

Abstract
Nitrogen mustard is a vesicant that causes damage to the respiratory tract. In these studies, we characterized the acute effects of nitrogen mustard on lung structure, inflammatory mediator expression, and pulmonary function, with the goal of identifying mediators potentially involved in toxicity. Treatment of rats (male Wistar, 200–225 g) with nitrogen mustard (mechlorethamine hydrochloride, i.t., 0.25 mg/kg) resulted in marked histological changes in the respiratory tract, including necrotizing bronchiolitis, thickening of alveolar septa, and inflammation which was evident within 24 h. This was associated with increases in bronchoalveolar lavage protein and cells, confirming injury to alveolar epithelial regions of the lung. Nitrogen mustard administration also resulted in increased expression of inducible nitric oxide synthase and cyclooxygenase-2, pro-inflammatory proteins implicated in lung injury, in alveolar macrophages and alveolar and bronchial epithelial cells. Expression of connective tissue growth factor and matrix metalloproteinase-9, mediators regulating extracellular matrix turnover was also increased, suggesting that pathways leading to chronic lung disease are initiated early in the pathogenic process. Following nitrogen mustard exposure, alterations in lung mechanics and function were also observed. These included decreases in baseline static compliance, end-tidal volume and airway resistance, and a pronounced loss of methacholine responsiveness in resistance, tissue damping and elastance. Taken together, these data demonstrate that nitrogen mustard induces rapid structural and inflammatory changes in the lung which are associated with altered lung functioning. Understanding the nature of the injury induced by nitrogen mustard and related analogs may aid in the development of efficacious therapies for treatment of pulmonary injury resulting from exposure to vesicants.
Keywords: Nitrogen mustard, iNOS, COX-2, CTGF, MMP-9, Lung function
Introduction
Bifunctional alkylating agents including sulfur mustard and nitrogen mustard were first synthesized for use in chemical warfare in the early part of the 20th century (Smith et al., 1995; Wattana and Bey, 2009). Although nitrogen mustard was never used in combat, it was stockpiled during World War II. Subsequently, nitrogen mustard and related analogs were developed as anticancer agents, and they remain a major therapeutic approach for treating lymphoma, as well as lung and breast cancer (Colvin and Hait, 2010). Nitrogen mustard, like sulfur mustard, is a potent vesicant, that causes severe and debilitating damage to target organs (Shakarjian et al., 2010; Wang and Xia, 2007). The lung appears to be particularly susceptible to the effects of these vesicants and pulmonary toxicity is the major cause of mortality and long-term morbidity (Ghanei and Harandi, 2007). Toxicity is thought to be initiated by DNA cross-linking resulting in DNA damage. However, it has been suggested that oxidative and nitrosative stress play a role in the pathogenic process (Korkmaz et al., 2006). This is supported by findings that exposure to mustards results in rapid depletion of glutathione and other cellular antioxidants, key events in lipid, protein, and DNA damage, and that lung injury induced by these agents is ameliorated in animals by treatment with antioxidants (Kumar et al., 2001; McClintock et al., 2002; Ucar et al., 2007; Wigenstam et al., 2009; Yaren et al., 2007).
Accumulating evidence suggests that inflammatory macrophages contribute to oxidative stress and toxicity associated with exposure to pulmonary irritants, and they may play a similar role in the pathogenic response to nitrogen mustard (Laskin et al., 2011). Data demonstrating that exposure of rodents to nitrogen mustard are associated with a marked inflammatory response in the lung and that anti-inflammatory corticosteroids protect against vesicant-induced toxicity are consistent with this idea (Wigenstam et al., 2009). The specific inflammatory mediators released in the lungs after nitrogen mustard exposure and their impact on pulmonary functioning are unknown. Activated macrophages generate a number of mediators that have been implicated in oxidative stress and tissue injury including reactive oxygen and nitrogen species, eicosanoids, and proteolytic enzymes as well as pro-inflammatory cytokines and growth factors (Laskin, 2009; Laskin et al., 2010a, 2011). Aberrant or excessive production of these inflammatory products can lead to chronic inflammation and fibrosis, two long-term consequences of nitrogen mustard intoxication (Dusenbery et al., 1988).
In the present studies, we characterized pulmonary injury, inflammation, and functional responses of rats to mechlorethamine hydrochloride, a prototypical nitrogen mustard. Identification of inflammatory mediators released following exposure to vesicants and potential functional consequences may be useful in developing efficacious pharmacologic approaches to mitigating the toxicity of these agents.
Materials and methods
Animals and treatments
Male specific pathogen-free Wistar rats (200–225 g) were obtained from Harlan Laboratories (Indianapolis IN). Animals were housed in filter top microisolation cages and maintained on food and water ad libitum. All animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Animals were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg) and then placed on a tilting rodent work stand (Hallowell EMC, Pittsfield, MA) in a supine position and restrained using an incisor loop. The tongue was extruded using a cotton tip applicator and the larynx visualized by a hemi-sectioned 4-mm speculum attached to an operating head of an otoscope (Welch Allyn, Skaneateles Falls, NY). Nitrogen mustard (mechlorethamine hydrochloride, Sigma-Aldrich, St. Louis, MO) was administered via Clay Adams Intramedic PE-60 (I.D. 0.76 mm, O.D. 1.22 mm) polyethylene tubing (Becton, Dickinson and Co., Franklin Lakes, NJ) attached to a 20-gauge hypodermic needle. The tubing was advanced approximately 20 mm past the epiglottis, and 0.1 ml of sterile PBS or nitrogen mustard prepared in PBS immediately before use was instilled into the trachea. The tubing and speculum were withdrawn immediately after instillation. Animals were then removed from the work stand and maintained in a vertical position until normal respiration was observed (less than 1 min). In preliminary studies, dose response (1–0.25 mg/kg) and time course (1, 3, and 7 days post-exposure) experiments were performed with nitrogen mustard. We found that mortality was >75% 1 day after administration of nitrogen mustard at doses greater than 0.25 mg/kg. Therefore, in all subsequent studies, a dose of 0.25 mg/kg nitrogen mustard and a post-exposure time of 1 day were used. At this dose and time, all animals survived and appeared clinically normal. Preparation and instillation of nitrogen mustard, which included the use of double gloves, safety glasses, and masks, were performed in a designated room under a chemical hood by personnel who followed Rutgers University Environmental Health and Safety guidelines.
Sample collection
Animals were euthanized by intraperitoneal injection of Nembutal (250 mg/kg). PBS (10 ml) was instilled into the lungs through a cannula in the trachea. Bronchoalveolar lavage (BAL) was collected by slowly withdrawing the fluid. BAL fluid was centrifuged (300×g, 8 min), supernatants collected, aliquoted, and stored in diethyl triamine pentaacetic acid (DTPA, 5 mM) at −80 °C until analysis. Cell pellets were resuspended in 1 ml PBS and viable cells (10 μl) counted on a hemocytometer using trypan blue dye exclusion. For differential analysis, cytospin preparations of BAL cells were fixed in methanol and stained with Giemsa (Labchem Inc., Pittsburgh, PA). A total of 300 cells were counted by light microscopy.
Measurement of BAL protein
Total protein was quantified in cell-free BAL using a BCA Protein Assay kit (Pierce Biotechnologies Inc., Rockford, IL) with bovine serum albumin (BSA) as the standard. Samples (25 μl) were analyzed in triplicate and plates evaluated at 560 nm on a Vmax MAXline™ microplate reader (Molecular Devices, Sunnyvale, CA).
Histology
Lungs were perfused, removed, fixed in 3% paraformaldehyde in PBS for 4 h on ice, and then transferred to 50% ethanol. The lung was cut longitudinally into anterior, middle, and posterior sections. Histological sections (4 μm) were stained with hematoxylin and eosin. Specimens were analyzed by light microscopy using an Olympus BX51 microscope (Olympus America Inc., Center Valley, PA). The extent of inflammatory changes including macrophage and neutrophil localization, alterations in alveolar epithelial barriers, and edema, were assessed blindly by a veterinary pathologist (Sherritta Ridgely, D.V.M., Ph.D.). Sections from three rats per treatment group were evaluated.
Immunohistochemistry
Tissue sections were deparaffinized. After antigen retrieval using citrate buffer (10.2 mM sodium citrate, 0.05% Tween 20, pH 6.0) and quenching of endogenous peroxidase with 3% H2O2 for 15 min, sections were incubated with 10% rabbit serum (room temperature, 1 h) to block nonspecific binding. This was followed by overnight incubation at 4 °C with rabbit IgG or polyclonal anti-inducible nitric oxide synthase (iNOS, 1:150), polyclonal anti-cycloxygenase-2 (COX-2, 1:400), polyclonal anti-connective tissue growth factor (CTGF, 1:200) or monoclonal anti-matrix metalloproteinase-9 (MMP-9, 1:150) antibody (Abcam, Cambridge, MA). Sections were then incubated with biotinylated secondary antibody (Vector Labs, Burlingame, CA) for 30 min at room temperature. Binding was visualized using a Peroxidase Substrate Kit DAB (Vector Labs). Sections from three rats per treatment group were analyzed for each antibody.
Measurement of lung mechanics and function
Animals were anesthetized with ketamine (80 mg/kg) and xylazine (10 mg/kg). After 5 min, tracheotomy was performed using a 15-gauge cannula, the animals attached to a SciReq flexiVent (Montreal, Canada), and baseline lung mechanics and function assessed. Animals were then challenged intratracheally with increasing doses of methacholine (0–96 mg/ml) and measurements of lung mechanics and function repeated. Data were analyzed using flexiVent software version 5.2.
Statistical analysis
All experiments were repeated 3–4 times. Data were analyzed using Student’s t-test; a p value ≤0.05 was considered statistically significant.
Results
Effects of nitrogen mustard on lung histology and BAL protein and cell content
Fig. 1 shows representative regions of bronchioles and alveoli from anterior, middle, and posterior longitudinal sections of the lung. Nitrogen mustard exposure resulted in moderate to severe histological changes in the lung relative to control (Fig. 1, upper panels A, E, and I), which were observed throughout the tissue, indicating uniformity of the injury (Fig. 1, upper panels B–D, F–H, and J–L). These changes included a marked accumulation of neutrophils and macrophages in peribronchiolar regions of the lung, as well as in alveolar spaces, vessels, and perivascular interstitium. Alveolar macrophages were also increased in size following exposure of animals to nitrogen mustard (Fig. 1, lower panels). Multifocal thickening of alveolar septa (Fig. 1, upper panels F, G, and H) and necrotizing bronchiolitis were also evident, along with necrotic debris in the bronchioles (Fig. 1, upper panels J, K, and L). Exposure of rats to nitrogen mustard also resulted in a six-fold increase in protein in BAL indicating that histological changes were accompanied by damage to the alveolar epithelial barrier (Fig. 2, upper panel). Additionally, a three-fold increase in the number of BAL cells was observed after nitrogen mustard exposure (Fig. 2, lower panel). Whereas in control animals, >99% of BAL cells were alveolar macrophages and <0.3% neutrophils, following nitrogen mustard exposure, neutrophil content increased to 6.3±0.9%.
Fig. 1.

Effects of nitrogen mustard on lung histology. Anterior (Ant), middle (Mid), and posterior (Pos) lung sections from animals-treated with PBS (CTL) or nitrogen mustard (NM) were stained with H and E. Sections from three rats per treatment group were evaluated. Representative sections from each portion of the lung are shown. Upper panels, original magnification, ×400; lower panels, original magnification, ×1000. Asterisk, necrotic debris in bronchiole; arrowhead, alveolar macrophages; arrow, neutrophils.
Fig. 2.
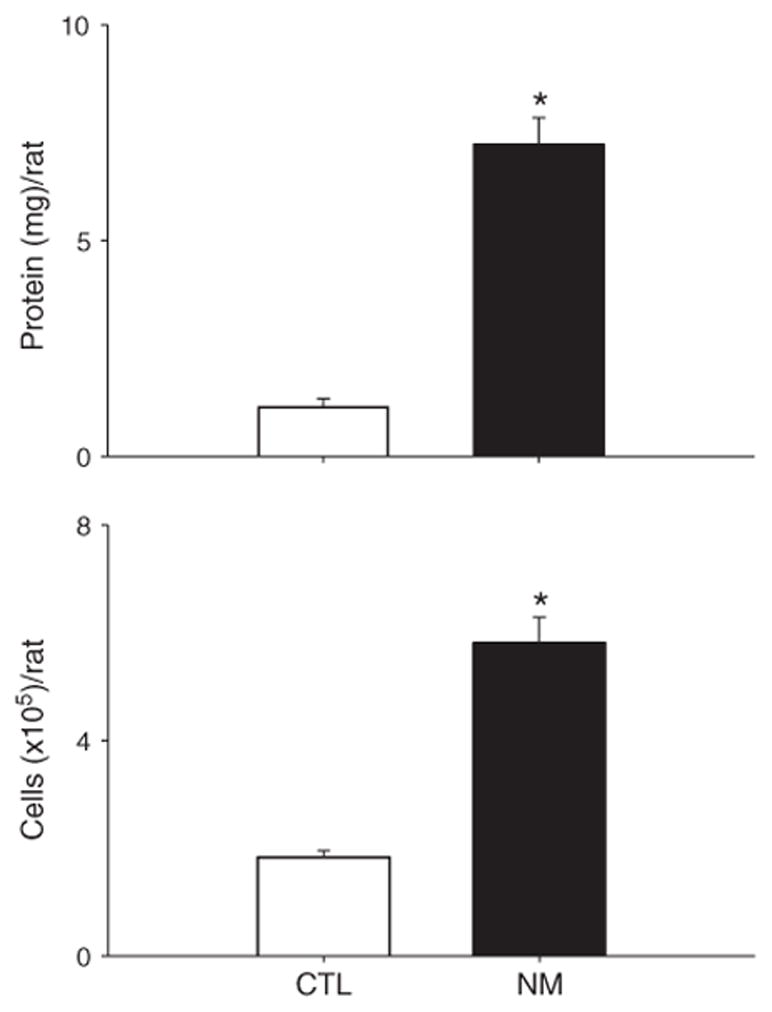
Effects of nitrogen mustard on BAL protein and cell number. BAL fluid was collected 24 h after treatment of rats with PBS (CTL) or nitrogen mustard (NM). Cell-free supernatants were analyzed in triplicate for protein using a BCA protein assay kit (Upper panel). Viable cells were enumerated by trypan blue dye exclusion (Lower panel). Each bar is the average±SE (n=6–9 rats). *Significantly different (p≤0.001) from CTL.
Effects of nitrogen mustard on expression of pro-inflammatory and pro-fibrotic mediators
Expression of iNOS and COX-2, two pro-inflammatory mediators implicated in lung injury (Laskin et al., 2010b, 2011), was next assessed. Low background levels of these proteins were observed in lungs of control animals, predominantly in epithelial cells of terminal bronchioles (Fig. 3). Nitrogen mustard administration resulted in a marked upregulation of both iNOS and COX-2. This was evident in alveolar macrophages as well as in alveolar and bronchial epithelial cells (Fig. 3). COX-2 staining was particularly pronounced in areas of thickened airway epithelium, presumably one of the major sites of nitrogen mustard-induced injury.
Fig. 3.

Effects of nitrogen mustard on iNOS and COX-2 expression. Lung sections from animals-treated with PBS (CTL) or nitrogen mustard (NM) were stained with antibody to iNOS (Upper panels) or COX-2 (Lower panels). Binding was visualized using a peroxidase DAB substrate kit. One representative section from 3 independent experiments is shown (original magnification, ×1000).
MMPs and transforming growth factor (TGF) β regulate extracellular matrix turnover, a key step in tissue remodeling and fibrosis following injury (Crosby and Waters, 2010). One downstream target of TGFβ is CTGF, an extracellular matrix associated protein that directly stimulates collagen production (Tall et al., 2010). In further studies, we analyzed the effects of nitrogen mustard on expression of MMP-9 and CTGF. Low level expression of both of these proteins was evident in alveolar and bronchial epithelia of control animals (Fig. 4). Nitrogen mustard exposure resulted in a dramatic increase in expression of MMP-9 and CTGF in alveolar macrophages as well as in alveolar and bronchial epithelia (Fig. 4). In contrast, expression of MMP-2, an extracellular protease, was not detected in the lung, even after nitrogen mustard administration (data not shown).
Fig. 4.
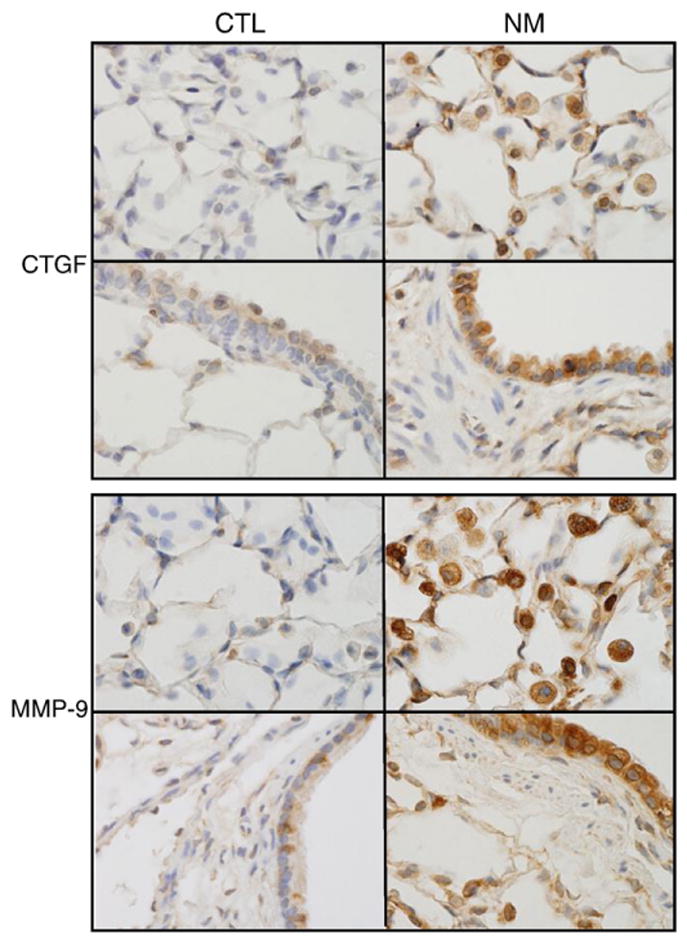
Effects of nitrogen mustard on MMP-9 and CTGF expression. Lung sections from animals-treated with PBS (CTL) or nitrogen mustard (NM) were stained with antibody to CTGF (upper panels) or MMP-9 (lower panels). Binding was visualized using a peroxidase DAB substrate kit. One representative section from 3 independent experiments is shown (original magnification, ×1000).
Effects of nitrogen mustard on lung mechanics and function
The data described above suggest that nitrogen mustard rapidly induces a complex pattern of injury, inflammation, and pre-fibrotic changes in the lung. To understand the consequences of these effects, a Scireq flexiVent was used to assess pulmonary function. Using a series of perturbations including single frequency and broadband forced oscillation, pressure–volume loops, and lung volume measurements, a range of functional parameters was measured. Whereas under non-stressed baseline conditions, total lung resistance was not altered following nitrogen mustard exposure, total lung compliance decreased (Fig. 5). A significant loss of static compliance, as well as end-tidal volume was also observed in nitrogen mustard-treated animals relative to controls. Although airway resistance also decreased after exposure to nitrogen mustard, this failed to reach statistical significance due to animal to animal variability (Fig. 5). These changes are consistent with increases in lung friction and a decline in airspace, presumably as a result of the inflammatory process.
Fig. 5.

Effects of nitrogen mustard on baseline lung function. Total lung resistance (R), total lung compliance (C), central airway resistance (Raw), static compliance (Cst), tissue damping (G), elastance (H), hysteresivity (eta), and end-tidal volume (ETV) were evaluated following exposure of rats to PBS (CTL) or nitrogen mustard (NM). Each bar represents the absolute value, mean±SE (n=5 rats). *Significantly different (p≤0.05) from CTL.
To further evaluate the functional consequences of nitrogen mustard intoxication, relative changes in these parameters were measured in response to administration of the bronchoconstrictive agent, methacholine. As expected, a significant increase in total lung resistance with a concomitant loss of compliance was observed in control animals following methacholine challenge (Fig. 6). Nitrogen mustard treatment of rats resulted in a pronounced loss of methacholine responsiveness in resistance. However, this loss was only partially reflected by changes in total and static compliance. In contrast, methacholine-induced increases in tissue damping and elastance, which were observed in control animals, were significantly blunted after nitrogen mustard administration (Fig. 6). This was also reflected by a decrease in eta, the ratio of tissue damping to elastance in nitrogen mustard-treated animals. No significant changes in end-tidal volume were noted with increasing doses of methacholine. These observations indicate that the principal functional effects of nitrogen mustard are at the parenchymal level and are consistent with a pronounced increase in fluid content of the lung.
Fig. 6.

Effects of nitrogen mustard on lung function in response to methacholine challenge. Total lung resistance (R), total lung compliance (C), central airway resistance (Raw), static compliance (Cst), tissue damping (G), elastance (H), hysteresivity (eta), and end-tidal volume (ETV) were evaluated in response to increasing doses of methacholine following exposure of rats to PBS (CTL) or nitrogen mustard (NM). Values are normalized and expressed as percentage change from baseline. Each point is the average±SE (n=5 rats). *Significantly different (p≤0.05) from CTL.
Discussion
Vesicant-induced lung injury involves damage to both the upper and lower airways (Malaviya et al., 2010; van Helden et al., 2004; Weber et al., 2010; Wigenstam et al., 2009). It has been suggested that inflammatory cells contribute to this pathology, releasing mediators that enhance oxidative stress, proteolytic damage, and cause aberrant wound healing (Laskin et al., 2011; Wigenstam et al., 2009). Elucidating the specific mediators involved in the pathogenic response and consequent functional alterations in the lung may aid in the development of pharmacotherapies effective in mitigating toxicity induced by vesicants such as nitrogen mustard.
Twenty-four hours following exposure of rats to nitrogen mustard, moderate to severe histological changes were observed in the lung; most notable was a marked increase in inflammatory cells (macrophages and neutrophils) in peribronchiolar areas, as well as in alveolar spaces, vessels, and perivascular interstitium. This was correlated with a 3-fold increase in BAL cells recovered from the animals. There was also evidence of necrotizing bronchiolitis, which was associated with necrotic debris in the bronchioles and multifocal thickening of alveolar septa. Histological changes in the lungs of rats have been previously described 6 days following nitrogen mustard exposure suggesting that the untoward effects of this vesicant are persistent (Ucar et al., 2007; Yaren et al., 2007). Similar persistent histological changes in the lung have also been reported up to 14 days after exposure of rodents to sulfur mustard (Calvet et al., 1999a, 1994; Guignabert et al., 2005). The results presented in this study are novel, as they demonstrate that both parenchymal and airway inflammation occur as early as 24 h after the initial exposure.
Increases in protein in BAL fluid are generally considered to be a marker of damage to the alveolar epithelial barrier in the lower lung. In accord with previous reports in rodents treated with sulfur mustard or the half mustard, 2-chloroethy ethyl sulfide (Allon et al., 2009; O’Neill et al., 2010), we found that BAL protein levels were significantly increased 24 h after exposure of animals to nitrogen mustard. These findings are consistent with nitrogen mustard-induced histological changes in alveolar epithelial regions of the lung, as well as functional alterations which indicate an increase in friction within the lower lung. The appearance of protein in BAL at this early post-exposure time point also suggests that it is a sensitive marker of vesicant-induced injury.
Macrophages play an important role in both the initiation and resolution of the inflammatory response. Whereas initially macrophages release chemotactic and cytotoxic mediators that promote the inflammatory response, later, they generate mediators involved in down regulating inflammation and initiating wound repair (Laskin, 2009; Stout and Suttles, 2004). Key proteins involved in the early pro-inflammatory and cytotoxic response are iNOS and COX-2, enzymes mediating the generation of reactive nitrogen species and prostaglandins, respectively (Laskin et al., 2010b, 2011). Following nitrogen mustard exposure, expression of iNOS and COX-2 was rapidly upregulated in alveolar macrophages, as well as in alveolar and bronchial epithelia. These findings are in accord with previous reports on the pulmonary effects of sulfur mustard and related analogs (Gao et al., 2010; Malaviya et al., 2010; Sunil et al., 2010; Ucar et al., 2007). The observation that nitrogen mustard-induced toxicity is ameliorated by blocking or reducing production of reactive nitrogen species provides support for a role of these cytotoxic oxidants in the pathogenesis of nitrogen mustard-induced lung injury (Yaren et al., 2007). The contribution of COX-2 to the pulmonary toxicity of mustards is unknown. At early stages of inflammation, COX-2 generates pro-inflammatory prostaglandins from arachidonic acid. These eicosanoids are thought to be important in the pathogenesis of diseases such as asthma and chronic obstructive pulmonary disease (Rolin et al., 2006), which are long-term consequences of mustard gas poisoning, and they may play a similar role in lung injury induced by nitrogen mustard and related vesicants. This is supported by findings that COX-2 knockout mice, or mice pretreated with the selective COX-2 inhibitor, celecoxib, are protected from sulfur mustard-induced skin injury (Wormser et al., 2004).
In the lung, MMPs are generated by macrophages, neutrophils, and epithelial cells (Chakrabarti and Patel, 2005). These proteases degrade extracellular matrix components, an important step in alveolar epithelial injury and detachment of cells from basement membranes following sulfur mustard intoxication (Calvet et al., 1999b; Guignabert et al., 2005). The present studies demonstrate that MMP-9, but not MMP-2, is rapidly upregulated in lung macrophages and epithelial cells after nitrogen mustard exposure. Similar increases in MMP-9 have been described in the lung and respiratory tract after exposure of rodents to sulfur mustard (Anderson et al., 2009; Calvet et al., 1999b; Guignabert et al., 2005; Malaviya et al., 2010), and in BAL cells from exposed humans (Radomska-Lesniewska et al., 2010). In rats, MMP inhibitors such as doxycyline and illomastat have been reported to ameliorate sulfur mustard-induced respiratory lesions (Anderson et al., 2009), demonstrating the importance of these proteases in vesicant-induced lung injury. The specific cytotoxic actions of MMP-9 have not been established. By inducing breakdown of extracellular matrix, MMP-9 may play a role in the reduced alveolar epithelial barrier function observed following nitrogen mustard exposure. Increases in MMP-9 may also be important in fibrogenesis and resultant lung restriction. However, the fact that there is no functional evidence of lung restriction 24 h after exposure of animals to nitrogen mustard suggests that at this early time point in the pathogenic process, the actions of MMP-9 are primarily cytotoxic. Further longer-term studies are necessary to determine if MMP-9 also acts as an initiating signal for airway remodeling.
If inflammation persists without resolution, macrophages and epithelial cells may release excessive amounts of mediators such as TGFβ which stimulate production of collagen leading to the development of fibrosis (Crosby and Waters, 2010). The actions of TGFβ are mediated in part, by CTGF, a heparin binding protein thought to play a pivotal role in lung fibrosis (Ponticos et al., 2009). Exposure of rats to nitrogen mustard resulted in increased expression of CTGF in alveolar macrophages, and in alveolar and bronchial epithelia. The fact that this occurred within 24 h of nitrogen mustard exposure suggests that the fibrogenic process leading to chronic lung diseases is initiated early in the pathogenic response to this vesicant. These findings are novel and indicate that CTGF may be a rational target for therapeutic intervention in fibrotic diseases induced by vesicants.
Nitrogen mustard exposure also resulted in a number of changes in pulmonary function including a decrease in static compliance, suggesting an increase in static elastic recoil within the lung; interestingly, this occurred without significant changes in total lung resistance. This is most likely due to an accumulation of inflammatory material within the lung parenchyma, causing an increase in friction. This is supported by findings that end-tidal volume was significantly reduced following nitrogen mustard exposure. Loss of parenchymal integrity, which occurs in emphysema, results in a reduced airway radius at rest. This is typically reflected by an increase in airway or Newtonian resistance. Following nitrogen mustard exposure, we noted a decrease in airway resistance. This indicates that despite increases in MMP-9 and CTGF, the parenchyma remains sufficiently intact to support airway structure. Since long-term vesicant intoxication is associated with the development of emphysema-like changes in the lung, we speculate that a significant rise in airway resistance will be observed at later times post-exposure and this remains to be investigated. Taken together, these observations indicate that a lung-remodeling event, although possibly initiated, has not occurred at the functional level 24 h after nitrogen mustard exposure.
Methacholine is a potent bronchoconstrictor used in animal models to examine hyper-responsiveness of the airway, and to assess airway wall stiffness and parenchymal elasticity (Bates and Lauzon, 2007). In accord with previous findings (Nagase et al., 1994), the present studies show a marked increase in total lung resistance and a decrease in compliance in response to methacholine in control rats. These changes were accompanied by disproportionate increases in both tissue damping and elastance, resulting in an increase in eta. Under homeostatic conditions, the lung behaves in a homogeneous fashion, despite its structural complexity (Bates and Lutchen, 2005; Bates and Suki, 2008). The rise in eta, in response to methacholine, indicates an increase in lung heterogeneity with concomitant airway restriction. This may result from regional differences in elasticity within the lung, which become apparent under conditions of stress induced by methacholine challenge. The most remarkable functional alteration noted following nitrogen mustard exposure was a reduction in methacholine-induced increases in total lung resistance, with no major effects on lung compliance. This blunting of lung resistance in response to methacholine can best be explained by a loss of active airway tone. Whether this results from a loss of smooth muscle function or a failure of sympathetic control cannot be ascertained from these data. A significant blunting of methacholine-induced increases in tissue damping and elastance were also noted in nitrogen mustard-treated animals. The reduced responses in tissue damping, elastance, and eta most likely result from the inability of the airways to respond actively to the bronchoconstrictive agent. It is important to note that the loss of compliance in lungs from control and nitrogen mustard-treated rats is, for the most part similar, despite the reduced constriction in vesicant-injured animals. This may be explained by parenchymal injury and an accumulation of inflammatory mediators in the lower lung, which results in a reduced static elastic recoil. These observations are consistent with both our histological observations, and baseline lung function data.
In summary, the present studies demonstrate that exposure of rats to nitrogen mustard results in significant histological changes and increases in BAL protein and inflammatory cells, along with increases in expression of pro-inflammatory and pro-fibrotic proteins. These changes are directly associated with altered lung mechanics and function. These studies are novel and provide support for pulmonary function assessment in understanding the adverse effects of vesicants such as nitrogen mustard on the respiratory system. Results from these studies may lead to the development of efficacious treatments for nitrogen mustard-induced pulmonary injury.
Acknowledgments
The authors wish to thank Sherritta Ridgely, D.V.M., Ph.D., for histological evaluation of H&E-stained sections.
Funding information
This work was supported by the National Institutes of Health grant nos. AR055073, ES004738, ES005022, CA132624, GM03430, and HL086621.
Contributor Information
Vasanthi R. Sunil, Email: sunilvr@eohsi.rutgers.edu.
Kinal J. Patel, Email: kinalv5@gmail.com.
Jianliang Shen, Email: jianliangs@gmail.com.
David Reimer, Email: reimerd@las.rutgers.edu.
Andrew J. Gow, Email: gow@rci.rutgers.edu.
Jeffrey D. Laskin, Email: jlaskin@eohsi.rutgers.edu.
Debra L. Laskin, Email: laskin@eohsi.rutgers.edu.
References
- Allon N, Amir A, Manisterski E, Rabinovitz I, Dachir S, Kadar T. Inhalation exposure to sulfur mustard in the guinea pig model: clinical, biochemical and histopathological characterization of respiratory injuries. Toxicol Appl Pharmacol. 2009;241:154–162. doi: 10.1016/j.taap.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Taylor SL, Fetterer DP, Holmes WW. Evaluation of protease inhibitors and an antioxidant for treatment of sulfur mustard-induced toxic lung injury. Toxicology. 2009;263:41–46. doi: 10.1016/j.tox.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Bates JH, Lauzon AM. Parenchymal tethering, airway wall stiffness, and the dynamics of bronchoconstriction. J Appl Physiol. 2007;102:1912–1920. doi: 10.1152/japplphysiol.00980.2006. [DOI] [PubMed] [Google Scholar]
- Bates JH, Lutchen KR. The interface between measurement and modeling of peripheral lung mechanics. Respir Physiol Neurobiol. 2005;148:153–164. doi: 10.1016/j.resp.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Bates JH, Suki B. Assessment of peripheral lung mechanics. Respir Physiol Neurobiol. 2008;163:54–63. doi: 10.1016/j.resp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet JH, Jarreau PH, Levame M, D’Ortho MP, Lorino H, Harf A, Macquin-Mavier I. Acute and chronic respiratory effects of sulfur mustard intoxication in guinea pig. J Appl Physiol. 1994;76:681–688. doi: 10.1152/jappl.1994.76.2.681. [DOI] [PubMed] [Google Scholar]
- Calvet JH, Gascard JP, Delamanche S, Brink C. Airway epithelial damage and release of inflammatory mediators in human lung parenchyma after sulfur mustard exposure. Hum Exp Toxicol. 1999a;18:77–81. doi: 10.1177/096032719901800203. [DOI] [PubMed] [Google Scholar]
- Calvet JH, Planus E, Rouet P, Pezet S, Levame M, Lafuma C, Harf A, D’Ortho MP. Matrix metalloproteinase gelatinases in sulfur mustard-induced acute airway injury in guinea pigs. Am J Physiol. 1999b;276:L754–L762. doi: 10.1152/ajplung.1999.276.5.L754. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Patel KD. Matrix metalloproteinase-2 (MMP-2) and MMP-9 in pulmonary pathology. Exp Lung Res. 2005;31:599–621. doi: 10.1080/019021490944232. [DOI] [PubMed] [Google Scholar]
- Colvin M, Hait WN. Alkylating agents and platinum antitumor compounds. In: Hong WK, Bast RC Jr, Hait WN, Kufe DW, Pollock RE, Weichselbaum RR, Holland JE, Frei E III, editors. Cancer Medicine. People’s Medical Publishing House-USA; Shelton, CT: 2010. pp. 633–644. [Google Scholar]
- Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–L731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusenbery KE, Peterson BA, Bloomfield CD. Chemotherapy with cyclophosphamide, vinblastine, procarbazine, and prednisone (CVPP) for Hodgkin disease: fourteen-year follow-up results. Am J Hematol. 1988;28:246–251. doi: 10.1002/ajh.2830280407. [DOI] [PubMed] [Google Scholar]
- Gao X, Ray R, Xiao Y, Ishida K, Ray P. Macrolide antibiotics improve chemotactic and phagocytic capacity as well as reduce inflammation in sulfur mustard-exposed monocytes. Pulm Pharmacol Ther. 2010;23:97–106. doi: 10.1016/j.pupt.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Ghanei M, Harandi AA. Long term consequences from exposure to sulfur mustard: a review. Inhal Toxicol. 2007;19:451–456. doi: 10.1080/08958370601174990. [DOI] [PubMed] [Google Scholar]
- Guignabert C, Taysse L, Calvet JH, Planus E, Delamanche S, Galiacy S, d’Ortho MP. Effect of doxycycline on sulfur mustard-induced respiratory lesions in guinea pigs. Am J Physiol Lung Cell Mol Physiol. 2005;289:L67–L74. doi: 10.1152/ajplung.00475.2004. [DOI] [PubMed] [Google Scholar]
- Korkmaz A, Yaren H, Topal T, Oter S. Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch Toxicol. 2006;80:662–670. doi: 10.1007/s00204-006-0089-x. [DOI] [PubMed] [Google Scholar]
- Kumar O, Sugendran K, Vijayaraghavan R. Protective effect of various antioxidants on the toxicity of sulphur mustard administered to mice by inhalation or percutaneous routes. Chem Biol Interact. 2001;134:1–12. doi: 10.1016/s0009-2797(00)00209-x. [DOI] [PubMed] [Google Scholar]
- Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol. 2009;22:1376–1385. doi: 10.1021/tx900086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin DL, Gardner CR, Laskin JD. Phagocytes. In: Lawrence D, editor. Comprehensive Toxicology. Elsevier; UK: 2010a. pp. 133–153. [Google Scholar]
- Laskin JD, Heck DE, Laskin DL. Nitric oxide pathways in toxic responses. In: Ballantyne B, Marrs T, Syversen T, editors. General and Applied Toxicology. Wiley-Blackwell; UK: 2010b. pp. 425–438. [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil V, Cervelli J, Anderson D, Holmes W, Conti M, Gordon R, Laskin J, Laskin DL. Inflammatory effects of inhaled sulfur mustard in rat lungs. Toxicol Appl Pharmacol. 2010;248:89–99. doi: 10.1016/j.taap.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock SD, Till GO, Smith MG, Ward PA. Protection from half-mustard-gas-induced acute lung injury in the rat. J Appl Toxicol. 2002;22:257–262. doi: 10.1002/jat.856. [DOI] [PubMed] [Google Scholar]
- Nagase T, Moretto A, Ludwig MS. Airway and tissue behavior during induced constriction in rats: intravenous vs. aerosol administration. J Appl Physiol. 1994;76:830–838. doi: 10.1152/jappl.1994.76.2.830. [DOI] [PubMed] [Google Scholar]
- O’Neill HC, White CW, Veress LA, Hendry-Hofer TB, Loader JE, Min E, Huang J, Rancourt RC, Day BJ. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic Biol Med. 2010;48:1188–1196. doi: 10.1016/j.freeradbiomed.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009;60:2142–2155. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- Radomska-Lesniewska DM, Skopinska-Rozewska E, Jankowska-Steifer E, Sobiecka M, Sadowska AM, Hevelke A, Malejczyk J. N-acetylcysteine inhibits IL-8 and MMP-9 release and ICAM-1 expression by bronchoalveolar cells from interstitial lung disease patients. Pharmacol Rep. 2010;62:131–138. doi: 10.1016/s1734-1140(10)70250-4. [DOI] [PubMed] [Google Scholar]
- Rolin S, Masereel B, Dogne JM. Prostanoids as pharmacological targets in COPD and asthma. Eur J Pharmacol. 2006;533:89–100. doi: 10.1016/j.ejphar.2005.12.058. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Hurst CG, Moeller RB, Skelton HG, Sidell FR. Sulfur mustard: its continuing threat as a chemical warfare agent, the cutaneous lesions induced, progress in understanding its mechanism of action, its long-term health effects, and new developments for protection and therapy. J Am Acad Dermatol. 1995;32:765–776. doi: 10.1016/0190-9622(95)91457-9. [DOI] [PubMed] [Google Scholar]
- Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel KJ, Shen JL, Reimer D, Gow AJ, Laskin JD, Laskin DL. Role of reactive nitrogen species in vesicant-induced lung injury and altered lung functioning. The Toxicologist. 2010;114:73. (A336) [Google Scholar]
- Tall EG, Bernstein AM, Oliver N, Gray JL, Masur SK. TGF-beta-stimulated CTGF production is enhanced by collagen and associated with biogenesis of a novel 31 kDa CTGF form in human corneal fibroblasts. Invest Ophthalmol Vis Sci. 2010;51:5002–5011. doi: 10.1167/iovs.09-5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucar M, Korkmaz A, Reiter RJ, Yaren H, Oter S, Kurt B, Topal T. Melatonin alleviates lung damage induced by the chemical warfare agent nitrogen mustard. Toxicol Lett. 2007;173:124–131. doi: 10.1016/j.toxlet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- van Helden HP, Kuijpers WC, Diemel RV. Asthma-like symptoms following intratracheal exposure of guinea pigs to sulfur mustard aerosol: therapeutic efficacy of exogenous lung surfactant curosurf and salbutamol. Inhal Toxicol. 2004;16:537–548. doi: 10.1080/08958370490442520. [DOI] [PubMed] [Google Scholar]
- Wang GQ, Xia ZF. Tissue injury by hot fluid containing nitrogen mustard. Burns. 2007;33:923–926. doi: 10.1016/j.burns.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Wattana M, Bey T. Mustard gas or sulfur mustard: an old chemical agent as a new terrorist threat. Prehosp Disaster Med. 2009;24:19–29. doi: 10.1017/s1049023x0000649x. [DOI] [PubMed] [Google Scholar]
- Weber WM, Kracko DA, Lehman MR, Irvin CM, Blair LF, White RK, Benson JM, Grotendorst GR, Cheng YS, McDonald JD. Inhalation exposure systems for the development of rodent models of sulfur mustard-induced pulmonary injury. Toxicol Mech Methods. 2010;20:14–24. doi: 10.3109/15376510903483730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigenstam E, Rocksen D, Ekstrand-Hammarstrom B, Bucht A. Treatment with dexamethasone or liposome-encapsuled vitamin E provides beneficial effects after chemical-induced lung injury. Inhal Toxicol. 2009;21:958–964. doi: 10.1080/08958370802596298. [DOI] [PubMed] [Google Scholar]
- Wormser U, Langenbach R, Peddada S, Sintov A, Brodsky B, Nyska A. Reduced sulfur mustard-induced skin toxicity in cyclooxygenase-2 knockout and celecoxib-treated mice. Toxicol Appl Pharmacol. 2004;200:40–47. doi: 10.1016/j.taap.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Yaren H, Mollaoglu H, Kurt B, Korkmaz A, Oter S, Topal T, Karayilanoglu T. Lung toxicity of nitrogen mustard may be mediated by nitric oxide and peroxynitrite in rats. Res Vet Sci. 2007;83:116–122. doi: 10.1016/j.rvsc.2006.11.004. [DOI] [PubMed] [Google Scholar]