

Abstract

Caeruleanone A (1), a novel rotenoid with an unprecedented arrangement of the D-ring, was isolated with another two new analogues, caeruleanones B (2) and C (3), together with 11 known rotenoids from the fruits of Millettia caerulea. The structures of the new compounds were determined by spectroscopic data analysis, with that of 1 being confirmed by single-crystal X-ray diffraction. Compounds 2 and 3 displayed potent mitochondrial transmembrane potential inhibitory and quinone reductase induction activities.
Millettia caerulea (Graham) Baker (Fabaceae-Papilionoideae) is a large shrub growing in dry, thorny, and stunted forests and is native to Myanmar, Thailand, and Vietnam.1 The stems and leaves of this plant are applied to wounds in Thai folkloric medicine to reduce infection.2 No previous phytochemical investigation has been performed on M. caerulea. Millettia spp., such as M. brandisiana Kurz and M. dura Dunn, contain rotenoids with anti-inflammatory and insecticidal activities.3,4
As part of an anticancer drug discovery program,5 a chloroform-soluble partition of a methanol extract from the fruits of M. caerulea, collected in Vietnam, showed potent cytotoxicity against the HT-29 human colon cancer cell line. Due to the observed cytotoxicity of the original extract, a recollection of the fruits of M. caerulea was undertaken and the chloroform extract exhibited an IC50 value of 2.0 μg/mL against HT-29 cells. The cytotoxicity-guided isolation of compounds from M. caerulea resulted in the purification of 14 rotenoids, inclusive of one novel (1) and two new (2 and 3) compounds (Figure 1). Caeruleanone A (1) is the first example of a rotenoid-type compound in which the aromaticity of the D-ring is disrupted.6,7 The isolation and structure elucidation of compounds 1–3 are presented herein, along with a proposed biogenetic pathway and the biological evaluation of these new isolates.
Figure 1.

Compounds 1–3 (with one of two possible cis configurations shown for 1 and 2).
The plant material of Millettia caerulea was collected at Nui Chua National Park in Vietnam in July 2011. The dried and powdered fruits (1.3 kg) were extracted by maceration with MeOH overnight at rt (3 × 3L). The methanol solvent was removed in vacuo (120 g) and then partitioned sequentially to give dried hexane (15.6 g) and CHCl3 extracts (30.4 g) (Supporting Information). Separation of the CHCl3-soluble extract by silica gel column chromatography using hexanes/acetone in a linear gradient led to 10 pooled fractions (Fr. 1.1–1.10). Fraction 1.4 (6.3 g) was chromatographed over a silica gel column using hexanes/acetone in a gradient (10:1 to 1:1) to yield seven subfractions (Fr. 2.1–2.7). Subfraction 2.2 (1.1 g) was chromatographed over a Sephadex LH-20 column, using MeOH/H2O (9:1) as solvent, to afford four subfractions (Fr. 3.1–3.4) and impure crystals in Fr. 3.2. The impure crystalline solid (77 mg) in Fr. 3.2 was further purified by semipreparative RP-18 HPLC, using MeOH/H2O (73:27) as solvent, to yield compound 1 (38.1 mg) as colorless crystals and an impure mixture (4.7 mg), which was purified in a separate semipreparative RP-18 HPLC experiment using MeOH/H2O (65:35) as solvent to yield compounds 2 (1.1 mg) and 3 (1.3 mg). Fr. 2.3 (3.1 g) was chromatographed on a silica gel column using hexanes/acetone (10:1 to 0:1) in a gradient to give 10 subfractions (Fr. 4.1–4.10). Fr. 4.4 (509.1 mg) was purified by preparative RP-18 HPLC using CH3CN/H2O (65:35) as solvent. A fraction (49.6 mg) from this HPLC separation was purified further by a separate semipreparative RP-18 HPLC procedure using MeOH/H2O (55:45) to afford a further amount of 3 (4.4 mg).
Caeruleanone A (1) was obtained as colorless fine crystals that were recrystallized by slow evaporation using hexanes/CH2Cl2 (10:1) to yield colorless plates. The molecular formula of 1 was assigned as C28H30O7 based on the sodiated molecular ion peak at m/z 501.1890 [M + Na]+ (calcd m/z 501.1889) in the HRESIMS. The 1H NMR spectrum of 1 displayed typical signals for a rotenoid-type compound with chemical shifts at δH 4.57 (dd, J = 12.3, 3.0 Hz, H-6ax), 4.15 (d, J = 12.3 Hz, H-6eq), 4.84 (t, J = 3.1 Hz, H-6a), and 3.72 (d, J = 4.3 Hz, H-12a) ppm (Table 1).8,9 The chemical shifts of C-8 (δC 60.2 ppm) and C-9 (δC 195.9 ppm) observed in the 13C NMR data of 1 indicated that the C8–C9 sp2 bond in ring D normally present in rotenoids6−9 was replaced with an sp3 bond, suggesting the presence of a cyclohexa-2,4-dienone ring system (Table 2). The diprenyl substituents at C-8 were supported by HMBC correlations of H-1′ and H-1″ with C-8, C-7a, and C-9. The singlet observed for H-11, and its HMBC correlations to C-7a, C-9, and C-12, along with the NOESY correlation between the methoxy protons at C-10 and H-11, provided additional evidence for the D-ring substitution (Figure 2). Griffonianone C, an isoflavone isolated from Millettia griffoniana Baill., and xanthones purified from Garcinia and Allanblackia spp. (Clusiaceae), contain the same substituents as 1.10−12 A deshielded methylene carbon (δC 101.5 ppm, C-2a) was observed in the 13C DEPT NMR spectrum, characteristic of a methylenedioxy substituent, as commonly seen in rotenoids.7,9 The relative configurations of H-6a and H-12a were proposed based on the chemical shift for H-1 at δH 6.64 ppm, which indicated a cis-B/C ring junction (6.4–6.8 ppm) versus the deshielded trans-form (7.6–7.9 ppm).9,13 In addition, compound 1 exhibited a zero specific rotation indicative of being a racemate. The cis-racemate configuration and structure of 1 were confirmed by X-ray diffraction analysis.14 The crystal structure of 1 contains two independent molecules in the asymmetric unit (Z′ = 2), and these two molecules differ in the conformations of the diprenyl substituents at C-8 (Figure 3). The crystal contains a racemic mixture of 1 because the space group, P21/c, is centrosymmetric.
Table 1. 1H NMR (400 MHz, CDCl3, J in Hz) Spectroscopic Data for 1–3.
| position | 1 | 2 | 3 |
|---|---|---|---|
| 1 | 6.64, s | 6.52, s | 6.53, s |
| 2a | 5.81, d | 5.85, br d | 5.82, d |
| (1.2) | (4.0) | (1.1) | |
| 5.87, d | 5.84, d | ||
| (1.2) | (1.1) | ||
| 4 | 6.39, s | 6.45, s | 6.44, s |
| 6ax | 4.57, dd | 4.57a | 4.59, dd |
| (12.3, 3.0) | (12.0, 2.2) | ||
| 6eq | 4.15, d | 4.45, d | 4.46, d |
| (12.3) | (12.0) | (12.0) | |
| 6a | 4.84, t | 4.51a | 4.51, br d |
| (3.1) | (1.4) | ||
| 11 | 6.67, s | 7.21, s | 7.17, s |
| 12a | 3.72, d | ||
| (4.3) | |||
| 1′ | 2.42, dd | 3.30, t | 3.30, t |
| (13.5, 7.2) | (7.6) | (7.9) | |
| 2.64a | |||
| 2′ | 4.20, d | 5.06, t | 5.15, t |
| (7.5) | (7.1) | (7.4) | |
| 4′ | 1.24, s | 1.74, s | 1.75, s |
| 5′ | 1.34, s | 1.62, s | 1.64, s |
| 1″ | 2.64a | 4.51a | |
| 4.57a | |||
| 2″ | 4.75, br t | 5.48, t | |
| (7.6) | (7.0) | ||
| 4″ | 1.57, s | 1.68, s | |
| 5″ | 1.58, s | 1.75, s | |
| OH-12a | 4.35, s | 4.40, br s |
Overlapping signals.
Table 2. 13C NMR (100 MHz, CDCl3) Spectroscopic Data for 1–3.
| position | 1 | 2 | 3 |
|---|---|---|---|
| 1 | 106.8 | 106.1 | 106.1 |
| 1a | 105.8 | 109.9 | 110.1 |
| 2 | 142.7 | 142.4 | 142.4 |
| 2a | 101.5 | 101.6 | 101.6 |
| 3 | 148.3 | 149.7 | 150.0 |
| 4 | 99.1 | 99.3 | 99.3 |
| 4a | 148.6 | 150.1 | 149.6 |
| 6 | 65.9 | 64.3 | 64.3 |
| 6a | 73.8 | 76.2 | 76.2 |
| 7a | 174.1 | 154.8 | 152.4 |
| 8 | 60.2 | 125.3 | 116.4 |
| 9 | 195.9 | 154.9 | 155.7 |
| 10 | 148.7 | 148.9 | 142.8 |
| 11 | 108.4 | 106.2 | 104.5 |
| 11a | 108.6 | 112.6 | 109.5 |
| 12 | 187.4 | 192.6 | 192.0 |
| 12a | 43.9 | 68.2 | 67.9 |
| 1′ | 39.3 | 23.4 | 22.5 |
| 2′ | 116.8 | 122.1 | 121.2 |
| 3′ | 135.7 | 132.4 | 133.0 |
| 4′ | 25.6 | 18.1 | 18.0 |
| 5′ | 17.9 | 26.1 | 26.1 |
| 1″ | 38.6 | 70.3 | |
| 2″ | 118.0 | 120.7 | |
| 3″ | 135.4 | 138.8 | |
| 4″ | 26.1 | 18.3 | |
| 5″ | 18.3 | 26.2 | |
| OMe | 55.9 | 56.3 | 56.6 |
Figure 2.
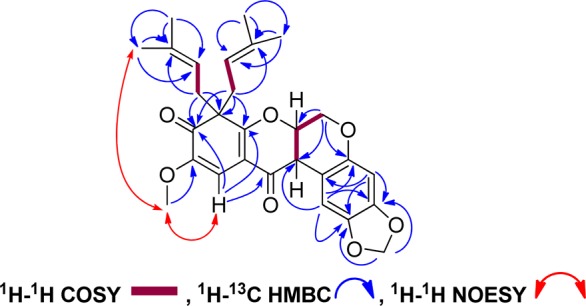
Selected NMR correlations of 1.
Figure 3.
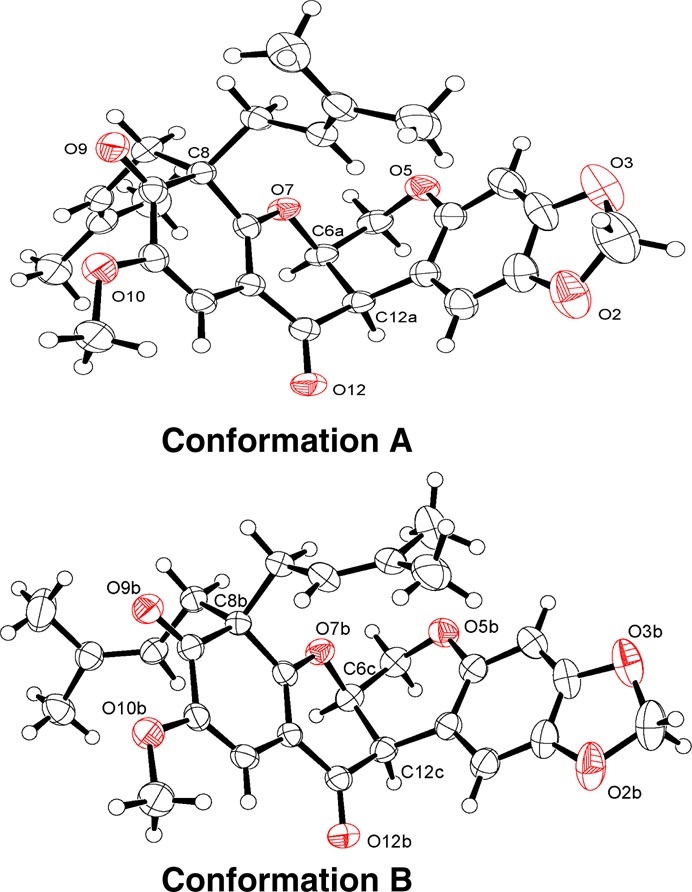
ORTEP plots for conformations A and B of 1 showing partial atom labeling and drawn with 50% probability displacement ellipsoids. Oxygen atoms are displayed in red.
Caeruleanone B (2) was isolated as a colorless solid, and its molecular formula was assigned as C28H30O8 based on the sodiated molecular ion peak at m/z 517.1844 [M + Na]+ (calcd m/z 517.1838) in the HRESIMS. The 1H NMR spectrum of 2 was closely comparable to that of 1, indicating the presence of a rotenoid-type compound with a methylenedioxy substituent at C-2a. In contrast to the 1H and 13C NMR spectra of 1, the absence of a proton signal at H-12a and the presence of a deshielded quaternary carbon at C-12a (δC 68.2 ppm) were consistent with a hydroxy substituent at this carbon for compound 2. The presence of a prenyl group and its substitution at C-8 were supported by HMBC correlations between the gem-dimethyl protons at H-4′ and H-5′ to C-2′ and C-3′ and the correlation of H-1′ with C-8, in addition to the COSY correlation between H-1′ and H-2′. The presence of an O-prenylated substituent at C-9 was supported by the observed deshielded methylene at δC 70.3 ppm (C-1″), HMBC correlations of the protons along the oxygenated prenyl chain, and the NOESY correlation observed between H-1′ and H-1″ further suggesting the position of the prenylated substituents at adjacent carbons. The singlet at H-11 exhibited the same HMBC and NOESY correlations as observed for 1. An additional NOESY correlation was observed for 2 between the methoxy protons at C-10 and one of the methylene protons at H-1″. The relative configuration for H-6a and the hydroxy group at C-12a were proposed based on the chemical shift of H-1 at δH 6.54 ppm indicating a cis-B/C ring junction.9,13 The zero specific rotation suggested that 2 is also a racemate. Compound 2 is a rare rotenoid since this is the first time that a member of this compound class containing both prenyl and O-prenylated substituents on the D-ring has been described. Similar substitutions have been observed previously for synthesized xanthones.15,16
Caeruleanone C (3) was isolated as a white powder, and its molecular formula was assigned as C23H22O8 based on the sodiated molecular ion peak at m/z 449.1216 [M + Na]+ (calcd m/z 449.1212) in the HRESIMS. The proton and carbon NMR spectra of 3 strongly suggested the presence of a rotenoid-type compound with a methylenedioxy substituent at C-2a as in 1 and 2. The presence of only one prenyl substituent was evident from the observation of two shielded methyl protons at δH 1.75 (H-4′) and 1.64 (H-5′), one nonaromatic methine proton at δH 5.15 (H-2′), and a methylene proton at δH 3.30 (H-1′). Compound 3 exhibited very similar 2D-NMR correlations to compounds 1 and 2, pointing toward the position of the prenyl substituent at C-8 and the methoxy group at C-10. The relative configuration of H-6a and the hydroxy group at C-12a were proposed based on the chemical shift of H-1 at δH 6.53 ppm, indicative of a cis-B/C ring junction.9,13 The dextrorotatory specific rotation, along with a positive Cotton effect at 311 nm and a negative Cotton effect at 359 nm observed in the ECD spectrum, corresponded to a (6aS,12aS) absolute configuration for 3.13 Two structurally closely related rotenoid analogues, (±)-villosinol and (−)-cis-12a-hydroxyrotenone, have been isolated previously from Lonchocarpus fluvialis of the family Fabaceae.17
It has been postulated previously that prenylation of rotenoids occurs after the formation of the A/B/C/D ring system and that rot-2′-enonic acid, a closely related analogue of 3, is the precursor that leads to the E-ring formation of chromenes, dihydrofurans, and furans in rotenoids.6,18 Dimethylallyl diphosphate (DMAPP) is the source for the isoprene unit being incorporated at the D-ring.6,18 With this taken into account, a proposed biogenetic route for the D-ring substitution of compounds 1–3 is postulated from rot-2′-enonic acid (Scheme 1).
Scheme 1. Proposed Biogenetic Pathways for the D-Ring Substitutions of 1–3.

Only the D-ring is shown.
Eleven known rotenoids inclusive of deguelin,8 6a,12a-dehydrodeguelin,8 11-hydroxy-6a,12a-dehydrodeguelin,8 erythynone,19 12a-hydroxyerythynone,19 millettosin,3,20 12a-hydroxyisomillettone,21 rotenone,22cis-(6aβ,12aβ)-hydroxyrotenone,22 tephrosin,8 and 11-hydroxytephrosin8 were also purified in this study. This corresponds to the second isolation of erythynone, 12a-hydroxyerythynone, and 12a-hydroxyisomillettone. Their complete spectroscopic data and those of millettosin have not been reported previously (Supporting Information). cis-(6aβ,12aβ)-Hydroxyrotenone and rotenone showed submicromolar cytotoxicities against HT-29 cells with IC50 values of 0.1 and 0.3 μM, respectively,23 and were primarily responsible for the initial cytotoxicity shown by the M. caerulea fruit chloroform-soluble extract.
The loss of the mitochondrial transmembrane potential (MTP) is an indication of apoptosis, and an agent that inhibits MTP may be useful in cancer chemotherapy.24 Also, the induction of quinone reductase (QR) is predictive of cancer chemopreventive activity.25 Therefore, compounds 1–3 were evaluated in these assays. Caeruleanone C (3) exhibited potent MTP inhibition with an IC50 value of 0.07 μM. Also, both caeruleanones B (2) and C (3) potently induced QR activity with a concentration required to double (CD) QR activity values of 0.9 and 1.0 μM and displayed low host cell cytotoxicity (IC50 values 34.6 and 27.7 μM, respectively). The results revealed the potent in vitro MTP inhibitory activity of 3 and also the strong in vitro quinone reductase induction activities of 2 and 3. The disruption of aromaticity of the D-ring in 1 that gives its singular rotenoid core may be responsible for its lack of activity in these bioassays.
Acknowledgments
This study was supported by NCI, NIH Grant P01 CA125066, awarded to A.D.K. We thank the Director of Nui Chua National Park, for permission to collect the plant material, and the Director of the Institute of Ecology and Biological Resources (IEBR) of the Vietnam Academy of Science and Technology, for overseeing the plant collection operation. The collection was executed under a UIC-IEBR collaborative agreement. We acknowledge Dr. C. McElroy, College of Pharmacy, and Mr. M. E. Apsega, Campus Chemical Instrument Center (CCIC), The Ohio State University, for maintenance and access to the NMR and mass spectrometers used herein.
Supporting Information Available
The extraction scheme, compound characterization, spectroscopic data, the X-ray data, and CIF file for 1 are included herein. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pham H. H. Cayco Vietnam [Flora of Vietnam] 1999, 1(894), #3581. [Google Scholar]
- Anderson E. F. Econ. Bot. 1986, 40, 38–53. [Google Scholar]
- Yenesew A.; Derese S.; Midiwo J. O.; Heydenreich M.; Peter M. G. Pest Manag. Sci. 2003, 59, 1159–1161. [DOI] [PubMed] [Google Scholar]
- Pancharoen O.; Athipornchai A.; Panthong A.; Taylor W. C. Chem. Pharm. Bull. 2008, 56, 835–838. [DOI] [PubMed] [Google Scholar]
- Bueno Pérez L.; Still P. C.; Naman B.; Ren Y.; Pan L.; Chai H.-B.; Carcache de Blanco E. J.; Ninh T. N.; Thanh B. V.; Swanson S. M.; Soejarto D. D.; Kinghorn A. D.. Phytochem. Rev.2014, 13, DOI 10.1007/s11101-014-9335-7. [DOI] [PMC free article] [PubMed]
- Crombie L.; Whiting D. A. Phytochemistry 1998, 49, 1479–1507. [DOI] [PubMed] [Google Scholar]
- Paz Parente J.; Pereira Da Silva B. Recent Res. Devel. Phytochemistry 2001, 5, 153–167. [Google Scholar]
- Andrei C. C.; Vieira P. C.; Fernandes J. B.; Da Silva M. F.; Das G. F.; Rodrigues Fo E. Phytochemistry 1997, 46, 1081–1085. [Google Scholar]
- Yenesew A.; Midiwo J. O.; Waterman P. G. Phytochemistry 1998, 47, 295–300. [Google Scholar]
- Yankep E.; Mbafor J. T.; Fomum Z. T.; Steinbeck C.; Messanga B. B.; Nyasse B.; Budzikiewicz H.; Lenz C.; Schmickler H. Phytochemistry 2001, 56, 363–368. [DOI] [PubMed] [Google Scholar]
- Azebaze A. G. B.; Meyer M.; Valentin A.; Nguemfo E. L.; Fomum Z. T.; Nkengfack A. E. Chem. Pharm. Bull. 2006, 54, 111–113. [DOI] [PubMed] [Google Scholar]
- Han Q.-B.; Yang N.-Y.; Tian H.-L.; Qiao C.-F.; Song J.-Z.; Chang D. C.; Chen S.-L.; Luo K. Q.; Xu H.-X. Phytochemistry 2008, 69, 2187–2192. [DOI] [PubMed] [Google Scholar]
- Slade D.; Ferreira D.; Marais J. P. J. Phytochemistry 2005, 66, 2177–2215. [DOI] [PubMed] [Google Scholar]
- The X-ray data of 1 were deposited at Cambridge Crystallographic Data Centre (CCDC 974061).
- Castanheiro R. A. P.; Pinto M. M. M.; Silva A. M. S.; Cravo S. M. M.; Gales L.; Damas A. M.; Nazareth N.; Nascimiento M. S. J.; Eaton G. Bioorg. Med. Chem. 2007, 15, 6080–6088. [DOI] [PubMed] [Google Scholar]
- Castanheiro R. A. P.; Pinto M. M. M.; Cravo S. M. M.; Pinto D. C. G. A.; Silva A. M. S.; Kijjoa A. Tetrahedron 2009, 65, 3848–3857. [Google Scholar]
- Blatt C. T. T.; Chávez D.; Chai H.-B.; Graham J. G.; Cabieses F.; Farnsworth N. R.; Cordell G. A.; Pezzuto J. M.; Kinghorn A. D. Phytother. Res. 2002, 16, 320–325. [DOI] [PubMed] [Google Scholar]
- Dewick P. M.Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons, Ltd.: Chichester, U.K., 2009; pp 175–177. [Google Scholar]
- Tahara S.; Narita E.; Ingham J. L.; Mizutani J. Z. Naturforsch. C: J. Biosci. 1990, 45, 154–160. [Google Scholar]
- Ollis W. D.; Rhodes C. A.; Sutherland I. O. Tetrahedron 1967, 23, 4741–4760. [Google Scholar]
- Oberholzer M. E.; Rall G. J. H.; Roux D. G. Phytochemistry 1976, 15, 1283–1284. [Google Scholar]
- Abidi S. L.; Abidi M. S. J. Heterocycl. Chem. 1983, 20, 1687–1692. [Google Scholar]
- Bueno Pérez L.; Li J.; Lantvit D. D.; Pan L.; Ninh T. N.; Chai H.-B.; Soejarto D. D.; Swanson S. M.; Lucas D. M.; Kinghorn A. D. J. Nat. Prod. 2013, 76, 1498–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y.; Balunas M. J.; Kim J.-A.; Lantvit D. D.; Chin Y.-W.; Chai H.-B.; Sugiarso S.; Kardono L. B. S.; Fong H. H. S.; Pezzuto J. M.; Swanson S. M.; Carcache de Blanco E. J.; Kinghorn A. D. J. Nat. Prod. 2009, 72, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang D. S.; Park E. J.; Kang Y.-H.; Hawthorne M. E.; Vigo J. S.; Graham J. G.; Cabieses F.; Fong H. H. S.; Mehta R. G.; Pezzuto J. M.; Kinghorn A. D. J. Nat. Prod. 2003, 66, 1166–1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.