

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Choroideremia (Tapetochoroidal dystrophy).
1.2 OMIM# of the disease
303100.
1.3 Name of the analysed genes or DNA/chromosome segments
CHM (formerly REP1, GGTA, RAB geranylgeranyl transferase component A or RAB GG transferase).
1.4 OMIM# of the gene(s)
300390.
1.5 Mutational spectrum
Choroideremia is an X-linked recessive inherited chorioretinal dystrophy caused by mutations in the CHM gene, this spans a genomic sequence of ∼150 kb on chromosome Xq 21.2, contains 15 exons and encodes a ubiquitously expressed protein of 653 amino acids; Rab Escort Protein 1 (REP1). REP1 is an essential component of the catalytic Rab geranyl-geranyl transferase (RGGTase) II complex, and is involved in the regulation of intracellular membrane transport traffic.
There have been over 130 unique mutations in CHM reported to date (web-based database http://www.lovd.nl/CHM). Characterisation of the mutation spectrum reveals deletions, insertions, duplications, translocations, nonsense, splice-site, frameshift and missense mutations. Full gene and partial deletions represent 25–50% of mutations, and a further 30% are nonsense mutations resulting in premature termination codons. Deletions vary in size from a few kilobases removing a single exon to ∼15 Mb comprising the entire CHM gene and large parts of chromosome Xq21.1 Two missense mutations have been reported: using in silico analysis, the c.1679 T>C (p.L550P) mutation was predicted to destabilise the β-structural elements and tertiary structure resulting in absence of REP1 in patient lymphocytes;2 the c.1520A>G (p.H507R) missense was found to generate a functionally inactive REP1 variant that was not capable of interacting with RGGTase.3
Carrier females are generally asymptomatic but funduscopic examination often shows patchy areas of chorioretinal atrophy that represent clonal areas of the disease due to random X-inactivation. However, later in life, carrier females can often develop night blindness and field loss because of expanding areas of chorioretinal atrophy. Translocations between the X-chromosome and an autosome, disrupting CHM have been detected in females (but not males), displaying mild clinical signs of choroideremia and ovarian dysgenesis; t(X;7)(q21.2;p12), t(X;13)(q21.2;p12) and t(X;4)(q21.2; p16.3).4, 5, 6 A fourth complex translocation involving chromosomes X, 1 and 3: t(X;1;3)(q13;q24;q21),inv(9)(p11q13) has been identified in a female carrier with a severe choroideremia phenotype (thought to be due to nonrandom inactivation of the normal X-chromosome) and ectodermal dysplasia, with no comment of gonadal dysgenesis.7
1.6 Analytical methods
Bi-directional fluorescent Sanger sequencing of coding and intron–exon boundaries of CHM is the mainstay analytical method.8 However, a multiplex ligation-dependent probe amplification assay (MLPA) has been developed to test for deletions and duplications within the CHM gene, and this is particularly useful in suspected heterozygotes.9 Sequence variants are described following HGVS nomenclature guidelines (http://www.hgvs.org/) relative to the NCBI reference sequence NM_000390.2.
1.7 Analytical validation
Parallel bi-directional fluorescent Sanger sequencing of known controls is required to validate procedures. Diagnostic testing must be carried out within a laboratory environment working to standards compliant with the ISO 15189. All mutations reported to date in the CHM gene result in nonfunctional or apparent complete absence of REP-1 in affected males, and this can be validated by immunoblot analysis of protein from peripheral blood lymphocytes.10
1.8 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence)
If known to be variable between ethnic groups, please report):
1.9 If applicable, prevalence in the ethnic group of the investigated person
Not applicable.
1.10 Diagnostic setting

Comment: Not applicable.
2. TEST CHARACTERISTICS
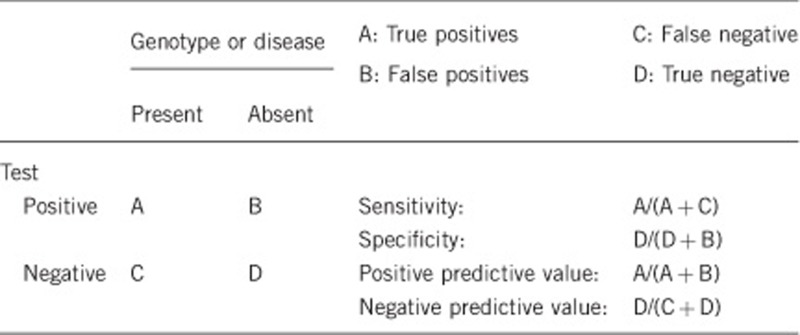
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
We estimate that the analytical sensitivity and specificity of the test used (bi-directional Sanger sequencing) will be >98%. A small number of families, with a typical phenotype in affected males and obligate female carriers, who show linkage to CHM, appear not to have mutations on testing described above. Hence, a small loss of sensitivity may be due to intronic or other mutations missed through exonic analysis. The proportion of such cases is not known, and in one cohort, 2 families out of 120 are in this category (AR Webster, personal communication, Moorfields Eye Hospital).
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
See above. We estimate analytical specificity of >98% given current testing methodologies, based on the false positives that can rarely occur in Sanger sequencing.
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
Choroideremia, unlike other retinal dystrophies, is not genetically heterogeneous and hence the clinical sensitivity and specificity are both high. Phenocopies do exist and include specific dominant alleles of RPE65 and RDS.11, 12 If the families are small, this can lead to an apparent reduction in sensitivity upon CHM testing. Otherwise, in large X-linked pedigrees with a typical phenotype, the clinical sensitivity will be the same as the analytical sensitivity declared, that is, >98%.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
A positive test in a male individual without signs of choroideremia is unlikely as the funduscopic abnormalities are usually apparent before the age of 5 years and hence the clinical specificity will be high.13 In females, however, carrier signs are variable and are more apparent with increasing age; occasionally they may not be apparent in adulthood. The clinical specificity will be low in young females, in which a positive genetic test will be useful in the context of a normal examination. Importantly, retinal examination and investigation, such as an electroretinogram, cannot be used to exclude carrier status in young at-risk females, and genetic testing will be required.
2.5 Positive clinical predictive value (life-time risk of developing the disease if the test is positive)
Estimated >99% for CHM mutations in males, although variable expressivity is recognised. Nonpenetrance has not been reported. Because of the rarity of missense changes (see above), the positive predictive value of a novel missense change remains low, until corroborative biochemical studies are performed on the specific mutant protein.
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
Assume an increased risk based on family history for a nonaffected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
For known pathogenic changes, or novel null mutations, the negative predictive value will be approaching 100%.
Index case in that family had not been tested:
In a male with a 1 in 2 prior risk of being affected, where clinical data are not available, a negative test result is highly predictive of unaffected status, but will fall short of 100% due to the analytical specificity noted above (a small proportion of families do not show CHM mutations). Hence, such a result can be interpreted more accurately, if the proband has already been tested.
3. Clinical Utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?
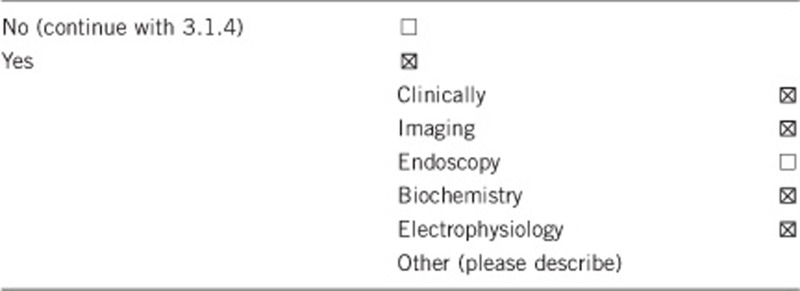
3.1.2 Describe the burden of alternative diagnostic methods to the patient
For patients with suspected choroideremia, a clinical diagnosis can be made based on medical history and fundus examination. It is characterised by a slowly progressive degeneration of the choroid, photoreceptors and retinal pigment epithelium (RPE), initially generating a peripheral pigmentary retinopathy, with gradually enlarging areas of RPE and choroidal atrophy resulting in exposure of the choroidal vessels in front of bare sclera. The macula initially remains intact, but undergoes atrophy in the late stages of the disease.
Supplementary clinical investigations include fundus autofluorescence, optical coherence tomography (OCT) and electrophysiology. Autofluorescence is decreased in the areas of chorioretinal atrophy with relatively high signal in the preserved retinal tissue of the macula. OCT reveals absence of the outer nuclear layer and outer segments, RPE and choroid. Electroretinograms (ERGs) show early loss of rod function in response to dim scotopic stimuli in affected male children, followed by deterioration of cone function (evoked by bright flashes and flicker stimuli) and, ultimately, a severely reduced or unrecordable ERG in later stages of disease.14, 15 However, there can be intrafamilial and interfamilial variabilities of ERG responses.16 Hence, presymptomatic testing may be undertaken with the aforementioned complement of investigations in association with genetic testing.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Choroideremia is a rare disorder, and its clinical recognition will be challenging to specialists who do not manage the disease regularly. Access to high-resolution imaging plus electroretinography is not always available and can be costly. Patients will often require tertiary referral for accurate diagnosis. The cost of an electroretinogram is in the order of £500. The cost of an autofluorescent camera, the most sensitive of imaging equipment for this disease, is in the order of £80 000, and the unit cost of imaging is £100. Although precise phenotyping is important for a proband, given the high sensitivity and specificity of genetic testing, a presymptomatic genetic test may be cost effective overall compared with the high cost of clinical assessment.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?

3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
As most patients with choroideremia suffer from night blindness in childhood with progressive loss of vision through their active adult life, professions requiring perfect vision are impossible. Hence, a clinically confirmed diagnosis can already help in providing guidance regarding career choice.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes. Choroideremia is an X-linked inherited chorioretinal dystrophy. A molecular diagnosis in an affected individual can resolve the genetic situation in that family, determine X-linked segregation unambiguously and is a prerequisite for genetic counselling of family members.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
If molecular testing has identified a CHM mutation in the index patient, examination can identify, and exclude disease in at-risk males. However, further genetic tests are required to determine the carrier status of females (see above), but this must be undertaken following genetic counselling and arguably when they are able to make their own decision.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Genetic testing for CHM mutations will provide a precise molecular diagnosis. This yields information regarding recurrence risk, carrier status and hence will provide choices that would not otherwise be available to facilitate decision making for the patient and their family. Gene testing is essential in defining inheritance patterns and enabling effective genetic counselling. A positive gene test will preclude the need for further genetic testing.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. MM gratefully acknowledges the support of the Choroideremia Research Foundation and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
The authors declare no conflict of interest.
References
- van den Hurk JA, Schwartz M, van Bokhoven H, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997;9:110–117. doi: 10.1002/(SICI)1098-1004(1997)9:2<110::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Sergeev YV, Smaoui N, Sui R, et al. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res. 2009;665:44–50. doi: 10.1016/j.mrfmmm.2009.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, De Falco F, Tinto N, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat. 2011;32:1460–1469. doi: 10.1002/humu.21591. [DOI] [PubMed] [Google Scholar]
- Cremers FP, van de Pol DJ, van Kerkhoff LP, Wieringa B, Ropers HH. Cloning of a gene that is rearranged in patients with choroideraemia. Nature. 1990;347:674–677. doi: 10.1038/347674a0. [DOI] [PubMed] [Google Scholar]
- van Bokhoven H, van den Hurk JA, Bogerd L, et al. Cloning and characterization of the human choroideremia gene. Hum Mol Genet. 1994;3:1041–1046. doi: 10.1093/hmg/3.7.1041. [DOI] [PubMed] [Google Scholar]
- Lorda-Sanchez IJ, Ibanez AJ, Sanz RJ, et al. Choroideremia, sensorineural deafness, and primary ovarian failure in a woman with a balanced X-4 translocation. Ophthalmic Genet. 2000;21:185–189. [PubMed] [Google Scholar]
- Mukkamala K, Gentile RC, Willner J, Tsang S. Choroideremia in a woman with ectodermal dysplasia and complex translocations involving chromosomes X, 1, and 3. Ophthalmic Genet. 2010;31:178–182. doi: 10.3109/13816810.2010.497529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald IM, Smaoui N, Seabra MC.Choroideremiain Pagon RA, Adam MP, Bird TD, et al (eds).: GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2013. Available from http://www.ncbi.nlm.nih.gov/books/NBK1337/ , 21 Feb 2003 [Updated 3 June 2010]. [Google Scholar]
- Chi JY, Macdonald IM, Hume S.Copy number variant analysis in CHM to detect duplications underlying Choroideremia Ophthalmic Genet 2012;. e-pub ahead of print 28 December 2012; doi: 10.3109/13816810.2012.752016 [DOI] [PubMed]
- MacDonald IM, Mah DY, Ho YK, Lewis RA, Seabra MC. A practical diagnostic test for choroideremia. Ophthalmology. 1998;105:1637–1640. doi: 10.1016/S0161-6420(98)99031-5. [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–1081. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelides M, Holder GE, Bradshaw K, Hunt DM, Moore AT. Cone-rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology. 2005;112:1592–1598. doi: 10.1016/j.ophtha.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Karna J. Choroideremia. A clinical and genetic study of 84 Finnish patients and 126 female carriers. Acta Ophthalmol Suppl. 1986;176:1–68. [PubMed] [Google Scholar]
- Mura M, Sereda C, Jablonski MM, MacDonald IM, Iannaccone A. Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. Arch Ophthalmol. 2007;125:1107–1113. doi: 10.1001/archopht.125.8.1107. [DOI] [PubMed] [Google Scholar]
- Sieving PA, Niffenegger JH, Berson EL. Electroretinographic findings in selected pedigrees with choroideremia. Am J Ophthalmol. 1986;101:361–367. doi: 10.1016/0002-9394(86)90832-9. [DOI] [PubMed] [Google Scholar]
- Renner AB, Kellner U, Cropp E, et al. Choroideremia: variability of clinical and electrophysiological characteristics and first report of a negative electroretinogram. Ophthalmology. 2006;113:2066, e1–e10. doi: 10.1016/j.ophtha.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Tolmachova T, Tolmachov OE, Wavre-Shapton ST, Tracey-White D, Futter CE, Seabra MC. CHM/REP1 cDNA delivery by lentiviral vectors provides functional expression of the transgene in the retinal pigment epithelium of choroideremia mice. J Gene Med. 2012;14:158–168. doi: 10.1002/jgm.1652. [DOI] [PubMed] [Google Scholar]
- Moosajee M, Gregory-Evans K, Ellis CD, Seabra MC, Gregory-Evans CY. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum Mol Genet. 2008;17:3987–4000. doi: 10.1093/hmg/ddn302. [DOI] [PubMed] [Google Scholar]