses in polycythemia vera [Letter]. N Engl J Med. 1974;290(24):1382. doi: 10.1056/nejm197406132902419. [DOI] [PubMed] [Google Scholar]
- 2.Zanjani ED, Lutton JD, Hoffman R, Wasserman LR. Erythroid colony formation by polycythemia vera bone marrow in vitro: dependence on erythropoietin. J Clin Invest. 1977;59(5):841–848. doi: 10.1172/JCI108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 4.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–3395. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 10.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107(11):4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaleskas VM, Krause DS, Lazarides K, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS ONE. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–142. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19(4):385–393. doi: 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Pradhan A, Lambert QT, Reuther GW. Transformation of hematopoietic cells and activation of JAK2-V617F by IL-27R, a component of a heterodimeric type I cytokine receptor. Proc Natl Acad Sci U S A. 2007;104(47):18502–18507. doi: 10.1073/pnas.0702388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang HM, Lin YL, Chen CH, Chang TW. Simultaneous activation of JAK1 and JAK2 confers IL-3 independent growth on Ba/F3 pro-B cells. J Cell Biochem. 2005;96(2):361–375. doi: 10.1002/jcb.20513. [DOI] [PubMed] [Google Scholar]
- 16.Jedidi A, Marty C, Oligo C, et al. Selective reduction of JAK2V617F-dependent cell growth by siRNA/shRNA and its reversal by cytokines. Blood. 2009;114(9):1842–1851. doi: 10.1182/blood-2008-09-176875. [DOI] [PubMed] [Google Scholar]
- 17.Wang JC, Chang TH, Goldberg A, Novetsky AD, Lichter S, Lipton J. Quantitative analysis of growth factor production in the mechanism of fibrosis in agnogenic myeloid metaplasia. Exp Hematol. 2006;34(12):1617–1623. doi: 10.1016/j.exphem.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt A, Jouault H, Guichard J, Wendling F, Drouin A, Cramer EM. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96(4):1342–1347. [PubMed] [Google Scholar]
- 19.Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105(11):4508–4515. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- 20.Panteli KE, Hatzimichael EC, Bouranta PK, et al. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br J Haematol. 2005;130(5):709–715. doi: 10.1111/j.1365-2141.2005.05674.x. [DOI] [PubMed] [Google Scholar]
- 21.Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342(17):1255–1265. doi: 10.1056/NEJM200004273421706. [DOI] [PubMed] [Google Scholar]
- 22.Lin Q, Meloni D, Pan Y, et al. Enantioselective synthesis of Janus kinase inhibitor INCB018424 via an organocatalytic aza-Michael reaction. Org Lett. 2009;11(9):1999–2002. doi: 10.1021/ol900350k. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102(52):18962–18967. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridman J, Nussenzveig R, Liu P, et al. Discovery and preclinical characterization of INCB018424, a selective JAK2 inhibitor for the treatment of myeloproliferative disorders [abstract]. Blood. 2007;110(11) Abstract 3538. [Google Scholar]
- 25.Pardanani A, Hood J, Lasho T, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21(8):1658–1668. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 26.Nosaka T, van Deursen JM, Tripp RA, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270(5237):800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 27.Russell SM, Tayebi N, Nakajima H, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270(5237):797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 28.Tefferi A, Kantarjian H, Pardanani AD, et al. The clinical phenotype of myelofibrosis encompasses a chronic inflammatory state that is favorably altered by INCB018424, a selective inhibitor of JAK1/2 [abstract]. Blood. 2008;112(11) Abstract 2804. [Google Scholar]
- 29.Jamieson CH, Gotlib J, Durocher JA, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A. 2006;103(16):6224–6229. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wendel HG, de Stanchina E, Cepero E, et al. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci U S A. 2006;103(19):7444–7449. doi: 10.1073/pnas.0602402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fridman JS, Caulder E, Wen X, et al. Small molecule inhibitors of JAK1/2 improve physiological and functional measures of cancer-associated cachexia [abstract]. Proc Am Assoc Cancer Res. 2009 Abstract 2848. [Google Scholar]
- 32.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270(5237):794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.