

Abstract
Background
Although reactive oxygen species (ROS) are believed to be involved in pathogenic mechanisms that underlie complex regional pain syndrome type I (CRPS-I), the role of ROS in the central mechanism of CRPS is not fully understood.
Objective
In this study we investigated whether ROS scavenger N-acetyl-l-cysteine (NAC) was capable of attenuating mechanical allodynia and whether pain was decreased through modulating N-methyl-d-aspartate (NMDA) receptor activation in a chronic postischemia pain (CPIP) animal model that mimics the symptoms of CRPS-I.
Methods
Thirty male Sprague-Dawley rats were randomly allocated to 5 different groups: (1) sham rats and CPIP rats treated with (2) vehicle; (3) NAC 30 mg/kg; (4) NAC 100 mg/kg; and (5) NAC 300 mg/kg intraperitoneally at 15 minutes before reperfusion. CPIP was generated after a 3-hour ischemia/reperfusion injury on the hind limb using a tight fitting O-ring. Then, mechanical paw-withdrawal thresholds to von Frey stimuli were assessed before ischemia (baseline), at 4 hours; 1, 3, and 5 days; and 1, 2, 3, and 4 weeks after reperfusion. Another set of 5 animal groups in the same categories was used to determine phosphorylated NMDA receptor 1 subunit (pNR1) immunoreactivity in the ipsilateral L4/6 spinal cord at 3 days after reperfusion.
Results
The sham group showed no significant difference in pain thresholds over 4 weeks. With NAC treatment, the pain thresholds measured after reperfusion increased significantly, and this increase lasted 4 weeks after reperfusion compared with the vehicle group (P < 0.01 on the ipsilateral side and P < 0.05 on the contralateral side). The relative density of pNR1 at 3 days after reperfusion in NAC-treated rats decreased significantly compared with that of the vehicle group (all, P < 0.001). The NAC dose was significantly correlated not only with paw-withdrawal threshold (ρ = 0.979; P < 0.001) but also with the relative density of pNR1 (ρ = –0.875; P < 0.001).
Conclusions
NAC, administered during the pre-reperfusion period, had a long-term antiallodynic effect through the attenuation of NMDA receptor phosphorylation, leading to central sensitization.
Key words: central sensitization, ischemia/reperfusion injury, NAC, NMDA receptor activation, pain, reactive oxygen species
Introduction
Complex regional pain syndrome (CRPS) is a chronic pain disorder with significant autonomic features; it typically develops in an extremity after tissue injury. In type I CRPS (CRPS-I), there is no obvious nerve injury. CRPS is one of the most challenging chronic pain conditions to treat successfully because of the incomplete understanding of its pathophysiologic mechanism(s).1
Consistent with growing evidence suggesting that oxygen free radicals may contribute to chronic pain, including neuropathic and inflammatory pain, several studies have reported that reactive oxygen species (ROS) are believed to be involved in the pathogenesis of CRPS. Serum lipid peroxidation products and salivary antioxidants, known to be induced by ROS, are increased in CRPS-I patients.2 CRPS-I symptoms are relieved after treatment with free-radical scavengers,3,4 and the incidence or duration of CRPS-I after wrist fractures may be reduced by preemptive treatment with the antioxidant, vitamin C.5,6
However, although these studies suggested the involvement of free radicals and/or oxidative stress in CRPS, little attention has been paid to the role of antioxidants in the central mechanism(s) of CRPS. Central nervous system plasticity in response to tissue injury may contribute to the development of chronic pain. It is well known that the N-methyl-d-aspartate (NMDA) receptor is important for the development of central sensitization in the dorsal horn.7–9 Thus, phosphorylation of the NMDA receptor is an important component in receptor facilitation, probably contributing to central sensitization as a basis for chronic pain.10–13
N-acetyl-l-cysteine (NAC) is a well-known and powerful antioxidant that has been used widely clinically as a mucolytic agent and as an antidote for paracetamol (acetaminophen) poisoning. In neurons, glutathione is important in preventing oxidative stress. By acting as a cysteine donor, NAC maintains the intracellular glutathione level and plays a neuroprotective role for a range of neuronal cell types against diverse stimuli.14 NAC has been reported to attenuate a significant reduction in hyperalgesia in chronic constriction injury-induced neuropathic rats.15 Also, systemic NAC administration reduced the nociception triggered by intrathecal injection of capsaicin, indicating a role in nociception at the level of the spinal cord.16
Thus, in the present study, we sought to evaluate the role of NAC in the central sensitization involved in the pathophysiology of CRPS. Specifically, we focused on the preventative role of NAC in modifying central neuroplastic changes. To test the hypothesis, we used chronic postischemia pain (CPIP) rats as reported by Coderre et al.17 Their initial report showed that 3-hour ischemia/reperfusion (IR) of the hind paw of CPIP rats induced long-term mechanical allodynia or CPIP, which was similar to the clinical features seen in patients with CRPS-I.17 In the present study, we investigated the antiallodynic effects of NAC administered before reperfusion in IR-induced CPIP rats and examined changes in the level of phosphorylated NMDA receptor 1 subunit (pNR1), which presumably indicates central sensitization, in the spinal dorsal horn of the CPIP model rats.
Methods
Animals
Male adult Sprague-Dawley rats (280–320 g) were used in this study. Animals were housed in groups, with food and water available ad libitum, on a 12:12 hour light:dark cycle. All animals were acclimated in their cages for 7 days before any experiments. All housing conditions and experimental procedures were approved by the Institutional Animal Care and Use Committee and in accordance with the National Institute of Health guidelines on laboratory animal welfare.
All rats were assessed for posture and righting reflexes based on the 5-point scales described by Kim et al,18 and all scored zero at all time points, showing that sedation or anesthesia did not occur.1
Hind Paw Ischemia/Reperfusion and Drug Treatment
Hind paw IR injury was produced according to Coderre et al17 In brief, rats were anesthetized over a 3-hour period with a sodium pentobarbital infusion. After induction of anesthesia, a Nitrile 70 Durometer O-ring (O-rings West, Seattle, Washington) with 7/32 inch internal diameter was placed around the rat's left hind limb just proximal to the ankle joint. The termination of sodium pentobarbital anesthesia was timed so that rats recovered fully within 30 minutes after reperfusion, which occurred immediately after removal of the O-ring. Sham rats received exactly the same treatment, except that the O-ring was cut so that it only loosely surrounded the ankle, and did not occlude blood flow to the hind paw.
Experiment I
To determine the effects of ROS scavengers on CPIP-induced mechanical allodynia and the dose response of a ROS scavenger, NAC, dissolved in luke warm normal saline (300 mg/mL, which was further diluted to 30 and 100 mg/mL) was administered intraperitoneally 15 minutes before reperfusion to rats with CPIP at 30, 100, and 300 mg/kg doses. Thirty male Sprague-Dawley rats were randomly allocated to 5 different groups: (1) sham rats and CPIP rats treated with (2) vehicle; (3) NAC 30 mg/kg; (4) NAC 100 mg/kg; and (5) NAC 300 mg/kg intraperitoneally at 15 minutes before reperfusion. The pain withdrawal threshold in mechanical allodynia was measured at different time intervals (before ischemia [baseline]; at 4 hours; at 1, 3, and 5 days; and at 1, 2, 3, and 4 weeks after reperfusion). The changes in the pain threshold in test groups were compared with that of sham- or vehicle-treated control rats. The whole procedures were conducted blindly, in that the experimenter did not know the nature of experimental manipulation.
Experiment II
To evaluate the role of NAC on the phosphorylation of NMDA receptors, another set of 5 animal groups consisting of 6 for each group was used: (1) sham rats and CPIP rats treated with (2) vehicle; (3) NAC 30 mg/kg; (4) NAC 100 mg/kg; and (5) NAC 300 mg/kg intraperitoneally at 15 minutes before reperfusion. Three days after reperfusion, pNR1 immunoreactivity was determined in the ipsilateral L4/6 spinal cord by Western blot after measuring the development of mechanical allodynia, which was assessed before ischemia and at 3 days after reperfusion.
Behavioral Test
Hind paw mechanical thresholds were assessed by measuring the withdrawal response to von Frey filament stimulation according to a modification of the up/down method described by Chaplan et al.19,20 In brief, rats were placed on a metal mesh floor covered with an inverted clear plastic cage (15 × 23 × 24 cm3) through which the von Frey filaments (nylon monofilaments; Stoelting, Woodale, Illinois) were applied to the plantar surface of the hind paw. Filaments were applied in either ascending or descending strength as necessary to determine the filament closest to the threshold of response. Each filament was applied once for 10 seconds to the center of the paw between the pads and a lower intensity hair after each positive response, whereas a higher intensity hair followed each negative response (until 6 responses were recorded after a first change in response). The minimum stimulus intensity was 0.25 g and the maximum was 15 g. Based on the response pattern and the force of the final filament, the 50% response threshold (grams) was calculated. The resulting pattern of positive and negative responses was tabulated, and the 50% response threshold was interpolated using the formula 50% g threshold = (10[xf + kδ])/10,000, where xf = the value (in log units) of the final von Frey hair used; k = tabular value19 for pattern of positive/negative responses; and δ = mean difference (in log units) between stimuli (here, 0.224).
Western Blot
To examine the levels of activated NMDA receptors in the dorsal horn, rats were sacrificed immediately after behavioral testing at 3 days after reperfusion. At the time of sacrifice, rats were anesthetized using sodium pentobarbital (50 mg/kg IP) and then perfused quickly through the ascending aorta with cold saline. The L4/6 spinal cord segments (ipsilateral and contralateral sides, separately) were quickly removed, frozen immediately on dry ice, and stored at −70°C until use. At the time of assay, each spinal cord sample was thawed and homogenized in a lysis buffer (20 mM Tris-hyrdocholoride pH 8.0, 150 mM sodium chloride 1 mM EDTA, 2 mM sodium orthovanadate, 0.5 mM dithiothreitol, 10% glycerol, 1% Nonidet P-40) containing a protease inhibitor cocktail (Roche, Mannheim, Germany). The homogenate was centrifuged at 12,000 rpm for 20 minutes at 4°C. The supernatant was decanted from the pellet and used for Western blot analyses. The concentration of protein in the homogenate was measured using the Bio-Rad Protein Assay Kit I (Bio-Rad, Hercules, California). The homogenates of equal amounts of protein (50 μg) were fractionated using 10% (w/v) SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membrane was then incubated with antiphospho-NR1 antibody (rabbit immunoaffinity purified immunoglobulin-G, 1:500, Upstate Biotechnology, Temecula, California) and monoclonal anti-β-actin antibody (1:50,000, Sigma, St. Louis, Missouri) for 2 hours at room temperature. The blots were washed 3 times for 20 minutes each with a washing buffer and then incubated with horseradish peroxidase conjugated immunoglobulin-G (1:2000, goat antirabbit, Upstate Biotechnology; 1:2000, donkey antimouse, Amersham Pharmacia Biotech, Arlington Heights, Illinois). The blots were exposed to autoradiographic film (Eastman Kodak Co., Rochester, New York); the films were scanned into a computer, and the intensity of the immunoreactive bands of interest was quantified using Meta Image series software (Molecular Devices, Downingtown, Pennsylvania). The density was calculated using the formula: Density = Log (255/Intensity). Using the β-actin as the internal standard, the ratios of the pNR1 to the β-actin density were calculated and compared among the different groups.
Statistical Analysis
All data are expressed as mean (SEM). Mechanical thresholds obtained as previously described were analyzed by 1-way ANOVA, followed by Dunnet t post hoc test at each time point. The data from the Western blot analysis were expressed as the ratios of pNR1 density to β-actin density. The changes of pNR1 ratio among different groups were also tested by 1-way ANOVA, followed by Dunnet t post hoc test. Correlation calculation was performed with Spearman's correlation analysis. A P value < 0.05 was considered significant.
Results
Experiment I: Anti-Allodynic Effect of NAC Over Time
Baseline values for the sham, vehicle, NAC 30, NAC 100, and NAC 300 groups were not significantly different. The vehicle-control group developed mechanical allodynia over a prolonged period in both the ipsilateral and contralateral hind paws, with more pronounced effects on the ipsilateral side as previously described.17 Ipsilateral and contralateral mechanical allodynia were present after reperfusion, peaked at 3 days, and lasted for at least 28 days after reperfusion (P < 0.001 from baseline or sham at all time points).
Compared with vehicle group, pain behavior was attenuated significantly, in a dose-dependent manner, in the CPIP-induced rats that received the antioxidant NAC. The pain threshold increased significantly at 3 and 5 days, and 1, 2, 3, and 4 weeks after reperfusion with the 30 mg/kg dose compared with the vehicle group on the ipsilateral side (P < 0.001 at all time points). At 100 mg/kg dose of NAC, the pain threshold increased significantly at 4 hours; 3 and 5 days; and 1, 2, 3, and 4 weeks after reperfusion (P = 0.007 at 4 hours and P < 0.001 at other time points), whereas at 300 mg/kg dose, the effect was observed at 4 hours; 1 3, and 5 days, and 1, 2, 3, and 4 weeks after reperfusion (P = 0.005 at 1 day and P < 0.001 at other time points). Significant main effects of group were observed at 4 hours (F3,20 = 21.6; P < 0.001), 1 day (F3,20 = 9.3; P < 0.001), 3 days (F3,20 = 484.6; P < 0.001), 5 days (F3,20 = 275.5; P < 0.001), 1 week (F3,20 = 176.4; P < 0.001), 2 weeks (F3,20 = 160.0; P < 0.001), 3 weeks (F3,20 = 202.3; P < 0.001), and 4 weeks (F3,20 = 139.2; P < 0.001) after reperfusion on the ipsilateral side. The contralateral ameliorative effect of NAC on mechanical allodynia at 30 mg/kg was significant only at 3 and 5 days, and 1, 2, and 3 weeks (P = 0.029 at 2 weeks, P = 0.022 at 3 weeks, and P < 0.001 at other time points). However, at 100 mg/kg, the effect was significant at 3 and 5 days, and 1, 2, 3, and 4 weeks after reperfusion (all, P < 0.001), whereas at 300 mg/kg dose, the effect was observed at 4 hours; 1, 3, and 5 days; and 1, 2, 3, and 4 weeks after reperfusion (P = 0.012 at 4 hours, P = 0.03 at 1 day, and P < 0.001 at other time points). Significant main effects of group were observed at 4 hours (F3,20 = 6.6; P = 0.003), 1 day (F3,20 = 3.6; P = 0.031), 3 days (F3,20 = 138.1; P < 0.001), 5 days (F3,20 = 269.0; P < 0.001), 1 week (F3,20 = 169.3; P < 0.001), 2 weeks (F3,20 = 109.5; P < 0.001), 3 weeks (F3,20 = 99.2; P < 0.001), and 4 weeks (F3,20 = 59.1; P < 0.001) after reperfusion on the contralateral side (Table I).
Table I.
Time course of the effects of N-acetyl-l-cysteine (NAC) on the development of mechanical allodynia in hind paws. Data are given in mean (SEM).
| Ipsilateral |
1-Way ANOVA |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Time Point | Sham | Vehicle | NAC 30 | NAC 100 | NAC 300 | F⁎ | P | F† | P |
| Before Ischemia | |||||||||
| Baseline | 14.50 (0.32) | 14.25 (0.34) | 13.42 (0.75) | 14.50 (0.32) | 14.50 (0.32) | 1.4 | 0.281 | 1.2 | 0.331 |
| After Reperfusion | |||||||||
| 4 h | 14.50 (0.32) | 2.18 (0.41) | 2.39 (0.09)‡ | 3.23 (0.05)‡§ | 4.38 (0.10)‡∥ | 1063.1 | <0.001 | 21.6 | <0.001 |
| 1 d | 13.67 (0.79) | 3.11 (0.34) | 2.79 (0.10)‡ | 2.97 (0.07)‡ | 4.03 (0.07) ‡§ | 169.7 | <0.001 | 9.3 | <0.001 |
| 3 d | 14.50 (0.32) | 0.71 (0.12) | 3.71 (0.10)‡∥ | 7.34 (0.23)‡∥ | 9.78 (0.22)‡∥ | 379.0 | <0.001 | 484.6 | <0.001 |
| 5 d | 14.75 (0.25) | 1.43 (0.21) | 3.99 (0.10)‡∥ | 7.16 (0.22)‡∥ | 9.55 (0.29)‡∥ | 404.0 | <0.001 | 275.5 | <0.001 |
| 1 wk | 14.50 (0.32) | 2.00 (0.30) | 4.06 (0.12)‡∥ | 6.51 (0.24)‡∥ | 8.51 (0.13)‡∥ | 417.0 | <0.001 | 176.4 | <0.001 |
| 2 wk | 13.92 (0.82) | 1.34 (0.23) | 3.76 (0.10)‡∥ | 5.97 (0.25)‡∥ | 7.95 (0.28)‡∥ | 92.8 | <0.001 | 160.0 | <0.001 |
| 3 wk | 14.25 (0.34) | 1.13 (0.16) | 3.83 (0.15)‡∥ | 6.31 (0.27)‡∥ | 8.74 (0.28)‡∥ | 273.4 | <0.001 | 202.3 | <0.001 |
| 4 wk | 13.67 (0.79) | 3.16 (0.31) | 5.31 (0.12)‡∥ | 7.29 (0.26)‡∥ | 9.78 (0.23)‡∥ | 68.4 | <0.001 | 139.2 | <0.001 |
| Contralateral | 1-Way ANOVA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Time Point | Sham | Vehicle | NAC 30 | NAC 100 | NAC 300 | F⁎ | P | F† | P |
| Before Ischemia | |||||||||
| Baseline | 14.75 (0.25) | 13.67 (0.79) | 13.92 (0.82) | 14.75 (0.25) | 14.50 (0.32) | 0.8 | 0.529 | 0.7 | 0.569 |
| After Reperfusion | |||||||||
| 4 h | 14.75 (0.25) | 4.72 (0.80) | 4.11 (0.09)‡ | 5.01 (0.14)‡ | 6.58 (0.05)¶‡ | 1031.8 | <0.001 | 6.6 | 0.003 |
| 1 d | 14.50 (0.32) | 4.40 (0.67) | 4.38 (0.10)‡ | 4.60 (0.06)‡ | 5.82 (0.23)¶‡ | 567.3 | <0.001 | 3.6 | 0.031 |
| 3 d | 13.67 (0.79) | 0.88 (0.04) | 4.85 (0.15)‡∥ | 8.25 (0.17)‡∥ | 10.36 (0.67)∥# | 48.8 | <0.001 | 138.1 | <0.001 |
| 5 d | 15.00 (0.00) | 2.19 (0.28) | 4.75 (0.12)‡∥ | 8.12 (0.16)‡∥ | 9.78 (0.23)‡∥ | 776.2 | <0.001 | 269.0 | <0.001 |
| 1 wk | 14.25 (0.33) | 2.28 (0.24) | 4.93 (0.15)‡∥ | 7.51 (0.22)‡∥ | 9.10 (0.29)‡∥ | 232.8 | <0.001 | 169.3 | <0.001 |
| 2 wk | 15.00 (0.00) | 4.23 (0.49) | 5.26 (0.12)¶‡ | 8.51 (0.13)‡∥ | 10.01 (0.00)‡∥ | 2173.0 | <0.001 | 109.5 | <0.001 |
| 3 wk | 14.17 (0.83) | 4.32 (0.44) | 5.39 (0.03)¶‡ | 8.51 (0.13)‡∥ | 9.78 (0.23)‡∥ | 69.6 | <0.001 | 99.2 | <0.001 |
| 4 wk | 14.50 (0.32) | 5.01 (0.19) | 5.82 (0.23)‡ | 9.55 (0.29)‡∥ | 10.59 (0.58)‡∥ | 89.1 | <0.001 | 59.1 | <0.001 |
NAC 30 = NAC 30 mg/kg treatment (n = 6); NAC 100 = NAC 100 mg/kg treatment (n = 6); NAC 300 = NAC 300mg/kg treatment (n = 6); Sham (n = 6); Vehicle = saline treatment (n = 6).
1-Way ANOVA between the sham and treatment groups (NAC 30, 100, 300); df = 3,20.
1-Way ANOVA between the vehicle and treatment groups (NAC 30, 100, 300); df = 3,20.
P < 0.001 versus the vehicle group.
P < 0.01 versus the vehicle group.
P < 0.001 versus the sham group.
P < 0.05 versus the vehicle group.
P < 0.01 versus the sham group.
However, mechanical thresholds for NAC groups were not normalized to the sham group (P < 0.001 at all time points, except P = 0.001 at 3 days on the contralateral side in NAC 300) on the ipsilateral and contralateral sides, thus indicating that NAC treatment did not abolish mechanical allodynia completely, although there was an unquestionable significant effect of the NAC treatment group compared with the vehicle group. Significant main effects of group were observed at 4 hours (F3,20 = 1063.1; P < 0.001), 1 day (F3,20 = 169.7; P = 0.031), 3 days (F3,20 = 379.0; P < 0.001), 5 days (F3,20 = 404.0; P < 0.001), 1 week (F3,20 = 417.0; P < 0.001), 2 weeks (F3,20 = 92.8; P < 0.001), 3 weeks (F3,20 = 273.4; P < 0.001), and 4 weeks (F3,20 = 68.4; P < 0.001) after reperfusion on the ipsilateral side. Significant main effects of group were observed at 4 hours (F3,20 = 1031.8; P < 0.001), 1 day (F3,20 = 567.3; P = 0.031), 3 days (F3,20 = 48.8; P < 0.001), 5 days (F3,20 = 776.2; P < 0.001), 1 week (F3,20 = 232.8; P < 0.001), 2 weeks (F3,20 = 2173.0; P < 0.001), 3 weeks (F3,20 = 69.6; P < 0.001), and 4 weeks (F3,20 = 89.1; P < 0.001) after reperfusion on the contralateral side (Table I).
Experiment II: Change in Pain Behavior and the Level of PNR1 Protein After NAC Administration
Baseline values of mechanical thresholds for the sham, vehicle, NAC 30, NAC 100, and NAC 300 groups were not significantly different (data not shown). Pain behavior and Western blot data measured at 3 days are presented in Figure 1. All NAC treatment groups showed significant antiallodynic effects relative to the saline-treated CPIP (vehicle) group (F3,20 = 521.0; P < 0.001; all, P < 0.001), although mechanical thresholds of NAC groups were not normalized to that of sham (F3,20 = 402.1; P < 0.001; all, P < 0.001). In vehicle rats, the relative levels of pNR1 protein were increased approximately 145% compared with sham rats, confirming that the amount of NMDA receptor phosphorylation was increased significantly in CPIP neuropathic rats (P < 0.001). The relative density of pNR1 in NAC-treated rats (NAC 30, 100, and 300 mg/kg) decreased significantly in the ipsilateral L4/6 spinal cord compared with that of the vehicle control group (all, P < 0.001). A significant main effect of group was observed (F3,20 = 260.7; P < 0.001). Compared with that of the sham group, relative density of pNR1 of NAC 300 mg/kg did not differ (P = 0.386), whereas the relative density of pNR1 in NAC-treated rats (NAC 30, 100 mg/kg) was significantly high (all, P < 0.001). A significant main effect of group was observed (F3,20 = 324.2; P < 0.001).
Figure 1.
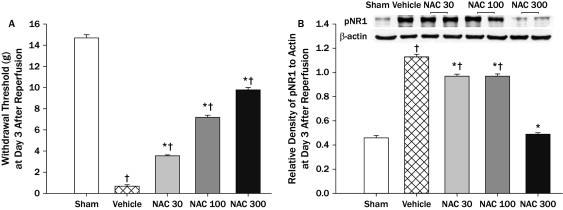
The effect of N-acetyl-l-cysteine (NAC) on mechanical allodynia (A) and the relative density of phosphorylated N-methyl-d-aspartate (NMDA) receptor 1 subunit (pNR1) protein in the spinal cord (L4/6) with an example of Western blot gel (B) at 3 days after reperfusion. In the left graph (A). all NAC groups showed significant antiallodynic effects relative to the vehicle group, although mechanical thresholds of NAC groups were not normalized to that of sham. The right graph (B) represents the pNR1 gel density ratio for each group. The relative density of pNR1 in all NAC groups decreased significantly compared with that of the vehicle group, although the relative density of pNR1 in NAC groups (NAC 30, 100 mg/kg) was significantly high compared with that of sham group. The data are presented as mean (SEM). NAC 30 = NAC 30 mg/kg treatment (n = 6); NAC 100 = NAC 100 mg/kg treatment (n = 6); NAC 300= NAC 300 mg/kg treatment (n = 6); sham rats (n = 6); vehicle = saline treatment (n = 6). *P < 0.001 versus the vehicle group; †P < 0.001 versus the sham group.
By Spearman's correlation analysis, the NAC dose was significantly correlated not only with paw-withdrawal threshold (ρ = 0.979; P < 0.001) but also with the relative density of pNR1 (ρ = –0.875; P < 0.001), indicating that NAC contributed, in a dose-dependent manner, to the analgesic effect and the block of phosphorylation of the NMDA receptor. Importantly, a significant correlation was found between the paw-withdrawal threshold and the relative density of pNR1 (ρ = –0.828; P < 0.001; Figure 2).
Figure 2.
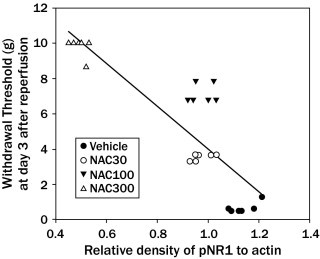
The relationship between mechanical allodynia (withdrawal threshold) and the relative density of phosphorylated N-methyl-d-aspartate (NMDA) receptor 1 subunit (pNR1) protein at 3 days after reperfusion. Rats as a whole showed the significant inverse relationship between the mechanical withdrawal threshold and the relative density of pNR1 protein (ρ = –0.828; P < 0.01). NAC 30 = NAC 30 mg/kg treatment (n = 6); NAC 100 = NAC 100 mg/kg treatment (n = 6); NAC 300 = NAC 300 mg/kg treatment (n = 6); vehicle = saline treatment (n = 6).
Discussion
The main finding of this study was that NAC, when administered in a pre-reperfusion period in CPIP rats, was capable of attenuating IR injury-induced mechanical allodynia for 4 weeks and attenuating the increase in NMDA-receptor phosphorylation in the spinal cord. Rats exposed to prolonged IR injury in the hind paw exhibited acute hyperemia and plasma extravasation, as well as chronic neuropathic pain-like symptoms, including mechanical allodynia and hyperalgesia in both the ischemic and, to a lesser extent, the contralateral hind paw. Prolonged hind limb IR was shown to produce a well-documented cascade of inflammatory events, with a key role for ROS, such as superoxide, hydrogen peroxide, and peroxynitrite anions.21 We demonstrated that mechanical allodynia among these symptoms, which are similar to those in CRPS patients, was attenuated by the administration of NAC, and this persisted for 4 weeks. The antinociceptive effect of NAC was initially evaluated in the CPIP model by Coderre et al,17 who reported that NAC alleviated mechanical allodynia in CPIP rats. This finding was consistent with other findings showing that NAC produced antinociception in the neurogenic phase of the formalin test in mice and the capsaicin test.16 These data supported the hypothesis that ROS were involved in the maintenance of chronic pain.
In contrast, the antinoceptive effect of NAC can be explained through several oxidative pathways. First, NAC treatment might have raised nerve glutathione levels because of its capacity to donate the amino acid cysteine, a component of glutathione.22 Because glutathione is one of the key defenses against ROS and oxidative stress, such as nitric oxide and superoxide within neurons, an increase in its levels might be protective.23 Furthermore, at high doses, NAC had direct reductant24 and antioxidant effects and could block lipid peroxidation by peroxinitrate.25 It might have also scavenged ROS and nitric oxide due to the free sulfhydryl group in NAC.15
Interestingly, we found a long-term antiallodynic effect of NAC injected during the early, formative phase of neuropathic pain. Unlike most previous studies, in which antioxidants were administered to rats with already developed neuropathic pain, NAC was administered just at pre-reperfusion, a critical phase of IR injury. This indicated that the modulation of oxidative stress through NAC administration might involve central sensitization. We confirmed this by demonstrating that NAC attenuated NMDA receptor phosphorylation in CPIP rats. Clearly, one should be cautious in interpreting animal data, but these findings might support a rationale for investigating the possible therapeutic use of NAC as an adjuvant in the treatment of patients with CRPS.
Central sensitization in the spinal cord is defined by an increased responsiveness of dorsal horn neurons to nociceptive peripheral stimulation.26 Glutamate, a major excitatory amino acid, is involved in central sensitization through NMDA receptor activation.27–29 Activation of NMDA receptors increases intercellular calcium levels, which, in turn, increases the activity of several free-radical generating enzymes, such as nitric oxide synthase.30 To activate NMDA receptors, phosphorylation of NMDA receptors, and more precisely, the functional subunit of NR1 is essential. Several studies showed that the amount of pNR1 protein was related to pain behavior in different animal models.13,31 In the present study, CPIP rats pretreated with NAC showed attenuation of the enhancement of NMDA-receptor phosphorylation, measured by the levels of pNR1 protein in the spinal cord in conjunction with the alleviation of mechanical allodynia. NAC is a low-molecular-weight compound that readily passes through the blood–brain barrier and reaches the spinal cord.32 Thus, systemically injected NAC can reduce central sensitization by acting directly on the spinal cord, in addition to peripheral modification of ROS in the IR injury tissue. However, considering the timing of pre-reperfusion administration of NAC, profound behavioral effect over a prolonged period of time would be related more closely to peripheral contribution of NAC to central sensitization. Thus, by increasing antioxidant defenses through increasing glutathione, NAC might reduce pain transmission and central sensitization.
There were several limitations to this study. First, systemically injected NAC acts peripherally and centrally. Therefore, additional study with various approaches, including intrathecal drug injection, is needed to unravel the relative contribution of both peripheral and central action of NAC in the modulation of central sensitization. Second, although the present experimental study was designed to resemble the clinical conditions of CRPS, all such experimental models have important differences compared with the setting of CRPS in humans. Additionally, this was a small experimental study in a CRPS model in rats; therefore, large and controlled studies in humans are needed to confirm the results.
Conclusions
This study demonstrated that ROS scavenger NAC effectively attenuated mechanical allodynia, and that this antiallodynia was achieved, in part, by inhibiting NMDA receptor phosphorylation, leading to central sensitization and resulting in decreased chronic pain in the CPIP rat model. Thus, this study identified oxidative stress as an important determinant of the neuropathologic and behavioral consequences of IR injury, providing support for the potential use of NAC as a therapeutic agent in the treatment of IR injury-related neuropathic pain in CRPS patients.
Acknowledgments
No grants or funding were provided for the performance of this study. The authors have indicated that they have no conflicts of interest regarding the content of this article. Dr. Baek participated in study design and implementation of experimental work. Dr. Lim participated in study design, implementation of experimental work, and manuscript preparation. Dr. Kwak participated in study design, implementation of experimental work, and manuscript preparation. The content of this article is a part of a doctorial thesis submitted to the Kyungpook National University in 2007.
References
- 1.Burton A.W., Bruehl S., Harden R.N. Current diagnosis and therapy of complex regional pain syndrome: refining diagnostic criteria and therapeutic options. Expert Rev Neurother. 2005;5:643–651. doi: 10.1586/14737175.5.5.643. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg E., Shtahl S., Geller R. Serum and salivary oxidative analysis in complex regional pain syndrome. Pain. 2008;138:226–232. doi: 10.1016/j.pain.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 3.Perez R.S., Zuurmond W.W., Bezemer P.D. The treatment of complex regional pain syndrome type I with free radical scavengers: a randomized controlled study. Pain. 2003;102:297–307. doi: 10.1016/S0304-3959(02)00414-1. [DOI] [PubMed] [Google Scholar]
- 4.Zuurmond W.W., Langendijk P.N., Bezemer P.D. Treatment of acute reflex sympathetic dystrophy with DMSO 50% in a fatty cream. Acta Anaesthesiol Scand. 1996;40:364–367. doi: 10.1111/j.1399-6576.1996.tb04446.x. [DOI] [PubMed] [Google Scholar]
- 5.Amadio C. Vitamin C reduced the incidence of reflex sympathetic dystrophy after wrist fracture. J Bone Joint Surg Am. 2000;82:873. doi: 10.2106/00004623-200006000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Cazeneuve J.F., Leborgne J.M., Kermad K. Vitamin C and prevention of reflex sympathetic dystrophy following surgical management of distal radius fractures. Acta Orthop Belg. 2002;68:481–484. [PubMed] [Google Scholar]
- 7.Ji R.R., Kohno T., Moore K.A. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;88:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]
- 10.Caudle R.M., Perez F.M., King C. N-Methyl-d-aspartate receptor subunit expression and phosphorylation following excitotoxic spinal cord injury in rats. Neurosci Lett. 2003;349:37–40. doi: 10.1016/s0304-3940(03)00700-6. [DOI] [PubMed] [Google Scholar]
- 11.Guo W., Zou S., Guan Y. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–6217. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slack S.E., Thompson S.W. Brain-derived neurotrophic factor induces NMDA receptor 1 phosphorylation in rat spinal cord. Neuroreport. 2002;13:1967–1970. doi: 10.1097/00001756-200210280-00027. [DOI] [PubMed] [Google Scholar]
- 13.Zou X., Lin Q., Willis W.D. Enhanced phosphorylation of NMDA receptor 1 subunits in spinal cord dorsal horn and spinothalamic tract neurons after intradermal injection of capsaicin in rats. J Neurosci. 2000;20:6989–6997. doi: 10.1523/JNEUROSCI.20-18-06989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seaton T.A., Cooper J.M., Schapira A.H. Free radical scavengers protect dopaminergic cell line from apoptosis induced complex I inhibitors. Brain Res. 1997;777:110–118. doi: 10.1016/s0006-8993(97)01034-2. [DOI] [PubMed] [Google Scholar]
- 15.Naik A.K., Tandan S.K., Dudhgaonkar S.P. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur J Pain. 2006;10:573–579. doi: 10.1016/j.ejpain.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Rossato M.F., Velloso N.A., de Oliveira Ferreira A.P. Spinal levels of nonprotein thiols are related to nociception in mice. J Pain. 2010;11:545–554. doi: 10.1016/j.jpain.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Coderre T.J., Xanthos D.N., Francis L. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain. 2004;112:94–105. doi: 10.1016/j.pain.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Kim H.K., Park S.K., Zhou J.L. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Chaplan S.R., Bach F.W., Pogrel J.W. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 20.Dixon W.J. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 21.Blaisdell F.W. The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: a review. Cardiovasc Surg. 2002;10:620–630. doi: 10.1016/s0967-2109(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 22.Wagner R., Heckman H.M., Myers R.R. Wallerian degeneration and hyperalgesia after peripheral nerve injury are glutathione-dependent. Pain. 1998;72:173–179. doi: 10.1016/S0304-3959(98)00091-8. [DOI] [PubMed] [Google Scholar]
- 23.Cooper A.J., Kristal B.S. Multiple roles of glutathione in central nervous system. Biol Chem. 1997;378:793–802. [PubMed] [Google Scholar]
- 24.Yan C.Y., Ferrari G., Greene L.A. N-acetylcysteine-promoted survival of PC12 cells is glutathione-independent but transcription-dependent. J Biol Chem. 1995;270:26827–26832. doi: 10.1074/jbc.270.45.26827. [DOI] [PubMed] [Google Scholar]
- 25.Han D., Sen C.K., Roy S. Protection against glutamate-induced cytotoxicity in C6 glial cells by thiol antioxidants. Am J Physiol. 1997;273:771–778. doi: 10.1152/ajpregu.1997.273.5.R1771. [DOI] [PubMed] [Google Scholar]
- 26.Woolf C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 27.Woolf C.J., Thompson S.W. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–299. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- 28.Coderre T.J., Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Q.P., Woolf C.J. Noxious stimuli induce an N-methyl-D-aspartate receptor-dependent hypersensitivity of the flexion withdrawal reflex to touch: implications for the treatment of mechanical allodynia. Pain. 1995;6:383–390. doi: 10.1016/0304-3959(94)00195-K. [DOI] [PubMed] [Google Scholar]
- 30.Barger S.W., Goodwin M.E., Porter M.M. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X., Kim H.K., Chung J.M. Enhancement of NMDA receptor phosphorylation of the spinal dorsal horn and nucleus gracilis neurons in neuropathic rats. Pain. 2005;116:62–72. doi: 10.1016/j.pain.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 32.Farr S.A., Poon H.F., Dogrukol-Ak D. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]