

Abstract
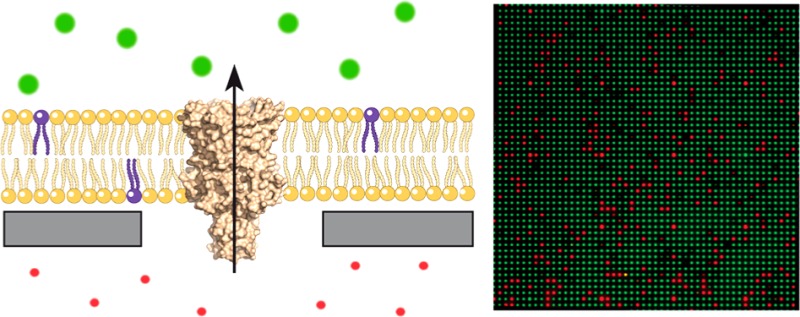
Membrane proteins are prime drug targets as they control the transit of information, ions, and solutes across membranes. Here, we present a membrane-on-nanopore platform to analyze nonelectrogenic channels and transporters that are typically not accessible by electrophysiological methods in a multiplexed manner. The silicon chip contains 250 000 femtoliter cavities, closed by a silicon dioxide top layer with defined nanopores. Lipid vesicles containing membrane proteins of interest are spread onto the nanopore-chip surface. Transport events of ligand-gated channels were recorded at single-molecule resolution by high-parallel fluorescence decoding.
Keywords: Biosensor, membrane proteins, nanopore, nanofabrication, lab-on-chip, suspended lipid bilayer
The human genome codes for approximately 6000 membrane proteins, accounting for 25–30% of all open reading frames.1−3 Despite their essential role in cell homeostasis and diseases, membrane proteins have persistently evaded efficient analyses due to the lack of suitable techniques and major challenges with regard to their fragile properties and low abundance. Their importance is underlined by the fact that more than 60% of all drugs prescribed today target membrane proteins.4 In particular, channels and transporters are of special interest as prime drug targets as they control the flux of ions, nutrients, and other solutes across cell membranes. In addition, membrane transporters are key secondary drug targets as they control the absorption, distribution, metabolism, and excretion of drugs targeting other proteins.5 Many medically important membrane proteins fulfill their function in intracellular membranes, which are difficult to access by conventional approaches and have so far evaded high-throughput approaches for their characterization.
Techniques to investigate the function of membrane transport proteins have been developed whereas some of them are commercially available.6,7 Systems that use solid-supported membranes present a promising tool to study channels and transporters,8−10 including solid-supported lipid bilayers,11−13 tethered bilayers,14,15 microblack lipid membranes,16 and native vesicle arrays.17 However, these methods are not yet able to combine highly parallel, semiautomated multiplexed analysis, small sample consumption, high sensitivity, and a single molecule resolution. The development of chip arrays consisting of spatially confined compartments enclosed by solid-state nanopores combines these desired features in a single platform.18,19 Considerable efforts have been made in this field using silicon20−22 or aluminum oxide.23 However, these chips are laborious as well as expensive in production and reproducible fabrication quality (at the nanoscale) is not always ensured.
Here, we present a chip architecture containing an array of 250 000 nanopores with well-defined femtoliter cavities. A 5-in. silicon-on-insulator (SOI) wafer was structured by reactive-ion etching, creating cylindrical cavities of approximately 1 μm in diameter and 10 μm in depth. Subsequently, a silicon dioxide top layer was formed by chemical vapor deposition, narrowing the cavity opening to a pore in the nanometer range. The mean nanopore diameter over an entire wafer was varied from 70 to 120 nm with an enclosed cavity volume of 6 fL (Figure 1). The standard deviation of the pore opening across individual chips is 9 nm (n = 84). Each wafer comprises approximately 1150 individual chips of identical quality with 250 000 homogeneous nanopores on a single chip. All cavities are arranged in a rectangular pattern with the ability to address each cavity individually by fluorescence readout. In addition, the chip possesses an opaque top layer and a transparent glass bottom, which allows the observation of export but also import processes.
Figure 1.

Design of multiplexed nanopore biochips. (A) An SOI wafer is structured by reactive-ion etching. Approximately 1150 individual chips are fabricated from each wafer with identical properties and quality. (B) Each chip comprises 250 000 individual microcavities with nanoapertures. Scale bar: 200 μm. (C) Each cavity is addressable via multispectral fluorescence read-out. An intransparent top layer blocks the fluorescent signals from the buffer reservoir, making the biochip compatible with inverted fluorescence microscopes. (D) AFM imaging reveals evenly arranged pore openings and surface roughness of the silicon dioxide layer of 3.6 nm (n = 40) optimal for vesicle fusion. Scale bar: 5 μm. (E) SEM image shows a cross-section through the nanopore allowing access to the femtoliter cavities inside the silicon chip.
Chips processed as described above were analyzed by atomic force microscopy (AFM) and scanning electron microscopy (SEM). All imaged chips (n = 5) showed perfectly ordered arrays of nanopores with a pitch of 4 μm (Figure 1D). A surface roughness of Rq = 3.6 nm was determined by AFM (n = 40). The accurate fabrication of the cavities and nanopore diameters was further confirmed via SEM for several cross sections of the chip, underlining the constricted opening of each pore (Figure 1E). In conclusion, the nanopore design offers a high throughput analysis of single transport events by simple fluorescence read-out.
Formation of pore-spanning lipid bilayers by spreading large unilamellar vesicles (LUVs) bears several advantages in comparison to alternative methods. Typically, LUVs with average diameters of 150–200 nm were employed to create pore-spanning suspended lipid bilayers (SLBs). Black lipid membranes in contrast contain organic solvents and may negatively affect the function of membrane proteins. Giant unilamellar vesicles (GUVs) can seal nanoporous openings by vesicle fusion,21,22 but reconstitution of membrane proteins into GUVs is very challenging. Compared to these methods, membrane proteins are efficiently reconstituted into LUVs under physiological conditions and without loss of their activity. Proteoliposomes can then directly be applied to the chip for SLB formation.24,25
In order to prepare suspended lipid bilayers, different lipids and lipid mixtures were tested for their ability to form stable pore-spanning lipid bilayers. In general, lipid mixtures containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), or cholesterol can be used. LUVs consisting of soybean lipids or a mixture of E. coli polar lipids and DOPC in a ratio of 7:3 (w/w) yielded the best result for our studies. Vesicle spreading to the chip surface is aided by adding 5 mM CaCl2 prior to LUV application, increasing the adherence of vesicles to the silicon dioxide chip surface. Liposomes and proteoliposomes were analyzed by nanoparticle tracking, yielding a monodisperse diameter of 214 ± 70 and 158 ± 60 nm, respectively (Supporting Information Figure S1A). The diameter of the liposomes must be well above the diameter of the nanopore to avoid vesicle contamination in the cavities. Nanopore suspended membranes created by this approach are stable for 48 h (Figure 2A and Movie M1) and seal a maximum of approximately 94% of the cavities, enclosing small and large solutes, such as organic fluorophores (0.7 kDa) or fluorescently labeled dextrans (10–70 kDa).
Figure 2.

Long-term stability and fluidity of nanopore suspended lipid bilayers. (A) Small fluorescent dyes (e.g., ATTO488) can be entrapped inside the cavities by SLB formation. All cavities that are sealed from the outside buffer reservoir by an intact lipid bilayer show a fluorescent signal, whereas all nonsealed cavities appear black. SLBs are stable for 20 h. Scale bar = 10 μm. (B) LUVs (0.5 mg/mL SoyPC20 + 0.1 mol % Bodipy-PE) spread on the nanoporous chip were studied by FRAP. A square of 7.4 × 7.4 μm2 was bleached and the recovery of the fluorescence was observed over time. Within 60 s, 90% recovery of the bleached area was observed. (C) The normalized recovery curves provide a diffusion constant of 2.0 ± 0.7 μm2 s–1 (n = 49) with an immobile fraction of 6 ± 3%, being in perfect agreement with previously reported values for silicon dioxide supported membranes.26 The images show an area of 60 × 60 μm2.
Next, the homogeneity, the continuous coverage, as well as the dynamic properties of the membrane covering the nanopore chip were examined by fluorescence recovery after photobleaching (FRAP) experiments. Liposomes supplemented with fluorescently labeled lipids (SoyPC20 plus 0.1 mol % Bodipy-PE) were spread on the chip surface, resulting in a nanopore suspended lipid bilayer (Figure 2B). A rectangular region of interest (7.4 × 7.4 μm2) was bleached by using an argon laser (488 nm, 25 mW). Notably, 90% fluorescence recovery was observed, demonstrating that the nanopore suspended lipid bilayer is intact, homogeneous, and fluid on the chip surface. The lateral diffusion coefficient for several independent FRAP experiments was 2.0 ± 0.7 μm2/s (n = 49) with an immobile fraction of 6 ± 3%. This result is in good agreement with the lateral diffusion coefficient of 1–3 μm2/s for lipid bilayers on glass surfaces.26 In the case of decreased liposome concentration on the chip, the fluorescent recovery is impaired (Supporting Information Figure S2), leading to mostly ill-defined membrane patches or free vesicles on the SiO2 surface. These results demonstrate that fluid lipid bilayers are assembled on nanopore chips.
Next, we examined the sealing and compartmentalization of various solutes by the nanopore suspended lipid bilayer. If no lipid bilayer is present, the cavities are freely accessible for fluorophores, which results in complete colocalization of two dyes inside the cavities (Figure 3A). In contrast, strict separation of fluorescent solutes is observed if a lipid bilayer seals the nanopore (Figure 3B). Prior to SLB formation, a fluorescent analyte (e.g., ATTO488) is added to the buffer reservoir, while all cavities are freely accessible. After SLB formation and buffer exchange, the lipid bilayer sealed the cavities and entrapped the analyte. In all nonsealed cavities, the fluorophore is diluted out. In a subsequent step, a differently labeled analyte is added (e.g., ATTO594). Notably, the fluorophore added later cannot enter those cavities sealed by the SLB, demonstrating the complete compartmentalization of both analytes by pore-spanning lipid bilayers.
Figure 3.

Compartmentalization of solutes by nanopore SLBs. (A) Fluorophores easily access chip cavities if no SLBs are present, resulting in perfect colocalization, when, for example, two dyes are added to the buffer reservoir (ATTO488 and ATTO594; Pearson’s coefficient rP = 0.97 ± 0.01). (B) Lipid bilayers spanning the nanopores retain small hydrophilic fluorophores inside the femtoliter cavities and seal them from the buffer reservoir by an impermeable barrier. If a second fluorophore is added after SLB formation, no colocalization is observed (rP = 0.03 ± 0.02). (C) By addition of Triton X-100 (0.5% (v/w) final concentration), the SLB is disrupted, leading to efflux of ATTO488, while the external fluorophore (ATTO594) will simultaneously fill the cavity. Scale bar = 10 μm.
Disruption of the SLB by adding detergent results in a rapid efflux of the entrapped solute out of the cavities. Simultaneously, the second analyte from the external reservoir entered the cavities, leading to ATTO594 signals in all compartments (Figure 3C). In summary, all results prove that nanopore-suspended lipid bilayers form stable impermeable barriers for small, hydrophilic solutes and that nonelectrogenic membrane transport processes mediated by transporters and channels can be followed via fluorescent readout.
In order to demonstrate the ability of the system to record transport events of reconstituted membrane proteins, the mechanosensitive channel of large-conductance (MscL) was used.27 In addition, chemically charging the pore constriction causes spontaneous opening of the channel.28 On the basis of this principle, MscL has been modified in its constriction site (MscLG22C) with rationally designed small chemical compounds to open specifically in response to external triggers. This generates an engineered on–off switch, which is closed in its default state.29−31 In the open state, however, engineered MscL allows the passage of molecules up to 6.5 kDa.32−34 Here, we used an engineered MscLG22C as membrane channel for the stimulus driven opening of the nanopore. Purified MscLG22C was reconstituted into liposomes composed of soybean lipids, and protein activity after reconstitution was verified by a fluorescence-dequenching assay (Supporting Information Figure S1B). The lipid-to-protein ratio chosen statistically yields 1.8 MscL channels per nanopore, implying a quantitative reconstitution. As some channel complexes may loose their activity, this ratio was considered optimal to achieve single channel recordings.
Proteoliposomes were spread to the nanopore chip as described before. Sealing rates of 80–90% were reached after spreading of proteoliposomes. Prior to SLB formation, Oy647 was entrapped inside the cavities as transported substrate. OregonGreen Dextran (70 kDa), which cannot cross the MscL pore, served as control. Furthermore, ATTO390-labeled DOPE was incorporated into the lipid bilayer to report on lipid contamination inside cavities during all experiments. Upon triggered opening of the right-side out MscLG22C channel by adding thiol specific reagent 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET), Oy647 efflux was exclusively observed for cavities where MscL was located inside the nanopore spanning SLB region (Figure 4A). MTSET by itself shows no such activity, excluding any destabilizing effect on the suspended lipid bilayer. Events derived from three fluorescence channels were classified and discriminated from each other. Only events displaying monoexponential efflux kinetics in the Oy647 channel (red) and constant signals for the control solute (green) as well as the lipid channel (violet) were chosen for final data analysis (Figure 4B). Hundreds of MTSET-triggered efflux events were recorded in one experiment (Figure 4C). Analysis of the translocation rate constant revealed two Gaussian populations, representing either one (kefflux = 1.36 × 10–3 s–1; 68% of all events) or two MscLG22C channels in the pore region (kefflux = 3.33 × 10–3 s–1; Figure 4D). This demonstrated the capacity of the technique to resolve single transport events.
Figure 4.

Ligand-gated solute translocation by MscL at single-molecule level. (A) Engineered MscLG22C channels are closed in their default state. Small solutes such as a fluorophore are unable to pass neither through the channel nor the lipid bilayer. Upon addition of MTSET, an engineered single cysteine of each MscLG22C protomer gets modified, introducing multiple positive charges in the constriction side of the pentameric channel complex. The MscLG22C channel is pushed open by charge repulsion and the entrapped dye can diffuse out of the cavity, leading to a decrease in fluorescence intensity. (B) Time traces of MscLG22C opening upon addition of MTSET. Proteoliposomes containing functionally reconstituted MscLG22C were spread on the chip surface. Oy647 (red) was entrapped inside the cavities as translocation substrate. OregonGreen Dextran (70 kDa, green) was added as channel impermeable control substrate to monitor the substrate specificity of the transport event as well as the membrane integrity during the experiment. LUVs were supplemented with ATTO390-DOPE (0.1 mol %; purple) to check for lipid contamination inside cavities. Addition of MTSET (3 mM final) at t = 0 s opens the channel, leading to Oy647 efflux from the cavities, recorded via fluorescence read-out. (C) Randomly picked MscLG22C efflux curves demonstrate typical efflux events. (D) Rate constants for translocation events (n = 242) cluster into two Gaussian populations, demonstrating that single- and multichannel transport events can be discriminated. (E) High-content screening and statistic analysis of 9046 sealed chip cavities by triple-color real-time readout. Eight percent of all analyzed chip cavities show exponential efflux events. Because of spectral decoding (red, translocated solute; green, control solute; violet, lipid), efflux events, complex kinetics, lipid intrusions, and membrane ruptures could be discriminated, thus eliminating false positives.
From all sealed cavities analyzed in this study (n = 9046), approximately 8% showed efflux activity. By multispectral decoding, these flux events can be classified into four categories: (i) monoexponential efflux events, (ii) complex kinetics, (iii) lipid intrusion, and (iv) membrane rupture (Figure 4E). Fifty percent of all efflux events are monoexponential with stable signals for the lipid probe and control solute. In contrast, complex efflux events are defined by exponential efflux of the translocated substrate but displaying unsteady signals for the control solute and/or the membrane dye. They are therefore not considered for final data analysis. Likewise, lipid intrusion events are discarded, where the lipid bilayer enters cavity space and reduces the cavity volume by displacing the entrapped dye (Supporting Information Figure S3). Membrane ruptures were also excluded, which lead to unspecific efflux as well as influx. By multicolor decoding, unbiased data selection prior to final analysis facilitates the elimination of false positive transport events.
In conclusion, we present a membrane-on-nanopore architecture to examine channels and transporters at single molecule level. Silicon-on-insulator based nanopore chips featuring cylindrical cavities and pore apertures below 100 nm can be reproducibly fabricated in high quantities and with identical properties. The transparent cavity floor makes it possible to use inverted fluorescence microscopes, which are more common than upright microscopes in life sciences. This makes the chip technology readily available to a larger number of potential users. Using air objectives, the system can be automated, another necessity for screening applications. In contrast to our previous nanopore chip design,22 both export and import events can be followed, as the fluorescence from the bulk reservoir is efficiently blocked by the opaque top layer. Proteoliposomes can easily be spread on the flat silicon surface of the chip and suspended lipid bilayers created by self-assembly in high yields. Recording of thousands of traces in a multiplex manner enables the comparison of ensemble properties. In contrast to other approaches, membrane proteins can readily be applied due to the usage of LUVs instead of GUVs or BLMs for SLB formation. Thus, the presented chip is compatible with a wide range of membrane proteins. Using our system, membrane translocation events can be monitored in a highly parallel fashion, creating statistically relevant data with minimal sample and chip consumption. The channel protein MscL used in this study demonstrates the capability of the system to analyze ligand-gated channels. In combination with a semiautomated microscope system, multiple biochips can be observed at the same time, thus offering a major advance in high-throughput screening of therapeutic drug targets.
Acknowledgments
We thank Alina Kollmannsperger, Christine Le Gal and Dr. Ralph Wieneke for helpful suggestions on the manuscript. The German-Israeli Project Cooperation (DIP) provided by the DFG and the Federal Ministry of Education and Research to R.T. as well as the Federal Ministry of Economics and Technology (ZIM R&D Project) to R.T. and Nanospot GmbH, and the European Research Council-Ideas Program Starting Grant (208814) to A.Ko. supported this work.
Supporting Information Available
Experimental procedures, size distribution of proteoliposomes and MscL function, and data selection by three-color experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ M.U. and A.K. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Fagerberg L.; Jonasson K.; von Heijne G.; Uhlen M.; Berglund L. Proteomics 2010, 1061141–9. [DOI] [PubMed] [Google Scholar]
- Wallin E.; von Heijne G. Protein Sci. 1998, 741029–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U.; Schlessinger A.; Kloppmann E.; Chang G. A.; Chou J. J.; Dumont M. E.; Fox B. G.; Fromme P.; Hendrickson W. A.; Malkowski M. G.; Rees D. C.; Stokes D. L.; Stowell M. H.; Wiener M. C.; Rost B.; Stroud R. M.; Stevens R. C.; Sali A. Nat. Struct. Mol. Biol. 2013, 202135–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim M. A.; Goh K. I.; Cusick M. E.; Barabasi A. L.; Vidal M. Nat. Biotechnol. 2007, 25101119–26. [DOI] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S. M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; Hoffmaster K. A.; Ishikawa T.; Keppler D.; Kim R. B.; Lee C. A.; Niemi M.; Polli J. W.; Sugiyama Y.; Swaan P. W.; Ware J. A.; Wright S. H.; Yee S. W.; Zamek-Gliszczynski M. J.; Zhang L. Nat. Rev. Drug Discovery 2010, 93215–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J.; Bowlby M.; Peri R.; Vasilyev D.; Arias R. Nat. Rev. Drug Discovery 2008, 74358–68. [DOI] [PubMed] [Google Scholar]
- Milligan C. J.; Li J.; Sukumar P.; Majeed Y.; Dallas M. L.; English A.; Emery P.; Porter K. E.; Smith A. M.; McFadzean I.; Beccano-Kelly D.; Bahnasi Y.; Cheong A.; Naylor J.; Zeng F.; Liu X.; Gamper N.; Jiang L. H.; Pearson H. A.; Peers C.; Robertson B.; Beech D. J. Nat. Protoc. 2009, 42244–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimhult E.; Kumar K. Trends Biotechnol. 2008, 26282–9. [DOI] [PubMed] [Google Scholar]
- Suzuki H.; Takeuchi S. Anal. Bioanal. Chem. 2008, 39182695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janshoff A.; Steinem C. Anal. Bioanal. Chem. 2006, 3853433–51. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; McConnell H. M. Biophys. J. 1985, 471105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E. Science 1996, 271524543–8. [DOI] [PubMed] [Google Scholar]
- Castellana E. T.; Cremer P. S. Surf. Sci. Rep. 2006, 6110429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M. L.; Tamm L. K. Biophys. J. 2000, 7931400–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann C. A.; Prucker O.; Lehmann T.; Ruhe J.; Knoll W.; Frank C. W. Biomacromolecules 2002, 3127–35. [DOI] [PubMed] [Google Scholar]
- Weiskopf D.; Schmitt E. K.; Kluhr M. H.; Dertinger S. K.; Steinem C. Langmuir 2007, 23189134–9. [DOI] [PubMed] [Google Scholar]
- Stamou D.; Duschl C.; Delamarche E.; Vogel H. Angew. Chem., Int. Ed. 2003, 42455580–3. [DOI] [PubMed] [Google Scholar]
- Howorka S.; Siwy Z. Chem. Soc. Rev. 2009, 3882360–84. [DOI] [PubMed] [Google Scholar]
- Dekker C. Nat. Nanotechnol. 2007, 24209–15. [DOI] [PubMed] [Google Scholar]
- Stutzmann M.; Garrido J. A.; Eickhoff M.; Brandt M. S. Phys. Status Solidi A 2006, 203143424–3437. [Google Scholar]
- Buchholz K.; Tinazli A.; Kleefen A.; Dorfner D.; Pedone D.; Rant U.; Tampé R.; Abstreiter G.; Tornow M. Nanotechnology 2008, 1944445305. [DOI] [PubMed] [Google Scholar]
- Kleefen A.; Pedone D.; Grunwald C.; Wei R.; Firnkes M.; Abstreiter G.; Rant U.; Tampé R. Nano Lett 2010, 10125080–5087. [DOI] [PubMed] [Google Scholar]
- Hennesthal C.; Drexler J.; Steinem C. ChemPhysChem 2002, 310885–9. [DOI] [PubMed] [Google Scholar]
- Cremer P. S.; Boxer S. G. J. Phys. Chem. B 1999, 103132554–2559. [Google Scholar]
- Richter R. P.; Berat R.; Brisson A. R. Langmuir 2006, 2283497–505. [DOI] [PubMed] [Google Scholar]
- Venkatesan B. M.; Polans J.; Comer J.; Sridhar S.; Wendell D.; Aksimentiev A.; Bashir R. Biomed. Microdevices 2011, 134671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukharev S. I.; Blount P.; Martinac B.; Blattner F. R.; Kung C. Nature 1994, 3686468265–8. [DOI] [PubMed] [Google Scholar]
- Yoshimura K.; Batiza A.; Kung C. Biophys. J. 2001, 8052198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koçer A.; Walko M.; Bulten E.; Halza E.; Feringa B. L.; Meijberg W. Angew. Chem., Int. Ed. 2006, 45193126–3130. [DOI] [PubMed] [Google Scholar]
- Koçer A.; Walko M.; Feringa B. L. Nat. Protocol 2007, 261426–1437. [DOI] [PubMed] [Google Scholar]
- Koçer A.; Walko M.; Meijberg W.; Feringa B. L. Science 2005, 3095735755–8. [DOI] [PubMed] [Google Scholar]
- Mika J. T.; Birkner J. P.; Poolman B.; Koçer A. FASEB J. 2013, 273882–92. [DOI] [PubMed] [Google Scholar]
- Cruickshank C. C.; Minchin R. F.; Le Dain A. C.; Martinac B. Estimation of the pore size of the large-conductance mechanosensitive ion channel of Escherichia coli. Biophys. J. 1997, 73, 1925–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G.; Krasnikov V.; Poolman B. Dual-color fluorescence-burst analysis to probe protein efflux through the mechanosensitive channel MscL. Biophys. J. 2007, 92, 1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.