

Abstract
Ischemia/reperfusion (I/R) injury to myocardium induces death of cardiomyocytes and destroys the vasculature, leading to cardiac fibrosis that is mainly mediated by the transdifferentiation of fibroblasts to myofibroblasts and the collagen deposition. Snail involvement in fibrosis is well known; however, the contribution of Snail to cardiac fibrosis during I/R injury and its underlying mechanisms have not been defined. We showed that I/R injury to mouse hearts significantly increases the expression of Snail. An in vitro hypoxia/reoxygenation (Hy/Reoxy) experiment showed that the cell source of Snail induction is endothelial cells rather than cardiac fibroblasts (cFibroblasts) or cardiomyoblasts. When Snail was overexpressed in endothelial cells, they underwent endothelial-to-mesenchymal transition (EndMT) but showed very poor capacity for collagen synthesis. Instead, reoxygenation- or Snail overexpression-mediated EndMT-like cells noticeably stimulated transdifferentiation of fibroblasts to myofibroblasts via secretion of connective tissue growth factor (CTGF). The injection of a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, a selective Snail inhibitor, remarkably suppressed collagen deposition and cardiac fibrosis in mouse I/R injury, and significantly improved cardiac function and reduced Snail and CTGF expression in vivo. Our findings suggested a new mechanism of cell-to-cell communication between EndMT-like cells and fibroblasts for fibrosis induction and implicated Snail as a potential target molecule in cardiac fibrosis after I/R injury.
Introduction
Early reperfusion after myocardial ischemia is an essential strategy for myocardial salvage. However, reperfusion causes cardiomyocyte death, microvasculature injury, and cardiac fibrosis, resulting in ventricular remodeling and myocardial dysfunction.1,2 After infarction, the myocardium develops scar tissue, which is a reparative process in which dead cardiac tissue is replaced by extracellular matrix proteins such as collagen types I and III.3,4 The excessive formation of fibrotic tissue in the heart in turn reduces cardiac muscle tissue, thereby decreasing cardiac function.3
Dai et al. have recently proposed that the reduction of collagen deposition in infarcted myocardium facilitates stem/progenitor cell engraftment and repair.5 The formation of scar tissue after myocardial infarction creates a barrier that impairs engraftment of reparative stem/progenitor cells mobilized from the distant organs. Reducing the collagen density in scar tissue could increase stem cell engraftment, promote cell penetration into the infarcted area, and enhance functional differentiation within the infarcted area, thus improving cardiac function.5 Thus, an understanding of cardiac fibrosis and mechanisms for reducing collagen deposition will be helpful in assessing the effectiveness of therapies.
During the process of fibrosis, fibroblasts differentiate to more active connective tissue cells known as myofibroblasts, which enhance the formation of α-smooth muscle actin (α-SMA) stress fibers and that can produce much more collagen than their fibroblast precursors do.6,7 Myofibroblasts in fibrosis are derived from several sources. Pre-existing resident fibroblasts proliferate and differentiate into myofibroblasts. Bone marrow-derived fibroblasts and epithelial cells undergoing an epithelial-to-mesenchymal transition (EMT) that may contribute to myofibroblast formation.8,9 Endothelial cells are also capable of endothelial-to-mesenchymal transition (EndMT) to generate fibroblasts.9,10
Snail, a zinc finger transcription factor, is best known for its capability to trigger EMT and EndMT.11,12 The Snail gene family influences tissue formation during embryonic development and plays a role in tumor invasion and metastasis.11,13 In addition, Snail activation induces fibrosis in various organs such as the kidneys,13,14 liver,15 and lungs.16 The pathological activation of Snail probably contributes to organ fibrosis, but its role in cardiac fibrosis has not been well defined. Therefore, we analyzed the involvement of Snail in cardiac fibrosis and its underlying mechanisms.
Herein, we found that Snail is significantly upregulated in the endothelial cells of the myocardium after ischemia/reperfusion (I/R) injury in mice. Snail-overexpressing endothelial cells undergo an EndMT-like process and activate the transdifferentiation of neighboring fibroblasts to myofibroblasts through profibrotic cytokine connective tissue growth factor (CTGF) in a paracrine manner. In addition, a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, a selective Snail suppressor,17 significantly reduced cardiac fibrosis but improved cardiac function in the mouse I/R injury model. Hence, communication between Snail-activated EndMT-like cells and neighboring fibroblasts would be pivotal in the development of cardiac fibrosis after I/R injury, and Snail might be a potential target molecule in the treatment of cardiac fibrosis.
Results
Snail is induced in endothelial cells in the heart after I/R injury
Pathological activation of Snail may be responsible for several fibrotic diseases.13,14,15,16 Therefore, we analyzed the possible involvement of Snail in cardiac fibrosis. We first examined Snail expression in an in vivo myocardial I/R model (Figure 1). Snail was strongly stained in the nucleus (brown) at injured sites in both endocardial and myocardial regions (Figure 1b). The immunoreactivity of Snail was very low in nonischemic areas (data not shown). In Western blotting, Snail protein in mouse hearts after I/R injury was significantly increased compared with that in sham-operated hearts (Figure 1c), suggesting that Snail might be involved in the disease process after I/R.
Figure 1.

Snail is induced by I/R injury in mouse heart. (a) Male C57BL/6 mice underwent I/R surgery by the occlusion of the LAD following reperfusion. (b) Detection of Snail protein (brown) in endocardial (Ba) and myocardial (Bb) regions. Many nuclei were positive for Snail (red arrows), whereas the other nuclei were negative (blue triangles). No signals in the IgG control group. Low magnification: ×12.5; high magnification: ×400 (n = 4 each). (c) Myocardial proteins at 7 days after I/R were immunoblotted with anti-Snail antibody. Snail was upregulated in I/R group (n = 4). I/R, ischemia/reperfusion; IgG, immunoglobulin G; LAD, left anterior descending artery; LV, left ventricle; RV, right ventricle.
We next investigated cell types that may be major sources of Snail expression in the heart (Figure 2). Three cell types – endothelial cells (human umbilical vein endothelial cell (HUVEC)), cardiac fibroblasts (cFibroblasts), and cardiomyoblasts (H9C2) – were cultured under hypoxia/reoxygenation (Hy/Reoxy),2 an in vitro condition mimicking in vivo I/R (Figure 2a). We assumed that cFibroblasts were the main source of Snail because fibroblasts are the main cells contributing to the fibrotic process. In addition, Snail expression has been reported in fibroblasts and tumor epithelial cells.18 Unexpectedly, Snail expression in endothelial cells was much higher than that in cFibroblasts or H9C2 cardiomyoblasts, and its expression was strongly upregulated by Hy/Reoxy (Figure 2a). To confirm these findings in vivo, we performed immunohistochemistry for Snail and vascular endothelial cadherin (VE-cadherin) (Figure 2b). Capillary endothelial cells stained with VE-cadherin (reddish brown) were located around cardiomyoblasts, and most Snail staining corresponded to VE-cadherin staining in capillary endothelial cells (Figure 2b). Moreover, endothelial cells stained with VE-cadherin (green) were colocalized with Snail immunofluorescence (red; Figure 2c).
Figure 2.

Hy/Reoxy stimulates the expression of Snail in endothelial cells, but not in CFibroblasts or myoblasts. (a) Three cell types – endothelial cells (HUVEC), cFibroblast, and cardiomyoblasts (H9C2) – were cultured under Hy for 16 hours and then reoxygenated for 2 hours (HR) (n = 3; *P < 0.05). (b) Immunohistochemical staining for Snail and VE-cad in ischemia/reperfusion cardiac tissue at postoperative day 7. Capillary endothelial cells are positive for VE-cad staining (reddish brown; arrows). Most Snail staining was observed in the nuclei of capillary endothelial cells (reddish brown; arrows). Original magnification ×200; scale bar: 50 µm. (c) Colocalization of Snail immunofluorescence (red) with VE-cad (green). Original magnification: ×630; scale bar: 10 µm. (d) PPAR-γ agonist (10 µmol/l) significantly diminished the Snail upregulation caused by Hy/Reoxy in HUVEC (n = 3; *P < 0.05). cFibroblast, cardiac fibroblast; DAPI, 4′,6-diamidino-2-phenylindole; Hy/Reoxy, hypoxia/reoxygenation; HUVEC, human umbilical vein endothelial cell; Hy, hypoxia; N, normoxia; NS, nonsignificant; PPAR-γ, peroxisome proliferator-activated receptor-γ VE-cad, vascular endothelial cadherin.
Next, we treated cells with a PPAR-γ agonist, rosiglitazone, under Hy/Reoxy conditions to test the possible involvement of Snail in the fibrotic process. PPAR-γ agonist has been reported to antagonize lung and kidney fibrosis,19,20 and inhibit lung cancer via selective suppression of Snail.17 PPAR-γ agonist treatment diminished the upregulation of Snail protein caused by Hy/Reoxy in endothelial cells (Figure 2d). Moreover, rosiglitazone specifically decreased Snail protein but had no significant effect on other Snail family proteins such as Slug, Twist, Zeb1/S1P1, and Zeb2 (data not shown). These data suggested that increased Snail protein in endothelial cells might play a role in cardiac fibrosis after I/R injury.
Snail overexpression induces an EndMT-like process in endothelial cells
Fibroblasts reportedly emerge as a result of EndMT in cardiac and renal fibrosis.10,21 We examined whether Snail overexpression in endothelial cells causes EndMT (Figure 3). During EndMT, endothelial cells lose their endothelial characteristics and intercellular adhesion while acquiring fibroblast-like properties and increased motility. Snail overexpression in endothelial cells reduced the endothelial markers – CD31, VE-cadherin, and von Willebrand factor – and increased the mesenchymal markers α-SMA and SM22α (Figure 3a). Snail overexpression in HUVEC accompanied a dramatic change in cell morphology from round cells in a cobblestone arrangement to elongated spindle-shaped cells characteristic of EndMT (Figure 3b). Snail overexpression had no effect on endothelial cell proliferation (data not shown). Similar to EndMT properties, Snail overexpression in HUVEC markedly increased migration capacity (Figure 3c), whereas it decreased the capacity to form capillary-like structures (Figure 3d).
Figure 3.

Snail overexpression promotes transition from endothelial cells toward mesenchymal ones (EndMT). (a) HUVEC was transfected with pSnail for 48 hours. Snail overexpression reduced the endothelial markers, CD31, VE-cad, and vWF, while increased the mesenchymal markers α-SMA and SM22α Quantitative graphs for Western blotting bottom, n = 3). (b) Phase bright imaging of HUVEC with Snail overexpression showed change in cell morphology from round cells in cobblestone arrangement to elongated spindle-shaped cells characteristic of EndMT. Magnification: ×100. Representative images are shown. Snail-overexpressing HUVEC showed (c) increased migration (16 hours) and (d) retarded tube formation on Matrigel (10 hours). Magnification: ×40. Con, nontransfected HUVEC. All experiments were repeated three times independently. HUVEC, human umbilical vein endothelial cell; SMA, smooth muscle actin; VE-cad, vascular endothelial cadherin; vWF, von Willebrand factor.
Conditioned media from Snail-induced EndMT-like cells activate cFibroblasts to myofibroblasts
We assumed that endothelial cells undergo EndMT after Hy/Reoxy via Snail induction and that they participate directly in fibrosis through collagen synthesis.4 Therefore, we measured the secretion of collagen type I by enzyme-linked immunosorbent assay (ELISA) assay (Figure 4a). Although collagen secretion was slightly increased, the amount of collagen produced by EndMT-cells was much lower than that from cFibroblasts or H9C2 cardiomyoblasts. Thus, we thought that direct contribution of EndMT-like cells to fibrosis might be minor and that another mechanism of EndMT-like cells may contribute to cardiac fibrosis after I/R injury.
Figure 4.
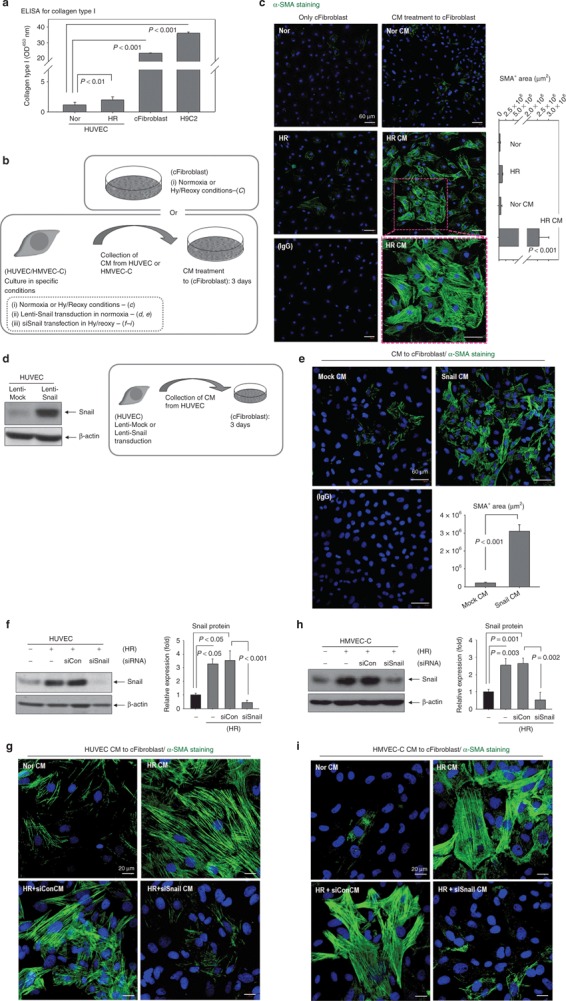
CM from endothelial-to-mesenchymal transition-like cells, after Hy/Reoxy or Snail overexpression, activate cFibroblasts to myofibroblasts. (a) ELISA for collagen type I in CM. The amount of collagen from HUVEC was much lower than that from cFibroblasts or H9C2 cardiomyoblasts (n = 3). (b) Schematic illustration of CM preparation and treatment. CM from various conditions (i, ii, iii conditions) it was treated to cFibroblast for 3 days. In parallel with CM treatment, cFibroblasts themselves were incubated under normoxia (3 days) or Hy/Reoxy conditions (hypoxia 16 hours plus reoxygenation 32 hours, total 3 days). (c) Phenotype switches from cFibroblasts to myofibroblasts by HR CM. Myofibroblasts display strong and fibrous α-SMA fluorescence (green). Nucleus for DAPI (blue). No fluorescence signal in the isotype IgG group. Magnification: ×100 (n = 3). (d) Snail was highly expressed in HUVEC via the lentiviral vector. (e) CM from Snail-overexpressing HUVEC (Snail CM) stimulated the formation of α-SMA filaments in cFibroblasts (green). Magnification: ×200 (n = 4). (f–i) The induction of Snail in HUVEC or in HMVEC-C by Hy/Reoxy was effectively blocked by siRNA against Snail (siSnail). (f,h) Western blotting for Snail (n = 3). (g,i) Snail knock-down with siSnail abolished the effect of HR CM on α-SMA fiber formation (green) in cFibroblasts. Magnification: ×400. siCon indicates control siRNA (n = 3). HR, cFibroblasts under Hy/Reoxy conditions; HR CM, CM from Hy/Reoxy HUVEC; Mock CM, CM from Mock-overexpressing HUVEC; Nor, cFibroblasts under normoxia; Nor CM, cFibroblasts treated with CM from normoxic HUVECs. α-SMA, α-smooth muscle actin; cFibroblast, cardiac fibroblast; CM, conditioned media; ELISA, enzyme-linked immunosorbent assay; HMVEC-C, human cardiac microvascular endothelial cell; HUVEC, human umbilical vein endothelial cell; HR, hypoxia/reoxygenation; IgG, immunoglobulin G; siRNA, small interfering RNA.
We alternately hypothesized that endothelial cells undergoing EndMT influence neighboring fibroblasts to participate actively in fibrosis (Figure 4b–i). To test this hypothesis, we obtained culture supernatants (conditioned media (CM)) from HUVEC under various conditions and used it to treat cFibroblasts (Figure 4b). We examined α-SMA immunofluorescence (Figure 4c) because the differentiation of fibroblasts to myofibroblasts is characterized by enhanced formation of α-SMA stress fibers.6,7 In cFibroblasts treated with CM from normoxic HUVEC, α-SMA immunofluorescence was punctuated or patchy, indicating rare fibrous structure. By contrast, in cFibroblasts treated with CM from Hy/Reoxy HUVEC, α-SMA immunofluorescence got much stronger and fibrous as typical stress fibers (Figure 4c).
In parallel with CM treatment, fibroblasts themselves were incubated under normoxia or Hy/Reoxy conditions without CM treatment to investigate whether Hy/Reoxy alone activated them to myofibroblasts. As shown with α-SMA stress fiber staining, a very low level of differentiation into myofibroblasts without CM treatment was observed under the Hy/Reoxy (Figure 4c). From these data, we can suggest that the CM from EndMT-undergoing cells after Hy/Reoxy strongly induces fibroblasts into fully differentiated myofibroblasts.
We next tested whether this effect is similarly achieved with CM from Snail-overexpressing HUVEC and whether Snail knock-down abolished the effect of CM from EndMT-undergoing cells after Hy/Reoxy (Figure 4d–i). For consistent Snail expression, we performed Lenti-Snail transduction instead of pSnail plasmid transfection. Snail was highly expressed in HUVEC via the lentiviral vector (Figure 4d). Similar to the Hy/Reoxy CM in Figure 4c, the CM of Snail-overexpressing HUVEC strongly stimulated the formation of α-SMA stress fibers in cFibroblasts (Figure 4e). By contrast, Snail knock-down with siSnail abolished the effect of Hy/Reoxy CM on α-SMA fiber formation in cFibroblasts (Figures 4f,g). We confirmed similar results by using human cardiac microvascular endothelial cells (HMVEC-C) (Figure 4h,i). These data indicated that Hy/Reoxy induces EndMT in endothelial cells through Snail upregulation, and Snail-mediated EndMT-like cells influence neighboring fibroblasts toward myofibroblast differentiation.
CTGF mediates the effect of Snail on transdifferentiation of cFibroblasts to myofibroblasts
CM from EndMT-like cells induces the differentiation of fibroblasts into myofibroblasts, which means that fibrosis-stimulating factor might be secreted from Snail-overexpressing EndMT-like cells. We investigated several key factors that encourage the transition of fibroblasts to myofibroblasts: CTGF, angiotensinogen, ET-1, and transforming growth factor-β1 (TGF-β1)22 (Figure 5a). Surprisingly, only CTGF expression was increased by Snail overexpression. Furthermore, Hy/Reoxy increased CTGF expression in endothelial cells (Figure 5b). Using ELISA, we confirmed CTGF secretion from HUVEC and HMVEC-C; CTGF levels were increased by Hy/Reoxy (Figure 5c).
Figure 5.

CTGF is a downstream target of Snail and mediates the activation of cFibroblasts to myofibroblasts. (a,b) Regulation of profibrosis factors by Snail. (a) Only CTGF expression was increased by Snail overexpression among several key factors that encourage the transition of fibroblasts to myofibroblasts. Quantification graph (bottom; n = 3). (b) CTGF expression was induced by HR in HUVEC and in HMVEC-C (n = 3). (c) ELISA for CTGF protein in culture supernatant (n = 4). (d) Schematic time table of Lenti-Snail transduction and siCTGF transfection. (e) Enhanced expression of CTGF by Snail overexpression in HUVEC was totally blocked by siCTGF. Quantitative graphs (bottom, n = 3). Silencing of (f) CTGF or (g) CTGF-neutralizing antibody (3 µg/ml) abolished the effect of CM from Snail-overexpressing HUVEC or HMVEC-C on the emergence of myofibroblasts (green; α-SMA) (n = 3). AGT, angiotensinogen; cFibroblast, cardiac fibroblast; CM, conditioned media; CTGF, connective tissue growth factor; ELISA, enzyme-linked immunosorbent assay; HMVEC-C, human cardiac microvascular endothelial cell; HR, hypoxia/reoxygenation; HUVEC, human umbilical vein endothelial cell; IgG, immunoglobulin G; N, normoxia; NS, nonsignificant; siCon, control small interfering RNA; siRNA, small interfering RNA; SMA, smooth muscle actin; TGF-β1, transforming growth factor-β1.
We confirmed the role of CTGF in fibroblast activation by treatment of recombinant human CTGF protein. Treatment of recombinant human CTGF strongly stimulated the formation of α-SMA stress fibers in cFibroblasts, indicating that recombinant human CTGF influences fibroblasts to differentiate into myofibroblasts (data not shown). To confirm a direct correlation between Snail and CTGF, we blocked CTGF by small interfering RNA against CTGF or neutralizing antibody against CTGF, under conditions of Snail overexpression (Figure 5d–g). siCTGF significantly decreased CTGF protein even under strong Snail expression (Figure 5e). Silencing of CTGF in Snail-overexpressing HUVEC abolished the influence of Snail on fibroblasts to form α-SMA stress fibers (Figure 5g). Moreover, CTGF-neutralizing antibody remarkably attenuated the α-SMA fiber formation caused by CM from Snail overexpression (Figure 5g), indicating that Snail increased by Hy/Reoxy in endothelial cells stimulates CTGF expression that is released into supernatant. This secreted CTGF, in turn, influences neighboring fibroblasts to differentiate into myofibroblasts.
Snail inhibition reduces cardiac fibrosis and improves cardiac function after I/R injury
Given these in vitro findings, we observed cardiac fibrosis, and Snail and CTGF immunoreactivity in vivo heart after I/R injury with or without treatment of PPAR-γ agonist (Figure 6). I/R injury increased myocardial fibrosis on Masson's Trichrome staining and deposition of collagen type I on immunohistochemistry, which was significantly decreased by treatment with the PPAR-γ agonist (Figure 6a–c).
Figure 6.

Snail inhibition reduces cardiac fibrosis and improves cardiac function after I/R injury. (a–c) Paraffin serial sections of mouse heart 4 weeks after I/R were analyzed by MT staining for cardiac fibrosis and immunohistochemistry for collagen type I (reddish brown). In MT staining, collagenous tissues are blue, whereas muscle fibers and the other tissue are red. Cardiac fibrosis induced by I/R injury was significantly reduced by PPAR-γ agonist. Magnification: ×400. Quantitative graph of (b) fibrotic area and (c) collagen+ area using ImageJ program is shown. (d,e) Echocardiographic results at 4 weeks after surgery. Left ventricular (LV) function was improved in PPAR-γ agonist-treated mice (n = 11) than in vehicle (DMSO)-treated mice (n = 8). For LV contractility, LV EF and LV FS were significantly improved. For dimension reduction, LVESD was decreased by PPAR-γ agonist. Sham-operated animals (n = 5). *P < 0.05, **P < 0.05. (f) Immunohistochemical staining of Snail and CTGF in adjacent thin sections from I/R heart at postoperative day 7. Strong Snail expression in nucleus and CTGF in cytosolic area were observed (arrows). The induction of Snail and CTGF was remarkably reduced by PPAR-γ agonist, the selective Snail inhibitor. Low magnification: ×20; high magnification: ×400. (g) CTGF expression was highly stimulated by Hy/Reoxy only in endothelial cells, and it was reduced by a Snail inhibitor, the PPAR-γ agonist (10 µmol/l) (n = 3). (h) Scheme of cardiac fibrosis after I/R injury. Hy/Reoxy induced Snail overexpression in endothelial cells and thus induced an EndMT-like process; the loss of endothelial markers and the gain of mesenchymal ones. EndMT-like cells secreted profibrotic cytokine CTGF, which might influence neighboring cardiac fibroblasts to differentiate into active myofibroblasts, resulting in collagen deposition and cardiac fibrosis. cFibroblast, cardiac fibroblast; CTGF, connective tissue growth factor; DMSO, dimethyl sulfoxide; EF, ejection fraction; EndMT, endothelial-to-mesenchymal transition; FS, fractional shortening; HUVEC, human umbilical vein endothelial cell; HR, hypoxia/reoxygenation; I/R, ischemia/reperfusion; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; MT, Masson's Trichrome; PPAR-γ, peroxisome proliferator-activated receptor-γ.
We then evaluated cardiac functional change by echocardiography 4 weeks after I/R injury2 (Figure 6d,e). Left ventricular (LV) remodeling in I/R + dimethyl sulfoxide group was worse than that in sham-operated animals with a reduction in LV ejection fraction and LV fractional shortening, and an increase in dimension, especially LV end-systolic dimension (LVESD). LV end-diastolic dimension (LVEDD) was unchanged. The PPAR-γ agonist significantly attenuated the decline in systolic function that occurred after myocardial I/R. LV ejection fraction and LV fractional shortening were better preserved and LVEDD was smaller in PPAR-γ agonist-treated animals than in dimethyl sulfoxide-treated animals with myocardial I/R (Figure 6d,e).
For Snail and CTGF expression patterns (Figure 6f), strong Snail immunoreactivity with a nuclear staining pattern was observed in the area with ischemic damage. Furthermore, Snail expression was remarkably decreased by PPAR-γ agonist, which is in accord with the effect of selective Snail suppressor PPAR-γ agonist.17 Interestingly, the pattern of CTGF expression correlated well with that of Snail expression, and secreted protein CTGF showed diffusible cytosolic staining. The induction of CTGF after I/R injury was effectively suppressed by the PPAR-γ agonist. To confirm Snail expression in endothelial cells in I/R injured hearts, we analyzed single cells from ischemic heart muscle with fluorescence-activated cell sorting on day 7 after I/R injury (Supplementary Figure S1). Anti-Snail-PE and anti-CD31-fluorescein isothiocyanate were used to evaluate the cell population. CD31(+)/Snail(+) cells were decreased by PPAR-γ agonist injection into I/R injury heart. These data suggested that PPAR-γ agonist reduced Snail induction in endothelial cells and thus attenuated postinfarction LV remodeling.
We then confirmed CTGF stimulation by reoxygenation in endothelial cells by comparing CTGF protein expression in HUVECs, cFibroblasts, and H9C2 cardiomyoblasts (Figure 6g). Similar to the results shown in Figures 1 and 2, Snail expression was highly stimulated by Hy/Reoxy only in endothelial cells (Figure 6g). CTGF protein was also strongly upregulated by Hy/Reoxy only in endothelial cells, and it was reduced by the PPAR-γ agonist, a Snail inhibitor. These results suggested that Snail overexpression in endothelial cells induces an EndMT-like process and, thus, CTGF release, which might participate in the activation of myofibroblasts and cardiac fibrosis after I/R injury (Figure 6h).
Discussion
Paracrine communication between EndMT-undergoing cells and fibroblasts for cardiac fibrosis
The fundamental biology of fibrosis is the transdifferentiation or activation of fibroblasts to myofibroblasts that express well-developed α-SMA stress fibers, produce collagen type I and III, and induce the deposition of extracellular matrix.6,7 An alternative fibroblast source is reportedly derived from EMT and EndMT.9,10 The process of EMT is important for normal embryonic development and tumor metastasis or tissue repair in adults.23,24 EndMT is a form of EMT that occurs during embryonic heart development.25 EndMT is also involved in cardiac fibrosis and tumor progression.10,26
Our experiments revealed dramatic induction of Snail selectively in endothelial cells after Hy/Reoxy (Figure 2), which can induce an EndMT-like phenotype. Snail-overexpressing endothelial cells expressed mesenchymal components, α-SMA, SM22α, and enhanced migration (Figure 3). We expected these EndMT-undergoing cells to contribute directly to fibrosis through complete differentiation to myofibroblasts and synthesis of collagen because local EMT and EndMT reportedly contribute to the progression of TGF-β1–induced fibrosis.8,9,10 Collagen production was slightly increased in EndMT-undergoing endothelial cells but was still extremely lower than that in cFibroblasts and H9C2 cardiomyoblasts (Figure 4). Thus, we suggest that EndMT-like cells induced by Hy/Reoxy participate in fibrosis directly in, at best, a limited area, leading to perivascular fibrosis, and that another mechanism of EndMT-like cells may contribute to cardiac fibrosis after I/R injury.
Alternatively, we propose that a new concept, such as cell cross-talk, is involved in fibrosis. We observed that EndMT-like cells indirectly contribute to cardiac fibrosis by releasing factors that stimulate the differentiation of cFibroblasts to myofibroblasts (Figure 5). We ascertained that fibroblasts by themselves did not efficiently differentiate to myofibroblasts even under Hy/Reoxy conditions without CM from EndMT-like cells, as shown in Figure 4c. In addition, quiet state endothelial cells were not as effective in fibroblast differentiation. These data emphasize the importance of paracrine communications between EndMT-like cells and fibroblasts for cardiac fibrosis. Our findings may imply that numerous roads reach fibrosis. Blocking just a single pathway of fibroblast activation is likely insufficient to halt fibrotic progression. Thus, understanding and targeting the various pathways of fibroblast activation is necessary to manage cardiac fibrosis.
Snail and CTGF axis: mediators of endothelial cell–fibroblast communication during cardiac fibrosis after I/R injury
CTGF, a potent profibrotic factor, was first discovered as a secreted protein from endothelial cells during angiogenesis under normal conditions.27,28 CTGF modulates the fibrotic process in skin disorders and tumor development,28 and CTGF expression is induced in infarcted myocardium.29 Snail is also known as an important regulator of fibrosis in the kidney,13,14 liver,15 and lung.16 However, the contribution of Snail to the pathogenesis of cardiac fibrosis after I/R injury or the relationship between Snail and CTGF has not been studied.
We found strong Snail expression in heart tissue after I/R injury (Figure 1). We had assumed that the main cell source of Snail induction under this condition would be cFibroblasts because previous reports mention that Snail expression is induced in activated fibroblasts, breast cancer cells, and epithelial cells18,30,31 during the TGF-β1–mediated EMT process. To our surprise, Snail expression in endothelial cells was much higher than that in cFibroblasts and H9C2 cardiomyoblasts (Figure 2), but this result is consistent with a previous report that TGF-β2, an important mediator of fibrosis, facilitates Snail-mediated EndMT.12 Thus, Snail induction in endothelial or epithelial cells during EndMT or EMT may be an important mechanism in fibrosis caused by Hy/Reoxy or TGF-β.
Snail activation in endothelial cells induced the EndMT process (Figure 3). Another unexpected and interesting finding was that EndMT-like cells stimulated the differentiation of cFibroblasts to myofibroblasts via CTGF secretion (Figure 5). We found that CTGF is a downstream factor of Snail (Figure 5). When Snail expression was interrupted with siSnail, CTGF expression also decreased. On the contrary, CTGF blockage using siCTGF did not affect Snail expression under Hy/Reoxy conditions (data not shown). Thus, targeting an upstream regulator such as Snail might be an effective approach for treating cardiac fibrosis after I/R injury.
Aside from considering these results, we are concerned about the safety of rosiglitazone, which has been withdrawn owing to increased risk of cardiovascular event. Although rosiglitazone is a failed drug, treatment with pioglitazone, another PPAR-γ agonist, provides protection from both micro- and macrocardiovascular events and plaque progression.32,33 Therefore, other PPAR-γ agonists such as pioglitazone might be useful for the treatment of heart fibrosis.
The physiological significance of cardiac fibrosis includes alteration of the structure and architecture of the heart as well as loss of cardiac function resulting from excessive extracellular matrix deposition. In addition, collagen deposition in the infarcted myocardium may become a barrier that impairs the homing and engraftment of stem cells or the delivery of therapeutic drugs. Thus, understanding the regulatory mechanisms of cardiac fibrosis after I/R injury that mimics emergent coronary intervention for patients with acute myocardial infarction is essential for the development of novel targeted cardioprotective or regenerative strategies for the infarcted heart.
Materials and Methods
Cell culture and preparation of CM. HUVECs (passages 4–6; Clonetics, Lonza, Allendale, NJ) were grown in M199 (Gibco, Carlsbad, CA) containing 20% FBS (Lonza), 3 ng/ml bFGF (Invitrogen, Carlsbad, CA), 5 U/ml heparin, and 1% penicillin/streptomycin (GIBCO). HMVEC-C (passages 4–6; Lonza) were cultured in EGM-2MV (Lonza). For preparation of CM, the medium of HUVECs or HMVEC-C was changed with M199 or basal medium containing 1% fetal bovine serum, collected after 16- or 18-hour culture, and filtered with 0.22-µm filter unit (Millipore, Billerica, MA). Primary rat cFibroblasts34 and rat cardiomyoblasts (H9C2; ATCC, Manassas, VA) were cultured in Dulbecco's modified eagle medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were incubated in hypoxic conditions for 16 hours using a BBL GasPac System (BD, Franklin Lakes, NJ), which can catalytically reduce oxygen levels to <1% oxygen within 15 minutes.2,35 Cells were then placed under normoxic conditions for 2 hours for reoxygenation treatment. PPAR-γ agonist (10 µmol/l), rosiglitazone, was added in reoxygenation conditions. CTGF-neutralizing antibody (3 µg/ml) or recombinant human CTGF (0.5 or 1 µg/ml; Fitzgerald Industries International, Acton, MA) was used.
Transfection and RNA interference. Snail full-length cDNA was cloned into the pcDNA3 vector, and transfection was done using Metafectene-Pro (Biontex, Planegg, Germany). For preparation of lentivirus, Lenti-Mock or Lenti-Snail vectors were transfected on HEK293T cells (ATCC). The viral supernatants were harvested 48 hours after transfection, filtered with 0.45-µm filter unit (Millipore), and the supernatants were concentrated with ultracentrifuge. For transduction to HUVEC or HMVEC-C, the concentrated lentivirus supernatants were added to culture medium. Cells were grown up to 80% confluence, and small interfering RNAs (50 µmol/l; Dharmacon, Lafayette, CO) were transfected using Metafectene-Pro (Biontex). Sequence of siSnail: 5′-GCGAGCUGCAGGACUCUAA-3′ siCTGF (5′-UAAAUUCUGUGGAGUAUGU-3′). Nontargeting siRNA pool (Dharmacon) was used as control.
Western blot analysis and ELISA. Cells were lysed by radioimmunoprecipitation assay buffer (50 mmol/l Tris–HCl, 150 mmol/l NaCl, 1% NP 40, 0.1% sodium dodecyl sulfate, 0.5% deoxycholate, and Protease Inhibitor Cocktail (Roche, Indianapolis, IN)). Mouse hearts were grinded in liquid nitrogen and then lysed by radio immunoprecipitation assay buffer. Total protein (15–30 µg) was fractionated in sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with specific antibodies against Snail (Cell signaling, Boston, MA), VE-Cadherin, CD31, von Willebrand factor, CTGF (SantaCruz, Santa Cruz, CA), α-SMA, and SM22α (Abcam, Cambridge, UK). Quantification of band intensity was analyzed using TINA 2.0 (RayTest, Straubenhardt, Germany) and normalized to the intensity of an internal control β-actin (SantaCruz). ELISA for collagen type I (Abcam) and CTGF (Abnova, Taipei City, Taiwan) was performed and the absorbance (450 nm) was measured on the ELISA reader (VERSAmax; AccuScan Instruments, Columbus, OH).
Endothelial function assay: wounding migration and tube formation. HUVECs were plated on 60-mm culture dishes at 90% confluence and the migration assay was done as described.36,37 HUVEC (1 × 105 cells) were seeded on polymerized Matrigel (BD Biosciences, Sparks, MD) and the tube formation assay was done as described.38,39
In vivo mouse myocardial I/R injury model and echocardiography. All animal experiments were performed under approved protocol by the Institutional Animal Care and Use Committee of Seoul National University Hospital and complied with the National Institutes of Health Guide for “The Care and Use of Laboratory Animals.” Male C57BL/6 mice (8–10 weeks) underwent I/R surgery by the occlusion of the left anterior descending artery following reperfusion.2 PPAR-γ agonist, rosiglitazone (10 mg/kg), was injected intraperitoneally after I/R surgery. For control, a sham injury was performed the same way, with the exception of left anterior descending artery ligation. Four weeks after I/R surgery, cardiac function was evaluated by two-dimensional, guided M-mode echocardiography with a 14 MHz linear probe (Toshiba, Tokyo, Japan). We measured LVEDD and LVESD, and then ejection fraction and fractional shortening were calculated according to the following formula: ejection fraction = (LVEDD2 − LVESD2) × 100/LVEDD2; fractional shortening = (LVEDD − LVESD) × 100/LVEDD.
Immunofluorescence, immunohistochemical, and Masson's Trichrome staining. Fibroblasts were grown on the cover glasses at monolayer and treated with CM. After 3 days, cells were fixed with 4% paraformaldehyde for 10 minutes at 4 °C, and blocked with 2% bovine serum albumin for 1 hour. Cells were labeled with anti-α-SMA antibody (Abcam) followed by Alexa-488 secondary antibody (Invitrogen). Mouse hearts were fixed in 10% buffered formaldehyde, embedded in paraffin blocks, and serially sectioned to 4 µm. Deparaffinized sections were blocked with 1% bovine serum albumin, and incubated with anti-Snail (Abcam) and anti-VE-cadherin (SantaCruz) followed by Alexa-488 or Alexa-555 secondary antibodies. The nuclei were stained with DAPI (Sigma, St Louis, MO) and mounted using fluorescent mounting medium (DAKO, Carpinteria, CA). The fluorescent image was obtained with a confocal microscope (Carl Zeiss LSM710; Carl Zeiss, Oberkochen, Germany). We performed immunohistochemistry using a Vector Labs RTU ABC kit (Vector Labs, Burlingame, CA). Deparaffinized sections were incubated with primary antibody against Snail, CTGF, VE-cadherin (SantaCruz), and collagen type I (Abcam). Sections were counterstained with Mayer's hematoxylin. To analyze fibrosis, tissue sections were stained for collagenous tissue with a Masson's Trichrome kit (Sigma). The images were obtained with a photomicroscope (Olympus BX50) and a DP50 camera (Olympus, Tokyo, Japan). At least three animals were used for each group.
Statistical analysis. The results are expressed as means ± SD. The differences between the groups were compared by the unpaired t test or one-way analysis of variance, followed by post hoc analysis with the Bonferroni test. P values ≤0.05 were considered statistically significant. All statistical analyses were performed using SPSS 17.0 (SPSS, Chicago, IL).
SUPPLEMENTARY MATERIAL Figure S1. FACS analysis was performed with cells from ischemic heart muscle. Materials and methods.
Acknowledgments
This research was supported by a grant from the Innovative Research Institute for Cell Therapy (A062260); National Research Foundation (NRF) grant funded by the Korea Government (2010-0024252 to S.-W.L.; 2010-0020258 to H.-S.K.); and grant No. R31-2008-000-10103-0 from the World Class University program of the NRF. The authors declared no conflict of interest.
Supplementary Material
References
- Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation. 2009;120:2105–2112. doi: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- Lee SW, Won JY, Lee HY, Lee HJ, Youn SW, Lee JY, et al. Angiopoietin-1 protects heart against ischemia/reperfusion injury through VE-cadherin dephosphorylation and myocardiac integrin-β1/ERK/caspase-9 phosphorylation cascade. Mol Med. 2011;17:1095–1106. doi: 10.2119/molmed.2011.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. Myocardial repair/remodelling following infarction: roles of local factors. Cardiovasc Res. 2009;81:482–490. doi: 10.1093/cvr/cvn333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B, Huang W, Xu M, Millard RW, Gao MH, Hammond HK, et al. Reduced collagen deposition in infarcted myocardium facilitates induced pluripotent stem cell engraftment and angiomyogenesis for improvement of left ventricular function. J Am Coll Cardiol. 2011;58:2118–2127. doi: 10.1016/j.jacc.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz F. How many different roads may a cell walk down in order to become a fibroblast. J Am Soc Nephrol. 2008;19:2246–2248. doi: 10.1681/ASN.2008101089. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- Medici D, Potenta S, Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. 2011;437:515–520. doi: 10.1042/BJ20101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet A, Esteban MA, Maxwell PH, Nieto MA. Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis. Cell Cycle. 2007;6:638–642. doi: 10.4161/cc.6.6.4022. [DOI] [PubMed] [Google Scholar]
- Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25:5603–5613. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RG, Lin Y, Shimizu-Hirota R, Hanada S, Neilson EG, Greenson JK, et al. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol Cell Biol. 2011;31:2392–2403. doi: 10.1128/MCB.01218-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan D, Melo T, Deng Z, Almeida C, Zhao W. ERK/GSK3β/Snail signaling mediates radiation-induced alveolar epithelial-to-mesenchymal transition. Free Radic Biol Med. 2012;52:983–992. doi: 10.1016/j.freeradbiomed.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary R, Li H, Winn RA, Sorenson AL, Weiser-Evans MC, Nemenoff RA. Peroxisome proliferator-activated receptor-γ inhibits transformed growth of non-small cell lung cancer cells through selective suppression of Snail. Neoplasia. 2010;12:224–234. doi: 10.1593/neo.91638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte J, Weidig M, Balzer P, Richter P, Franz M, Junker K, et al. Expression of the E-cadherin repressors Snail, Slug and Zeb1 in urothelial carcinoma of the urinary bladder: relation to stromal fibroblast activation and invasive behaviour of carcinoma cells. Histochem Cell Biol. 2012;138:847–860. doi: 10.1007/s00418-012-0998-0. [DOI] [PubMed] [Google Scholar]
- Samah M, El-Aidy Ael-R, Tawfik MK, Ewais MM. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur J Pharmacol. 2012;689:186–193. doi: 10.1016/j.ejphar.2012.05.026. [DOI] [PubMed] [Google Scholar]
- Kiss E, Popovic ZV, Bedke J, Adams J, Bonrouhi M, Babelova A, et al. Peroxisome proliferator-activated receptor (PPAR)γ can inhibit chronic renal allograft damage. Am J Pathol. 2010;176:2150–2162. doi: 10.2353/ajpath.2010.090370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol. 1991;114:1285–1294. doi: 10.1083/jcb.114.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Lam SC. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- Peinado H, Quintanilla M, Cano A. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- Romagnoli M, Belguise K, Yu Z, Wang X, Landesman-Bollag E, Seldin DC, et al. Epithelial-to-mesenchymal transition induced by TGF-β1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 2012;72:6268–6278. doi: 10.1158/0008-5472.CAN-12-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, PERISCOPE Investigators et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA. 2008;299:1561–1573. doi: 10.1001/jama.299.13.1561. [DOI] [PubMed] [Google Scholar]
- Mannucci E, Monami M, Lamanna C, Gensini GF, Marchionni N. Pioglitazone and cardiovascular risk. A comprehensive meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2008;10:1221–1238. doi: 10.1111/j.1463-1326.2008.00892.x. [DOI] [PubMed] [Google Scholar]
- Hahn JY, Cho HJ, Bae JW, Yuk HS, Kim KI, Park KW, et al. β-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2006;281:30979–30989. doi: 10.1074/jbc.M603916200. [DOI] [PubMed] [Google Scholar]
- Mestril R, Chi SH, Sayen MR, O'Reilly K, Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest. 1994;93:759–767. doi: 10.1172/JCI117030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Jung KH, Jeong CH, Seo JH, Yoon DK, Suh JK, et al. Inhibition of endothelial cell migration through the down-regulation of MMP-9 by A-kinase anchoring protein 12. Mol Med Rep. 2011;4:145–149. doi: 10.3892/mmr.2010.389. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Cho H, Lee SW, Kim YJ, Kim KW. Antisense-thioredoxin inhibits angiogenesis via pVHL-mediated hypoxia-inducible factor-1α degradation. Int J Oncol. 2005;26:1049–1052. [PubMed] [Google Scholar]
- Song HS, Son MJ, Lee YM, Kim WJ, Lee SW, Kim CW, et al. Oxygen tension regulates the maturation of the blood-brain barrier. Biochem Biophys Res Commun. 2002;290:325–331. doi: 10.1006/bbrc.2001.6205. [DOI] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.