

Abstract
Core myopathies (CM), the main non-dystrophic myopathies in childhood, remain genetically unexplained in many cases. Heart disease is not considered part of the typical CM spectrum. No congenital heart defect has been reported, and childhood-onset cardiomyopathy has been documented in only two CM families with homozygous mutations of the TTN gene. TTN encodes titin, a giant protein of striated muscles. Recently, heterozygous TTN truncating mutations have also been reported as a major cause of dominant dilated cardiomyopathy. However, relatively few TTN mutations and phenotypes are known, and titin pathophysiological role in cardiac and skeletal muscle conditions is incompletely understood. We analyzed a series of 23 families with congenital CM and primary heart disease using TTN M-line-targeted sequencing followed in selected patients by whole-exome sequencing and functional studies. We identified seven novel homozygous or compound heterozygous TTN mutations (five in the M-line, five truncating) in 17% patients. Heterozygous parents were healthy. Phenotype analysis identified four novel titinopathies, including cardiac septal defects, left ventricular non-compaction, Emery–Dreifuss muscular dystrophy or arthrogryposis. Additionally, in vitro studies documented the first-reported absence of a functional titin kinase domain in humans, leading to a severe antenatal phenotype. We establish that CM are associated with a large range of heart conditions of which TTN mutations are a major cause, thereby expanding the TTN mutational and phenotypic spectrum. Additionally, our results suggest titin kinase implication in cardiac morphogenesis and demonstrate that heterozygous TTN truncating mutations may not manifest unless associated with a second mutation, reassessing the paradigm of their dominant expression.
INTRODUCTION
Core myopathies (CM) are the most common form of inherited non-dystrophic muscle disorder in childhood. These congenital myopathies typically present with delayed motor development and generalized muscle weakness, sometimes associated with scoliosis and respiratory failure. Histopathologically, they are defined by areas of mitochondria depletion and sarcomere disorganization (cores) in muscle fibers. The recessive form of core myopathy, multi-minicore disease (MmD), is clinically and genetically heterogeneous (1). MmD has been mainly associated with RYR1 (2), SEPN1 (3) or MEGF10 (4) mutations, but for many cases, the underlying gene remains unknown.
While respiratory insufficiency is a common finding in MmD, primary myocardial involvement is reportedly exceptional (5) and congenital heart defects are not considered part of the MmD spectrum (6). Indeed, while congenital cardiopathies, including atrial and ventricular septal defects (ASD and VSD), are the most common human birth defects and a major cause of morbidity and mortality in childhood, to our knowledge they have never been definitely associated with any skeletal muscle condition (7). Conversely, inherited cardiomyopathies, defined by defective myocardial contractility, and left ventricular non-compaction (LVNC), a recently described condition which results from intrauterine arrest of myocardial development and abnormal compaction of the cardiac tissue (8), have been associated with different skeletal muscle or metabolic conditions (9). However, there are only a few case reports of patients presenting with the association of cardiomyopathy and congenital core myopathy, most of them from the pre-molecular era (9–14). Autosomal dominant forms of core myopathy with variable adult-onset cardiomyopathy have been associated with heterozygous mutations of the MYH7 (15,16) or the ACTA1 (17) genes in eight and one families, respectively. Additionally, we reported two families with recessive minicore myopathy and fatal dilated cardiomyopathy (DCM) starting from ages 5 to 20 years (also known as Salih myopathy or as early-onset myopathy with fatal cardiomyopathy (EOMFC, MIM [611705])) associated with homozygous truncating mutations of the titin gene TTN, which represented the first titinopathy involving both skeletal and heart muscles (5). No other similar cases have been reported; thus, this condition is probably under-recognized, and the association of CM and pediatric heart conditions is incompletely understood.
The 363-exon titin gene (TTN, MIM [188840]) encodes titin, the largest protein known (18). Titin is one of the main sarcomeric proteins and plays crucial roles in cardiac and skeletal muscle development, structure, elasticity and cell signaling. Each single titin filament bridges half of the entire sarcomere, from the Z-disk to the M-band (19), where it interacts with numerous protein partners. The molecular architecture of the human titin protein is organized into four distinct parts: the amino-terminal Z-disc, the I-band and A-band regions, and the carboxyl-terminal M-line extremity encoded by the last six exons (358–363, or Mex1 to Mex6).
Prior to next generation sequencing (NGS), routine analysis of the whole TTN gene was impossible because of its size and complexity. Thus, only a few TTN mutations had been reported, and the general incidence and spectrum of titinopathies was probably significantly underestimated. TTN mutations were identified in conditions involving exclusively skeletal muscles (tibial muscular dystrophy, TMD, hereditary myopathy with early respiratory failure, HMERF and limb girdle muscular dystrophy LGMD2J (20–22)), in isolated dilated or hypertrophic cardiomyopathy (DCM, HCM) (23–25) and in the rare cases of congenital multi-minicore myopathy with childhood-onset dilated cardiomyopathy mentioned earlier (5). In the last months, NGS has disclosed a plethora of novel TTN mutations and TTN is emerging as a key gene in human inherited disease (26–30). Virtually all the newly reported TTN mutations are heterozygous and associated with adult-onset dominant phenotypes involving either cardiac or skeletal muscles. Particularly, heterozygous TTN truncating mutations have been reported as the most common known genetic cause of dominant or sporadic DCM (31). However, truncating TTN mutations have been also reported in 3% of a nominally healthy control population (31), which illustrates the difficulties in interpreting the pathogenicity of novel TTN changes. Furthermore, the role in disease of several key titin domains remains to be established. This includes the unique titin serine–threonine kinase domain (TK) (18), a pivotal player in the control of muscle gene expression and protein turnover (22). Knock-out of the TK-encoding region and adjacent TTN exons (Mex1 and Mex2) results in embryonic death at mid-gestation in homozygous mice (32). In humans, only one TK mutation has been reported in the adult-onset dominant myopathy HMERF, which does not affect the heart (22). Surprisingly, despite the rising number of TTN mutations identified, no novel TTN-associated skeletal and cardiac phenotype has been reported until now.
Here we report results from targeted or exome-wide sequencing on a first large series of core myopathy with heart disease disclosing multiple titin new mutations and phenotypes. Furthermore, we characterize the first-reported absence of a functional TK in humans, leading to the first antenatal-onset titinopathy. Our work suggests a major role of titin defects in early-onset myopathies with cardiac dysfunction, expands the spectrum of TTN-associated muscular and cardiological phenotypes and demonstrates that TTN truncating mutations do not always produce disease unless associated with a second mutation. Complementary ex vivo and in vitro studies suggest a key role of TK in human skeletal and cardiac muscle.
RESULTS
Genetic studies
We collected and analyzed a first cohort of 31 patients (from 23 families), which presented with congenital core myopathy combined with primary heart disease. M-line TTN mutations having been associated with a skeletal and cardiac myopathy with minicores (5), we started genetic studies in the 23 families by Sanger sequencing of the 6 TTN M-line-encoding exons (Mex1–Mex6). Five pathogenic TTN sequence changes were identified in four families (Patients 1–5, P1–P5, Fig. 1, Table 1). These included one missense and four truncating mutations (two nonsense and two frameshift). Only one of these sequence changes (p.Trp34072Arg) was reported as a rare privative variant of unclear significance in dbSNP, 1000 Genomes Project, and none was observed in 120 unrelated Caucasian or 96 Turkish control samples.
Figure 1.
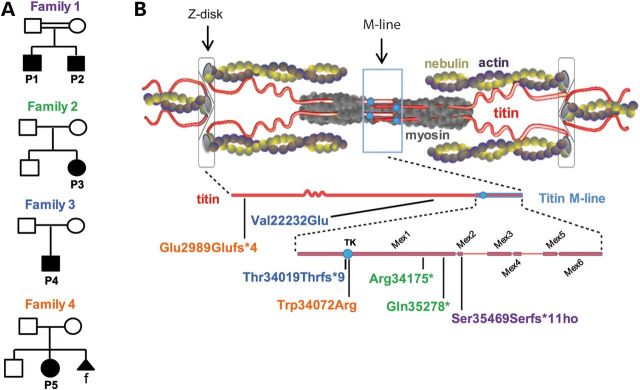
TTN mutations and distribution. (A) Pedigrees from Families 1 to 4. Solid symbols denote affected subjects. Double lines signify consanguinity. Triangular shape indicates pregnancy termination; f = female. (B) Schema of a sarcomere with its main proteins (top) including titin (middle) and zoom on the titin M-line region (bottom). TK = titin kinase. Patient's mutations are indicated using colors corresponding to their pedigrees.
Table 1.
Clinical features
| Family 1 (Patients 1 and 2) | Family 2 (Patient 3) | Family 3 (Patient 4) | Family 4 (Patient 5) | |
|---|---|---|---|---|
| Mutation | c.106632–106633delTG p.Ser35469Serfs*11 homozygous | c.C102748T p.Arg34175*; c.C106057T p.Gln35278* |
c.T66920A p.Val22232Glu; c.102282delT p.Asn34020Thrfs*9 |
c.G9388+1C p.Glu2989Glufs*4 c.T102439C p.Trp34072Arg |
| Cardiac septal defects | No | Neonatal ASD | ASD, VSD; surgical repair in the 1st year LV to RA shunt repaired at 4 years |
Small muscular VSD |
| Cardiomyopathy | Asymptomatic DCM detected at 19 years in the elder brother. Normal echocardiography in the younger brother at 16 years |
|
Arrhythmia and SVT at the age of 8 years DCM from the age of 13 years (SF 11%) Cardiac transplant at 14 years |
LVNC Slightly depressed LV function at birth At the age of 1 month, DCM with EF 10–15% Terminal heart failure and cardiac transplant at 4 years |
| Rhythm disturbances | No | Holter: ventricular extrasystoles++ | Arrhythmia and SVT at the age of 8 years | NA |
| Early motor development | Normal | Neonatal hypotonia, delayed head control, walked at the age of 2 years | Normal | Neonatal hypotonia, severe motor skill delay, head drop, sitting and standing with support at the age of 19 months |
| First musculoskeletal signs | From the age of 2 years: weakness, elbow and ankle contractures | Neonatal hypotonia, poor suckling (NG tube), never run or jumped, difficulties climbing stairs, steppage (7 years) | Never able to run From the age of 5 years: difficulties climbing stairs |
Decreased fetal movements, dislocated hips, bilateral hip dysplasia, thoracic kyphosis |
| Joint contractures | Moderate/severe, elbows and ankles | Severe, elbows and ankles, from the age of 6 years | Mild, ankles | Elbow, hip, knee, digit (distal AMC) |
| Rigid spine | NA | Yes, severe | Yes, moderate | Yes |
| Scoliosis | NA | Yes, from the age of 12 years Arthrodesis at the age of 13 years |
Yes, mild, thoracic | Yes, kyphosis from birth, progressive kyphoscoliosis in childhood |
| Respiratory failure | No | FVC 800 ml, FEV1 700 ml | No | NA |
| Ptosis | NA | No | Mild | No |
| CK | 4 × N | 2–5 × N | 195 U/l | NA |
| Muscle weakness evolution | Slowly progressive | Slowly progressive | Stable | Stable AMC: improving Intrasurgical spinal cord infarction resulting in paraplegia at the age of 4 years |
| Muscle biopsy | Minicores, central nuclei, dark oxidative areas, type 1 predominance (16 years) | Minicores, central nuclei +++, target-like fibers, central basophilic areas, type 1 predominance (2 years, 14 years and 25 years) | Minicores, central nuclei, central basophilic areas, dark oxidative areas, type 1 uniformity (12 years) | Minicores, central nuclei, type 1 predominance, mild endomysial fibrosis (15 months) |
| Walking ability | Good | 200 m | Good | Never walked |
| Others | Mild dysphagia Large neck Flat, retractile thorax |
Cleft soft palate Webbed short neck, hypertelorism Radioulnar synostosis |
Short neck Finger ulnar deviation, bilateral camptodactyly Low-set ears |
|
| Parents age at last echocardiogram and ECG (years) | 51 and 52 | 53 and 55 | 49 and 50 | 38 and 42 |
NCBI accession numbers: NM_001267550.1 and NP_001254479.
AMC, arthrogryposis multiplex congenita; ASD/VSD, atrial/ventricular septum defect; CK, creatine kinase; DCM, dilated cardiomyopathy; ECG, electrocardiogram; EF, ejection fraction; FEV1, forced expiratory volume 1; FVC, forced vital capacity; LV, left ventricle; LVNC, left ventricular non–compaction; MRI, magnetic resonance imaging; NA, non-available data; NG, nasogastric; PAH, pulmonary artery hypertension; RA, right atrium; SF, shortening fraction; SVT, Supraventricular tachycardia.
In Families 1 and 2, patients were, respectively, homozygous and compound heterozygous for TTN truncating mutations. The two affected brothers from Turkish consanguineous Family 1 (P1 and P2) were homozygous for a two base-pair deletion in Mex2, c.106632-106633delTG (p.Ser35469Serfs*11), which predicts a truncated protein deleted of 522 C-terminal amino acids. In Family 2, P3 harbored two heterozygous nonsense mutations, c.C102748T (p.Arg34175*) and c.C106057T (p.Gln35278*), in Mex1, downstream of the TK encoding residues. Both mutations predict premature termination codons (PTC) leading to a truncated protein lacking the C-terminal 1,796 and 689 amino acids, respectively. Immunohistochemical studies on P3 muscle confirmed expression of a truncated titin totally devoid of its C-terminus, which was integrated into apparently normal sarcomeres (Fig. 2).
Figure 2.

Expression and sarcomere integration of mutant titin in Patients 3 and 5. (A) Quadriceps from P3 and control (left panel, 63×) and myocardium from P5 and control (right panel, 63 × 4); longitudinal cryosections. Note total loss of labeling with T51 in P3. Dapi (blue) stains nuclei. (B) Recognition sites of Z1Z2 and T51 anti-titin antibodies, respectively, upstream and downstream of P5 and P3 mutations.
In contrast, in Families 3 and 4 (P4 and P5, respectively), M-line TTN screening identified only one private heterozygous Mex1 mutation in each patient, which was also heterozygous in each of their healthy fathers. Interestingly, both mutations affected the critical TK domain. P4 had a one-base-pair deletion at the beginning of the TK-encoding sequence (c.102282delT, p.Asn34020Thrfs*9 or p.N208Tfs*9 in the structure of the isolated TK domain (33)). This mutation predicts protein truncation after nine frame-shifted amino acids, leading to loss of the most C-terminal 100 amino acids of the 307 amino acid-TK domain and deleting 1963 C-terminal titin amino acids. P5 had a missense change (c.T102439C) affecting a highly conserved residue of the TK (p.Trp34072Arg of full-length titin, or p.Trp260Arg (p.TK-W260R) in the TK (33)). Given the paternal carrier status for each family, we suspected recessive inheritance and therefore a second TTN mutation in these patients. Whole-exome sequencing confirmed that both patients are compound heterozygous for two TTN mutations. In P4, exome data confirmed the paternally inherited TK deletion and identified an additional missense variant c.T66920A (p.Val22232Glu, exon 316), which impacts a Fn3 domain in the end of the titin I-band (Fig. 1), inherited from his heterozygous mother. In Family 4, exome sequencing confirmed the paternal p.Trp34072Arg change and identified a single nucleotide deletion c.G9388+1C (p.Glu2989Glufs*4) in the splice-donor site at 5′ of exon 38, inherited from the healthy mother and predicting titin truncation near its N-terminus. Immunolabelling of myocardium cryosections from P5 confirmed expression and normal sarcomere integration of a full-length titin, presumably encoded by the paternal TK-mutant allele (Fig. 2). No other frameshift, nonsense, splice or highly conserved residue changes were found in either patient.
In all families, the TTN mutations identified were carried by heterozygous parents. All parents (aged from 38 to 55 years) underwent neuromuscular and cardiac examinations (echocardiography and electrocardiogram) with normal results. There was no history of cardiomyopathy or sudden cardiac death in any of the parental families.
Phenotype findings
Analysis of clinical data in the mutant patients (Table 1, Fig. 3A) disclosed novel phenotypes not yet reported or associated with TTN, as well as an unexpectedly wide range of severity.
Figure 3.
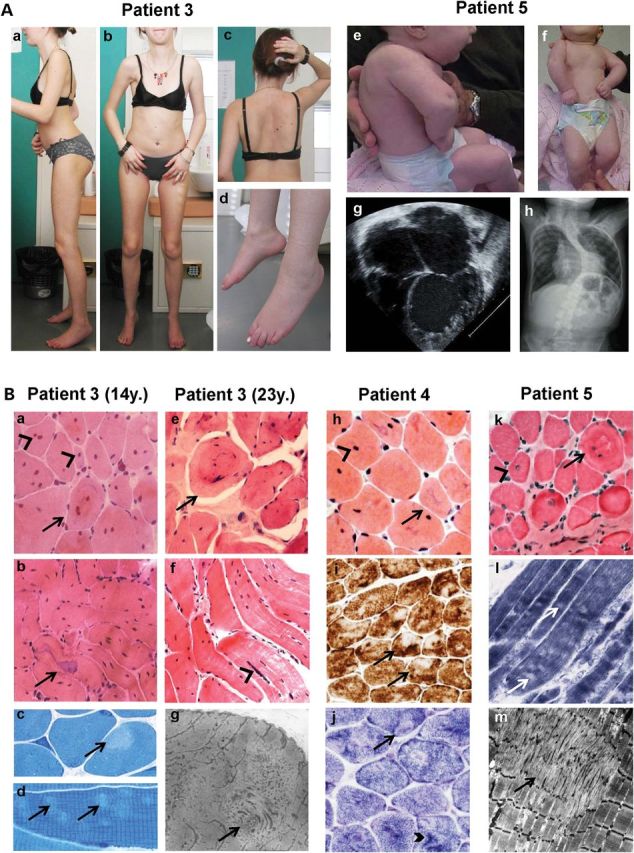
Clinical and histopathological findings. (A) P3 (23 years, left) and P5 (3 months, right). P3 showed global amyotrophy, relatively bulky calf muscles, severe elbow and heel contractures, flat retractile thorax (a, b), scoliosis with dorsal lordosis (c, after arthrodesis) and foot drop (d). Neck flexor weakness and cervical rigidity require head support when standing (c). P5 presented with shoulder, elbow, wrist, hand and lower limb contractures, short neck, low-set ears, retrognathia and kyphoscoliosis (e, f) which subsequently progressed (h). Echocardiography (four-chamber view) showed severe LVNC at 1 month (g). (B) Muscle biopsies from P3 (at 14 (a–d) and 23 years (e–g)), P4 (h–j) and P5 (k–m). A massive (a, b, e, f) or moderate (h, k) number of nuclear centralizations (arrowheads), sometimes forming nuclear chains (f, arrowhead) were associated with ring-like fibers (a, e, k; arrows) and irregular-shaped basophilic areas (b, h; arrows). Minicores were visible as small light foci of mitochondria depletion (i, j, l) and sarcomere disorganization (c, d, g, m). In some cases, they coexisted with dark areas visible on oxidative stainings (j, thick arrowhead). Endomysial fibrosis was absent (h) or mild (k). Sequential quadriceps biopsies from P3 at 2, 14 and 23 years were comparable. Transverse (a, e, h–k) and semi-longitudinal (b, f) cryosections stained with HE (a, b, e, f, h, k), Cox (i), SDH (j) or NADH-TR (l) (40× b, f, i; 60× a, e, h, j–l); transverse (c) and longitudinal (d) semithin sections; electron microscopy (g, m).
At the most severe end of the spectrum, P5 presented with a previously unreported phenotype, associating distal arthrogryposis multiplex congenita (AMC), congenital muscle weakness, kyphosis and neonatal cardiac failure. Muscle biopsy pathology was consistent with MmD (Fig. 3B). Echocardiography revealed a VSD and a trabeculated myocardial appearance typical of LVNC (Table 1, Fig. 3A). The patient developed terminal heart failure requiring transplantation at 4 years of age. Intrasurgical complications resulted in spinal cord infarction and paraplegia. Muscle weakness remains otherwise stable at 9 years, whereas arthrogryposis tends to improve gradually.
P3 and P4 presented with structural heart defects (ASD and/or VSD) leading to heart dysfunction from birth (Table 1). In P3, ASD resolved spontaneously. A retractile myopathy became the main clinical concern until the development at the age of 16 years of moderate DCM with rhythm disturbances. Her phenotype was considered typical of Emery–Dreifuss muscular dystrophy (EDMD, Fig. 3A); LMNA and FHL1 gene screens were negative. P4 had severe ASD and VSD, which required surgical repair at 1 and 4 years, and developed rhythm disturbances and terminal DCM from the age of 13 years, requiring heart transplantation. Conversely, his skeletal muscle phenotype remains mild and stable.
The mildest phenotype was observed in P1 and P2, who presented at 2 years with moderate muscle weakness and contractures. Systematic echocardiography detected early-stage DCM in the elder brother at 19 years and was normal in his brother aged 16. Both remain ambulant without cardiac symptoms.
Despite variability in severity, all patients shared particular phenotypical signs, namely congenital or infantile muscle weakness with axial (Rigid Spine) and distal joint contractures, relatively preserved respiratory function and CK levels <5 times normal associated with primary heart disease. Weakness was most severe in axial muscles (neck flexor weakness and/or scoliosis) and proximal muscles, although P3 had marked drop foot. Furthermore, the skeletal muscle histopathology was consistent and recognizable (Fig. 3B). All muscle biopsies showed type 1 fiber predominance or uniformity and multi-minicore lesions associated with a variable but sometimes extremely high number of internal nuclei. In addition, most samples showed nonspecific but typical abnormalities of the internal structure of the muscle fibers, including star-shaped basophilic areas, ring-like fibers and small zones of increased oxidative activity (Fig. 3B). Sequential biopsies of the quadriceps muscle taken from P3 at 2, 14 and 25 years suggested lack of significant morphological progression (Fig. 3B). No sample showed signs of necrosis/regeneration or immunohistochemical abnormalities of sarcolemma-associated proteins, including dystrophin or laminin-α2.
Ex vivo and in vitro studies
The molecular defects in P5 predicting the first human equivalent of a TK knock-out, we developed further mechanistic studies of these unique mutations.
For the maternally inherited mutation (p.Glu2989Glufs*4), in silico analysis predicted abnormal splicing and a PTC in exon 39 (Fig. 4). Deep sequencing on P5 myocardium confirmed the coexistence of a wild-type and a shorter transcript (36% reads) corresponding to use of an alternative splice-donor site and leading to titin truncation after 2950 N-terminal amino acids, therefore devoid of TK (Fig. 4). Semi-quantitative protein gels confirmed a moderate reduction of the global titin amount in the patient's samples. Consistent with the mRNA data, a correspondingly sized truncated protein band was identified by two different titin antibodies (data not shown).
Figure 4.

Consequences of P5 splice site mutation. ESE finder predicted major loss of strength of the mutant donor site (down to 7, 75%) and two alternative sites in exon 38. Use of one of these leads to coexistence of full-length and shorter exon 38 transcripts as revealed by deep sequencing (A) and cDNA amplification of TTN exon 38 (B). P and C: patient and control, respectively. A few reads suggested minor intron 38 retention (A).
The paternally inherited p.Trp34072Arg (TK-W260R) mutation involves a highly conserved amino acid in the core of the TK active site. We tested the effect of this mutation on interactions with the TK known ligands Nbr1 and the TK autoinhibitory tail (AI) in genetic interaction assays, comparatively with the dominant HMERF mutation TK-R279W (22). Both interactions were completely abrogated in TK-W260R, whereas only Nbr1 interaction was lost in HMERF TK-R279W (Fig. 5). Analysis of thermal stability by circular dichroism spectroscopy demonstrated reduced stability of the TK-W260R mutant (Fig. 5). We could also not detect catalytic activity of the mutant enzyme. These results together suggest that the TK-W260R (p.Trp34072Arg) mutation causes a complete loss-of-function of TK that abrogates M-line and Z-disk linked pathways via Nbr1, thereby potentially implicating this titin defect in myofibril turnover.
Figure 5.

Biochemical characterization of the TK-W260R mutant. (A) Molecular model of TK (PDB entry 1TKI) with the regulatory tail shown in red. The mutated tryptophane residue, Trp34072 or W260 in the TK sequence (TK-W260), occupies a hydrophobic pocket formed by alpha-helices C3, C5, C7 and C8. Mutation to arginine replaces this conserved hydrophobic residue with a large hydrophilic residue and results also in a predicted steric clash with alphaC7 (red volume), likely destabilizing part of the catalytic domain. (B) Mutations in TK abrogate Nbr1 binding. Yeast-two-hybrid reporter gene assay (growth on histidine-free media and activation of beta-galactosidase activity, blue color reaction). Wild-type TK (TK-WT) interacts with its own autoinhibitory tail (AI) and with Nbr1 in trans. Both interactions are abrogated in the TK-W260R mutant, whereas the TK-R279W mutant maintains AI interaction. (C) CD spectra recorded from 200 to 260 nm over 6–94°C reveal a single sharp secondary structure transition in wild-type TK with loss of negative ellipticity (Y) at 209 nm (Z) of ∼59°C (X). In contrast, the TK-W260R mutant shows partial melting (red asterisk) ∼42°C followed by a second transition at 57°C (C).
DISCUSSION
In this work, we establish for the first time that compound heterozygosity of non-symptomatic TTN truncated alleles and missense mutations is a cause of core myopathy with congenital heart disease. The association of heart disease with CM, and particularly with MmD, has been uncommonly recognized and characterized so far. While cardiac (mainly right ventricular) impairment secondary to respiratory failure is not unusual in the classic phenotype of MmD as a result of SEPN1 mutations, primary heart disease is not present in either SEPN1- or RYR1-mutant patients. Aside from two families with minicores, DCM and homozygous TTN mutations (5) and a few families with dominantly inherited core myopathy and adult-onset cardiomyopathy associated with MYH7 or ACTA1 mutations (15,17), most of the rare, mainly dominant cases reported to have both cardiomyopathy and core lesions remain genetically unresolved (6), and differential diagnosis with other conditions (particularly myofibrillar myopathies or laminopathies) can be considered in some of them (10,14). Additionally, while one of the original MmD cases described by Engel (34) had ASD and VSD, no other occurrences of congenital structural heart defects have been reported, and therefore, congenital cardiopathies were not associated with MmD. An exception is the common occurrence of subclinical mitral prolapsus in MmD and other myopathies, probably related to thorax and thoracic spine deformities rather than to primary heart involvement (35). To clarify this complex landscape, we collected and analyzed a first cohort of 23 families with primary heart disease and core myopathy in which the main genes causing other skeletal and cardiac conditions had been excluded, indicating for the first time that core myopathy with heart disease is not as uncommon as previously thought.
Molecular study of this series led to the identification of seven novel homozygous or compound heterozygous TTN mutations in five patients presenting with various forms of multi-minicore myopathy with heart disease. Screening of the last 6 TTN exons (6.5% of the gene) disclosed mutations in 4 of the 23 families studied (17%). This relatively high proportion suggests that TTN M-line mutations are a significant cause of MmD with pediatric heart disease. Interestingly, none of the 72 heterozygous truncating TTN mutations recently reported in a large cohort of familial cases of idiopathic DCM involves the M-line (31). This, together with our current and previous (5) results, suggests a particular role of M-line titin in skeletal muscle integrity. Nevertheless, TTN M-line sequencing identified no mutation in the remaining families in our series, implicating other titin domains or other defective proteins in this phenotype. Complementary exome studies are in progress.
This is the first description of TTN compound heterozygosity leading to phenotypical expression of recessive missense mutations on the background of recessive truncating mutations. Absence of phenotype in the heterozygous parents confirms that not all truncating TTN mutations, particularly if in the M-line, manifest unless associated with a second mutation (5). This supports the proposed recessivity of the TTN in frame insertions/deletions or truncating mutations (upstream of the M-line) lately reported in five patients, although some of their heterozygous parents had subclinical phenotypes or could not be specifically examined (30). Additionally, our results can potentially explain the identification by Herman and coworkers of truncating TTN mutations in 3% of a nominally healthy control population (31), which remained unsolved. Interestingly, in this study of a large idiopathic DCM cohort, the authors identified, besides the reportedly pathogenic truncating TTN mutations, approximately one rare TTN missense variant in every study subject, although these variants were not considered further (31). Our results suggest that some of these missense variants might play a role in disease expression. They also stress the importance of careful family studies to establish pathogenicity of each nonsense mutation and exclude compound heterozygosity with a pathogenic missense mutation, which may have serious impact on genetic counseling and prenatal diagnosis.
In addition, this work establishes that recessive TTN mutations cause novel skeletal and heart muscle phenotypes, including arthrogryposis, LVNC, Emery–Dreifuss syndrome, MmD without pediatric heart disease and cardiac septal defects. This represent a significant expansion of the spectrum of TTN-associated phenotypes. We define here a novel condition in one patient associating MmD with AMC and LVNC. AMC (MIM [208100]) is a relatively common disorder (1 in 3000) (36), characterized by multiple joint contractures from birth (37). The major cause of arthrogryposis is fetal akinesia owing to a variety of uterine or fetal abnormalities (neurogenic, muscular or connective tissue abnormalities), whose heterogeneity has hindered identification of the primary defects. Similarly, LVNC causes, pathogenesis and course remain poorly understood. Our results associate for the first time AMC and LVNC and identify TTN as a new culprit gene, thereby describing the first titinopathy leading to severe fetal abnormalities.
Other patients presented with MmD without pediatric heart disease, EDMD phenotype or septal defects, none of which has been so-far associated with titin. All cases showed also a variable degree of Rigid Spine syndrome (RSS). EDMD, characterized by myopathic changes in skeletal muscles, early contractures at the neck, elbows and Achilles tendons, and cardiac conduction defects, has been associated with mutations in the EMD (MIM [310300]) or the LMNA genes (MIM [181350]). RSS (limitation of spinal flexion owing to contractures of the spine extensors) can be isolated or associated with multiple early-onset myopathies (MIM [602771], (3)). As 60% of EDMD patients, 70% of arthrogryposis and many cases of RSS are without genetic diagnosis and represent a difficult diagnostic challenge, identification of TTN as a novel gene in these conditions may have significant diagnostic impact.
Septal defects are among the most common congenital cardiopathies worldwide. Their pathogenesis is largely unknown. Unexpectedly, they were present in three of the four TTN-mutant families in our series. Owing to their prevalence, one might consider whether their association with TTN mutations is merely accidental. A causal role of TTN in septal defects in this set of patients is supported by (i) the presence of ASD and/or VSD in the majority of cases and (ii) their coexistence with other forms of myocardial involvement imputable to titin dysfunction. While we cannot conclude from this study that TTN is a cause of isolated congenital cardiopathies, the large spectrum of cardiologic phenotypes observed suggests that titin defects underlie an unsuspected number of cardiac conditions, including potentially any congenital heart disease with or without skeletal muscle involvement.
The histopathological pattern in our recessive TTN-mutant patients was consistent and recognizable. While the histological criterion for inclusion in the initial series was the presence of cores of any type and length, the five patients with TTN mutations identified in this study showed typical minicores and fulfilled all the diagnostic criteria of MmD, including marked type 1 fiber predominance. Remarkably, in some patients, centrally located nuclei were present in massive numbers, rarely observed in any other muscle condition including centronuclear myopathy. Abundant internal nuclei were also reported in other TTN-mutant patients (5,30,38). Five of these cases had a histopathological pattern identical to the one observed in our patients, but internal nuclei were so conspicuous that they led to the diagnosis of centronuclear myopathy (30). Taken together, this morphological and/or molecular overlap between titinopathies and centronuclear myopathy raises the question of a potential role of titin (particularly its M-line) in determining and/or maintaining the normal subsarcolemmal nuclear positioning. Additionally, several types of changes in the internal structure of muscle fibers, particularly star-shaped basophilic areas, were observed in the recessive TTN-mutant patients characterized here as well as in those previously reported (5). These areas were relatively rare and therefore could not be characterized ultrastructuraly, but they might be comparable with the regions of myofibrillar disruption observed in TTN-mutant HMERF patients (38). Although none of the observed changes is pathognomonic in itself, the association of minicores, abundant centrally located nuclei and changes in the internal fiber structure seems to be a consistent histopathological hallmark. We propose that this pattern strongly suggests a titinopathy and therefore can help to orientate the genetic studies in MmD; while RYR1-associated CM can show abundant internal nuclei but typically no basophilic lesions, both are usually absent in SEPN1-related myopathy.
Thus, the extended range of cardiac phenotypes and severity in this set of TTN-mutant patients contrasts with the relatively homogeneous and recognizable neuromuscular and histopathologic pattern. Indeed, these patients have so much in common in terms of contractile skeletal muscle phenotype, histology and genetics that they are best understood as having a unique emerging condition, which we propose to term Autosomal Recessive Multi-minicore Disease with Heart Disease (AR MmD-HD) and which, aside from covering the Salih myopathy cases (5), could in the future expand to include other so-far unknown skeletal muscle and heart phenotypes.
The factors determining the presence of heart involvement associated with M-line TTN mutations are unclear. Phenotype–genotype analysis suggests that, in our series, the heart phenotype severity correlates with the localization of the TTN mutations. Patients having a TK mutation presented with a life-threatening cardiac phenotype, whereas those with more C-terminal mutations had less severe or no heart involvement. Moreover, one of these patients showed the first complete loss of a functional TK domain in humans. Her antenatal-onset phenotype combined with our in vitro results represents the first indication that the TK enzymatic or signaling role might have a significant importance for myogenesis, structural integrity and function of skeletal and cardiac muscles during human development. However, the two families with MmD and childhood-onset DCM previously reported had homozygous truncating mutations downstream of the TK-encoding regions (5). Furthermore, almost all the heterozygous TK or M-line mutations reported (20,21,22,39,40) affect exclusively the skeletal muscles. Therefore, the role of titin TK in the molecular pathogenesis of cardiac disease remains to be clarified. Misfolding, perturbed interactions with protein partners or loss of structural integrity and elasticity of sarcomeres containing truncated titins may contribute significantly to phenotypic expression of disease.
In conclusion, we expand the mutational and phenotypic spectrum of titin, delineating an emerging recessive condition (AR MmD-HD) and adding TTN as a novel candidate gene for AMC, Rigid Spine, EDMD, LVNC and MmD without pediatric heart disease. We conclude that TTN is involved in an unexpectedly broad range of cardiac phenotypes and that compound heterozygosity of non-symptomatic TTN truncated alleles and missense mutations is likely to underlie a wide spectrum and significant proportion of currently undiagnosed skeletal muscle conditions, with or without cardiac disease. We propose that sequencing of the six M-line-encoding TTN exons is a useful first-step diagnostic screen in patients with early-onset myopathies and/or pediatric heart conditions. Lastly, we believe that NGS will bring about a dramatic expansion of titinopathies, this report being among the first of what we think will be a plethora of novel TTN-associated phenotypes.
SUBJECTS AND METHODS
Patients
We studied 31 patients (23 families including 5 multiplex and 3 consanguineous) recruited through an international collaboration. Inclusion criteria were as follows: (i) congenital or infantile-onset muscle weakness; (ii) core lesions in the muscle biopsies; (iii) primary heart disease; (iv) exclusion of mutations in the main known genes causing myopathy with cardiopathy, by sequencing (FHL1, DES, LMNA, ACTA1, LAMP2, MYBPC3, MYH7, MYL2, MYL3, PRKAG2, TNNT2, TPM1, RYR1) or SNPs genotyping (Affymetrix, 250K Nsp array).
Biological samples were obtained with informed consent; the study was approved by local research ethics committees under NIH protocol 12-N-0095.
TTN genetics studies
PCR-based sequencing of TTN M-line-encoding exons Mex1–Mex6 was performed on genomic DNA as described in Carmignac et al. (5).
Exome sequencing was performed on genomic DNA from P4 and P5 using Agilent SureSelect All exon 50Mb kit, to capture ∼180 000 annotated protein-coding exons plus selected non-coding RNAs with the Life Technologies (ABI) protocol, including all the 312 exons of the TTN isoform N2A (NM_133378). Captured fragments were sequenced in single-end 50-base mode using SOLiD4 (Life Technologies) chemistry. Reads were aligned to the reference genome GRCh37 and analyzed with NextGene v.2.16 (Soft Genetics, Inc.). A mean of 78.8 million reads (mean length 48 nucleotides) were obtained for each patient, of which ∼60% mapped uniquely to targets (mean coverage 30×). The TTN gene had a mean coverage of 80 with a good repartition of the reads along the sequence, excepting the TTN exons encoding the N-terminal Z-line, for which the coverage was ∼40.
For transcriptome analysis, reads were generated from P5 myocardium total RNA, sequenced on a HiSeq 2000 instrument (Illumina) with 2 × 100 bp paired-end chemistry, mapped to GRCh37 using TopHat 1.4.1, normalized for exon length and scored percent-spliced in as described in Guo et al. (41).
Histopathological and immunofluorescence studies
Muscle biopsies and/or explanted myocardium (from P5) were frozen or fixed and processed for standard histological, histochemical and electron microscopy studies. Standard indirect immunohistochemical studies were performed using anti-titin antibodies T51 (1/100) (42) and Z1Z2 (1/100) (43). Confocal images were captured as described in Carmignac et al. (5).
Western blot
Proteins were extracted from myocardium cryosections from P5 and a control individual as described in Hackman et al. (20) and resolved in 6 or 12% linear SDS–PAGE gels, followed by transfer onto PVDF membrane for ECL detection using pAb m10-1 at 1:500 (20), TTN mAb T12 at 1:500 (44) or Z1Z2 at 1:1000 (43).
In vitro studies
Expression of recombinant TK and circular dichroism spectroscopy were performed as described using a construct comprising the autoinhibited TK domain and its preceding fibronectin-like domain (Fn-TK) (45). Yeast genetic interaction analysis was performed using constitutively open TK-kin3 as described (22).
WEB RESOURCES
ESE Finder, http://rulai.cshl.edu/tools/ESE2/
NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/), v.0.0.19. (date last accessed, 22 March 2013)
TTN NCBI accession numbers: NM_001267550.1 and NP_001254479.
All variants reported in this work are registered in the LOVD database on the TTN page: http://www.dmd.nl/nmdb2/home.php?select_db=TTN
FUNDING
This work was supported by the Institut National de la Santé Et de la Recherche Médicale, the Association Institut de Myologie and the Assistance Public-Hôpitaux de Paris (Contrat d'Interface, A.F.), the Leducq Foundation and the MRC (A.Y.K., A.A., M.G.), the DZHK, the BMBF (M.G. and N.H.), the Centre de Recherche du CHU Ste-Justine (M.E.S.), the Iowa Wellstone Muscular Dystrophy Cooperative Research Center (U54, NS053672, S.A.M.), and the Fonds Québec de recherche pour la Nature et Technology (Frontenac grant, C.C.).
ACKNOWLEDGEMENTS
We are grateful to the patients and families for participation in this study, to Siegfried Labeit (Heidelberg University, Mannheim Campus) and to Dieter Fürst (Institute for Cell Biology, University of Bonn) for kind gift of the Z1Z2 and the T51 antibodies respectively, and to Tam T.T. Bui and Alex Drake (Biomolecular Spectroscopy Centre, King's College London) for assistance with CD spectroscopy. M. Gautel holds the BHF Chair of Molecular Cardiology.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ferreiro A., Estournet B., Chateau D., Romero N.B., Laroche C., Odent S., Toutain A., Cabello A., Fontan D., dos Santos H.G., et al. Multi-minicore disease: searching for boundaries. Phenotypical analysis of 38 cases. Ann. Neurol. 2000;48:745–757. [PubMed] [Google Scholar]
- 2.Ferreiro A., Monnier N., Romero N.B., Leroy J.-P., Bönnemann C., Haenggeli C.-A., Straub V., Voss W.D., Nivoche Y., Jungbluth H., et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 2002;51:750–759. doi: 10.1002/ana.10231. [DOI] [PubMed] [Google Scholar]
- 3.Ferreiro A., Quijano-Roy S., Pichereau C., Moghadaszadeh B., Goemans N., Bönnemann C., Jungbluth H., Straub V., Villanova M., Leroy J.-P., et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am. J. Hum. Genet. 2002;71:739–749. doi: 10.1086/342719. doi:10.1086/342719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyden S.E., Mahoney L.J., Kawahara G., Myers J.A., Mitsuhashi S., Estrella E.A., Duncan A.R., Dey F., DeChene E.T., Blasko-Goehringer J.M., et al. Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics. 2012;13:115–124. doi: 10.1007/s10048-012-0315-z. doi:10.1007/s10048-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmignac V., Salih M.A.M., Quijano-Roy S., Marchand S., Al Rayess M.M., Mukhtar M.M., Urtizberea J.A., Labeit S., Guicheney P., Leturcq F., et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 2007;61:340–351. doi: 10.1002/ana.21089. doi:10.1002/ana.21089. [DOI] [PubMed] [Google Scholar]
- 6.Jungbluth H., Sewry C.A., Muntoni F. Core myopathies. Semin. Pediatr. Neurol. 2011;18:239–249. doi: 10.1016/j.spen.2011.10.005. doi:10.1016/j.spen.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Finsterer J., Stöllberger C. Cardiac involvement in primary myopathies. Cardiology. 2000;94:1–11. doi: 10.1159/000007039. doi:10.1159/000007039. [DOI] [PubMed] [Google Scholar]
- 8.Oechslin E., Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur. Heart J. 2011;32:1446–1456. doi: 10.1093/eurheartj/ehq508. doi:10.1093/eurheartj/ehq508. [DOI] [PubMed] [Google Scholar]
- 9.Finsterer J., Stöllberger C., Wahbi K. Cardiomyopathy in neurological disorders. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2013 doi: 10.1016/j.carpath.2012.12.008. doi:10.1016/j.carpath.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Bertini E., Bosman C., Bevilacqua M., Ricci E., Gagliardi G.M., Parisi F., Servidei S., Dionisi-Vici C., Ballerini L. Cardiomyopathy and multicore myopathy with accumulation of intermediate filaments. Eur. J. Pediatr. 1990;149:856–858. doi: 10.1007/BF02072073. doi:10.1007/BF02072073. [DOI] [PubMed] [Google Scholar]
- 11.Magliocco A.M., Mitchell L.B., Brownell A.K., Lester W.M. Dilated cardiomyopathy in multicore myopathy. Am. J. Cardiol. 1989;63:150–151. doi: 10.1016/0002-9149(89)91108-9. doi:10.1016/0002-9149(89)91108-9. [DOI] [PubMed] [Google Scholar]
- 12.Ohkubo M., Ino T., Shimazaki S., Yabuto K., Okada R., Sato T. Multicore myopathy associated with multiple pterygium syndrome and hypertrophic cardiomyopathy. Pediatr. Cardiol. 1996;17:53–56. doi: 10.1007/BF02505814. doi:10.1007/BF02505814. [DOI] [PubMed] [Google Scholar]
- 13.Shuaib A., Martin J.M., Mitchell L.B., Brownell A.K. Multicore myopathy: not always a benign entity. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1988;15:10–14. doi: 10.1017/s0317167100027098. [DOI] [PubMed] [Google Scholar]
- 14.Willemsen M.A., van Oort A.M., ter Laak H.J., Sengers R.C., Gabreëls F.J. Multicore myopathy with restrictive cardiomyopathy. Acta Paediatr. Oslo Nor. 1992. 1997;86:1271–1274. doi: 10.1111/j.1651-2227.1997.tb14862.x. doi:10.1111/j.1651-2227.1997.tb14862.x. [DOI] [PubMed] [Google Scholar]
- 15.Cullup T., Lamont P.J., Cirak S., Damian M.S., Wallefeld W., Gooding R., Tan S.V., Sheehan J., Muntoni F., Abbs S., et al. Mutations in MYH7 cause multi-minicore disease (MmD) with variable cardiac involvement. Neuromuscul. Disord. NMD. 2012;22:1096–1104. doi: 10.1016/j.nmd.2012.06.007. doi:10.1016/j.nmd.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Fananapazir L., Dalakas M.C., Cyran F., Cohn G., Epstein N.D. Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA. 1993;90:3993–3997. doi: 10.1073/pnas.90.9.3993. doi:10.1073/pnas.90.9.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaindl A.M., Rüschendorf F., Krause S., Goebel H.-H., Koehler K., Becker C., Pongratz D., Müller-Höcker J., Nürnberg P., Stoltenburg-Didinger G., et al. Missense mutations of ACTA1 cause dominant congenital myopathy with cores. J. Med. Genet. 2004;41:842–848. doi: 10.1136/jmg.2004.020271. doi:10.1136/jmg.2004.020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bang M.L., Centner T., Fornoff F., Geach A.J., Gotthardt M., McNabb M., Witt C.C., Labeit D., Gregorio C.C., Granzier H., et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. doi:10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 19.Labeit S., Barlow D.P., Gautel M., Gibson T., Holt J., Hsieh C.L., Francke U., Leonard K., Wardale J., Whiting A. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature. 1990;345:273–276. doi: 10.1038/345273a0. doi:10.1038/345273a0. [DOI] [PubMed] [Google Scholar]
- 20.Hackman P., Marchand S., Sarparanta J., Vihola A., Pénisson-Besnier I., Eymard B., Pardal-Fernández J.M., Hammouda E.-H., Richard I., Illa I., et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD) Neuromuscul. Disord. NMD. 2008;18:922–928. doi: 10.1016/j.nmd.2008.07.010. doi:10.1016/j.nmd.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 21.Hackman P., Vihola A., Haravuori H., Marchand S., Sarparanta J., De Seze J., Labeit S., Witt C., Peltonen L., Richard I., et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002;71:492–500. doi: 10.1086/342380. doi:10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange S., Xiang F., Yakovenko A., Vihola A., Hackman P., Rostkova E., Kristensen J., Brandmeier B., Franzen G., Hedberg B., et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. doi:10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 23.Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., Seidman J.G., Seidman C., Granzier H., Labeit S., et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002;30:201–204. doi: 10.1038/ng815. doi:10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 24.Itoh-Satoh M., Hayashi T., Nishi H., Koga Y., Arimura T., Koyanagi T., Takahashi M., Hohda S., Ueda K., Nouchi T., et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. doi:10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 25.Satoh M., Takahashi M., Sakamoto T., Hiroe M., Marumo F., Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem. Biophys. Res. Commun. 1999;262:411–417. doi: 10.1006/bbrc.1999.1221. doi:10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 26.Toro C., Olivé M., Dalakas M.C., Sivakumar K., Bilbao J.M., Tyndel F., Vidal N., Farrero E., Sambuughin N., Goldfarb L.G. Exome sequencing identifies titin mutations causing hereditary myopathy with early respiratory failure (HMERF) in families of diverse ethnic origins. BMC Neurol. 2013;13:29. doi: 10.1186/1471-2377-13-29. doi:10.1186/1471-2377-13-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmio J., Evilä A., Chapon F., Tasca G., Xiang F., Brådvik B., Eymard B., Echaniz-Laguna A., Laporte J., Kärppä M., et al. Hereditary myopathy with early respiratory failure: occurrence in various populations. J. Neurol. Neurosurg. Psychiatry. 2013 doi: 10.1136/jnnp-2013-304965. 10.1136/jnnp-2013–304965. [DOI] [PubMed] [Google Scholar]
- 28.Pfeffer G., Barresi R., Wilson I.J., Hardy S.A., Griffin H., Hudson J., Elliott H.R., Ramesh A.V., Radunovic A., Winer J.B., et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J. Neurol. Neurosurg. Psychiatry. 2013 doi: 10.1136/jnnp-2012-304728. 10.1136/jnnp-2012–304728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Izumi R., Niihori T., Aoki Y., Suzuki N., Kato M., Warita H., Takahashi T., Tateyama M., Nagashima T., Funayama R., et al. Exome sequencing identifies a novel TTN mutation in a family with hereditary myopathy with early respiratory failure. J. Hum. Genet. 2013 doi: 10.1038/jhg.2013.9. 10.1038/jhg.2013.9. [DOI] [PubMed] [Google Scholar]
- 30.Ceyhan-Birsoy O., Agrawal P.B., Hidalgo C., Schmitz-Abe K., Dechene E.T., Swanson L.C., Soemedi R., Vasli N., Iannaccone S.T., Shieh P.B., et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013 doi: 10.1212/WNL.0b013e3182a6ca62. 10.1212/WNL.0b013e3182a6ca62 (Published online before print 23 August 2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herman D.S., Lam L., Taylor M.R.G., Wang L., Teekakirikul P., Christodoulou D., Conner L., DePalma S.R., McDonough B., Sparks E., et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. doi:10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinert S., Bergmann N., Luo X., Erdmann B., Gotthardt M. M line-deficient titin causes cardiac lethality through impaired maturation of the sarcomere. J. Cell Biol. 2006;173:559–570. doi: 10.1083/jcb.200601014. doi:10.1083/jcb.200601014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayans O., van der Ven P.F., Wilm M., Mues A., Young P., Fürst D.O., Wilmanns M., Gautel M. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395:863–869. doi: 10.1038/27603. doi:10.1038/27603. [DOI] [PubMed] [Google Scholar]
- 34.Engel A.G., Gomez M.R., Groover R.V. Multicore disease. A recently recognized congenital myopathy associated with multifocal degeneration of muscle fibers. Mayo Clin. Proc. Mayo Clin. 1971;46:666–681. [PubMed] [Google Scholar]
- 35.Ferreiro A., Estournet B., Chateau D., Romero N.B., Laroche C., Odent S., Toutain A., Cabello A., Fontan D., dos Santos H.G., et al. Multi-minicore disease–searching for boundaries: phenotype analysis of 38 cases. Ann. Neurol. 2000;48:745–757. doi:10.1002/1531-8249(200011)48:5<745::AID-ANA8>3.0.CO;2-F. [PubMed] [Google Scholar]
- 36.Darin N., Kimber E., Kroksmark A.-K., Tulinius M. Multiple congenital contractures: birth prevalence, etiology, and outcome. J. Pediatr. 2002;140:61–67. doi: 10.1067/mpd.2002.121148. doi:10.1067/mpd.2002.121148. [DOI] [PubMed] [Google Scholar]
- 37.Bamshad M., Van Heest A.E., Pleasure D. Arthrogryposis: a review and update. J. Bone Joint Surg. Am. 2009;91(Suppl 4):40–46. doi: 10.2106/JBJS.I.00281. doi:10.2106/JBJS.I.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohlsson M., Hedberg C., Brådvik B., Lindberg C., Tajsharghi H., Danielsson O., Melberg A., Udd B., Martinsson T., Oldfors A. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain J. Neurol. 2012;135:1682–1694. doi: 10.1093/brain/aws103. doi:10.1093/brain/aws103. [DOI] [PubMed] [Google Scholar]
- 39.Pollazzon M., Suominen T., Penttilä S., Malandrini A., Carluccio M.A., Mondelli M., Marozza A., Federico A., Renieri A., Hackman P., et al. The first Italian family with tibial muscular dystrophy caused by a novel titin mutation. J. Neurol. 2010;257:575–579. doi: 10.1007/s00415-009-5372-3. doi:10.1007/s00415-009-5372-3. [DOI] [PubMed] [Google Scholar]
- 40.Van den Bergh P.Y.K., Bouquiaux O., Verellen C., Marchand S., Richard I., Hackman P., Udd B. Tibial muscular dystrophy in a Belgian family. Ann. Neurol. 2003;54:248–251. doi: 10.1002/ana.10647. doi:10.1002/ana.10647. [DOI] [PubMed] [Google Scholar]
- 41.Guo W., Schafer S., Greaser M.L., Radke M.H., Liss M., Govindarajan T., Maatz H., Schulz H., Li S., Parrish A.M., et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012;18:766–773. doi: 10.1038/nm.2693. doi:10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obermann W.M., Gautel M., Steiner F., van der Ven P.F., Weber K., Fürst D.O. The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J. Cell Biol. 1996;134:1441–1453. doi: 10.1083/jcb.134.6.1441. doi:10.1083/jcb.134.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi J., Chi L., Labeit S., Banes A.J. Nuclear localization of the titin Z1Z2Zr domain and role in regulating cell proliferation. Am. J. Physiol. Cell Physiol. 2008;295:C975–C985. doi: 10.1152/ajpcell.90619.2007. doi:10.1152/ajpcell.90619.2007. [DOI] [PubMed] [Google Scholar]
- 44.Fürst D.O., Osborn M., Nave R., Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 1988;106:1563–1572. doi: 10.1083/jcb.106.5.1563. doi:10.1083/jcb.106.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puchner E., Alexandrovich A., Kho A.L., Hensen U., Schäfer L.V., Brandmeier B., Gräter F., Grubmuller H., Gaub H.E., Gautel M. Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. USA. 2008;105:13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]