

Abstract
A second-generation synthesis of three structurally related chlorosulfolipids has been developed. Key advances include highly stereocontrolled additions to α,β-dichloroaldehydes, kinetic resolutions of complex chlorinated vinyl epoxide intermediates, and Z-selective alkene cross metatheses of cis-vinyl epoxides. This strategy facilitated the synthesis of enantioenriched danicalipin A, mytilipin A, and malhamensilipin A in nine, eight, and 11 steps, respectively.
Introduction and Background
Almost 40 years after the first report of their existence,1,2 intense activity aimed at the chemical synthesis of the chlorosulfolipids (1–5, Figure 1) began independently and essentially simultaneously in at least four research groups around the world. Apparently purely coincidental, this confluence of research might well have stemmed from the fact that truly novel and unstudied classes of natural product targets are extremely rare in current times and make very attractive research problems. Since 2009, the groups of Carreira,3 Yoshimitsu,4 Matsuda,5 and our own6 have contributed syntheses of chlorosulfolipids and, in so doing, have taken what once looked like intractable problems for synthesis and found multiple creative ways for their construction. With the exception of Carreira’s tour de force synthesis of the proposed structure2i of mytilipin C (5) that determined the incorrectness of that structure,3c all of the published work to date has been focused on the three structurally similar chlorosulfolipids mytilipin A (3),3a,4b,6e danicalipin A (1),4c,5,6b and malhamensilipin A (2).6d At the outset of our work, we sought a general strategy toward these three targets; however, the unknown relative configuration of danicalipin A and malhamensilipin A prevented the development of such an approach at the time. Our productive collaboration with the Gerwick group unveiled the relative and absolute configuration of these two lipids6b,6c and revealed that a “central” stereotriad was conserved among the three lipids (1–3), but that there were important differences at other centers. Indeed, the difference at C16 between danicalipin A and malhamensilipin A precluded the direct translation of our successful synthesis of the former to the latter.
Figure 1.
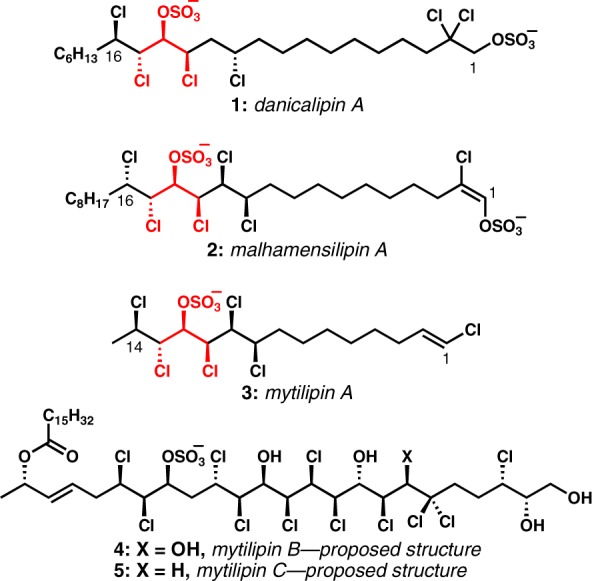
Representative chlorosulfolipids.
The approaches adopted for mytilipin A by Carreira3a and for danicalipin A and malhamensilipin A by our group6b,6d took advantage of alkene oxidation reactions for the introduction of all of the polar atoms in the stereochemically rich regions of these targets. However, since shortly after our interest in the chlorosulfolipids began, we have been keenly interested in an approach involving diastereoselective carbonyl additions to α,β-dichloroaldehydes. Conceptually, this approach was attractive because these starting materials are easy to access—at least in racemic form—and additions to the aldehyde should be highly stereocontrolled.7 However, the poor stability of the these aldehydes, which eliminate HCl easily, prevented our early attempts to use this tactic. To our knowledge, only Yoshimitsu and co-workers had successfully added nucleophiles to α,β-dichloroaldehydes4b,4c prior to the work that we describe here. It was that significant challenge in implementation that led us instead to the alkene oxidation approach that permitted the first syntheses of danicalipin A and malhamensilipin A.6b,6d
Although effective, our first-generation syntheses of 1 and 2 were fraught with the several problems: (1) the critical convergent Wittig reaction was both poorly stereocontrolled and somewhat erratic in terms of reproducibility; (2) the enantioselective route to malhamensilipin A could not be applied to danicalipin A because of a stereochemical difference in the targets; and (3) the routes were longer than we had hoped. In this article, we describe the evolution of our second-generation strategy that is applicable to chlorosulfolipids 1–3 in enantioenriched forms by virtue of an interesting kinetic resolution of chlorinated cis-vinyl epoxides. This approach also obviates the troublesome Wittig reaction, which is replaced by a convergent Z-selective alkene cross metathesis reaction. The results are (1) for danicalipin A, eight steps racemic, nine steps enantioselective (previous best 12 steps racemic6b or 13 steps enantioselective4c); (2) for mytilipin A, seven steps racemic, eight steps enantioselective (previous best 10 steps racemic3a,3d or 19 steps enantioselective4b); and (3) for malhamensilipin A, 11 steps formal enantioselective (previous best was our previous 12-step route, which was the only prior synthesis6d).
Synthesis Plan
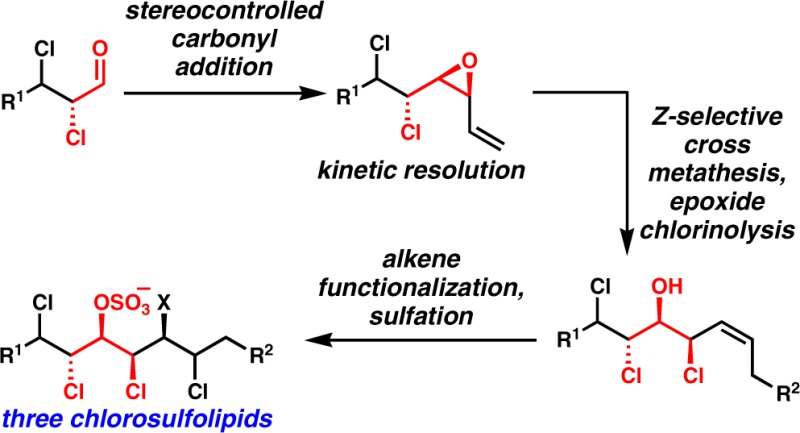
To put the second-generation approach into perspective, our first synthesis of racemic danicalipin A is shown in Scheme 1a. As alluded to above, we were aiming for a shorter synthesis that could be generalized to targets 1–3 that obviates the troublesome Wittig olefination and that takes advantage of the common stereotriad highlighted in Figure 1. The synthesis plan that was most attractive is shown in Scheme 1b. Stereospecific anti-dichlorination of either an (E)- or a (Z)-allylic alcohol will lead to anti- or syn-dichloroalcohol products, respectively. Assuming high levels of 1,2-stereoinduction,7,8 a haloallylation reaction would afford either syn-halohydrin 15 or cis-vinyl epoxide 16, depending upon workup conditions. Either of these intermediates could be productive substrates for Z-selective alkene cross metathesis as a replacement for the Wittig olefination; the products that result would intersect with the late stages of our previous syntheses. The major impediments to the implementation of this plan were: (1) it was not certain that an efficient and stereoselective carbonyl addition to dichloroaldehydes would be possible; (2) there was no obvious way to render the synthesis enantioselective; and (3) Z-selective alkene cross metathesis was, at the time we began this work, very much in its infancy and was not certain to work on such unusual and potentially reactive substrates.
Scheme 1. a. Previous Synthesis of Racemic Danicalipin A. b. General Approach to Danicalipin A, Malhamensilipin A, and Mytilipin A Featuring Carbonyl Additions to α,β-Dichloroaldehydes and Convergent Z-selective Alkene Cross Metathesis.

Results and Discussion
Because of our familiarity with its late-stage chemistry, we aimed to first apply our new strategy to an enantioselective synthesis of danicalipin A. As a result, in the following sections, we will first discuss the solutions to the three key unknowns described above in the context of this target. We will then demonstrate the generality of the approach with the syntheses of all three targets in subsequent sections.
Additions to α,β-Dichloroaldehydes
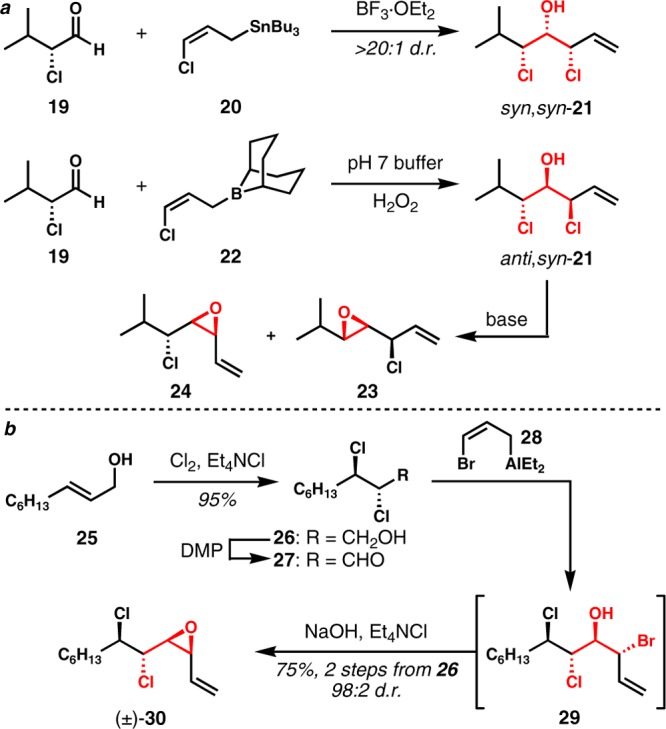
For our new synthetic route, it was necessary to develop conditions for mild and highly diastereoselective haloallylation of α,β-dichloroaldehydes to establish an efficient route toward the requisite cis-vinyl epoxide of type 16. In our earliest studies, attempted Grignard, organolithium, or alkali metal enolate additions to these aldehydes were met with failure, as were Lewis acid-catalyzed addition of π-nucleophiles. While the Yoshimitsu group had some success in this area,4b,4c Carreira alludes to similar problems in their disclosure of the mytilipin A synthesis.3a On the other hand, additions to α-chloroaldehydes were generally quite efficient and often stereoselective; these outcomes were not surprising given the lack of elimination pathways and the rather well-known stereocontrol imparted by α-acceptor groups on carbonyl additions.8 For example, the chloroallylation of α-chloroisovaleraldehyde (19) with (Z)-γ-chloroallylstannane 20(9) in the presence of BF3·OEt2 provided undesired syn,syn-21 with high diastereoselectivity (Scheme 2a). Not surprisingly, this reaction type was not successfully extended to electrophiles with β-chlorides. In contrast, 19 could be converted to desired anti,syn-21 by chloroallylation with (Z)-γ-chloroallylborane 22.10 However, the base-promoted epoxide formation surprisingly proceeded with poor site selectivity to give a mixture of constitutional isomers 23 and 24. Nonetheless, this haloallylborane reactivity could be extended to α,β-dichloroaldehydes (see below), and this outcome was the first hint that this type of electrophile tends to survive the milder conditions associated with closed transition structure allylations and related reactions. These observations were important in the eventual discovery that bromoallylaluminum reagents of type 28(9) were optimal from the perspectives of efficiency, stereoselectivity, and ease of preparation. An attractive sequence resulted: after dichlorination of (E)-2-nonen-1-ol (25) and careful oxidation with the Dess–Martin periodinane, bromoallylation followed by basic workup afforded vinyl epoxide 30 as a single regioisomer in high yield and with essentially perfect diastereoselectivity consistent with both the Felkin–Anh and Cornforth models (Scheme 2b). This sequence could produce racemic 30 in multigram scale in about 70% yield from the commercially available allylic alcohol precursor.
Scheme 2. a. Chloroallylation of α-Chloroaldehyde. b. Synthesis of Racemic cis-Vinyl Epoxide (±)-30.
Preparation of Enantioenriched Intermediates via Kinetic Resolution
Because we were clearly beholden to starting our synthesis from α,β-dichloroaldehydes, we required either enantioselective access to these key intermediates, or a means to resolve them, if we were to render our synthesis enantioselective. Although technology for asymmetric alkene chlorination is improving, with Nicolaou’s recent enantioselective dichlorination of allylic alcohols11 being particularly noteworthy, there is not currently a method that would prove economical enough in the preparation of highly enantioenriched dichloroalcohols to service a natural product synthesis endeavor of this type.
Certainly, we spent some time trying to develop just such a reaction, but with no success. Attempts to obtain enantioenriched material via enantioselective dichlorination with Cinchona alkaloid-derived chiral variants12 of Mioskowski’s reagent (Et4NCl3)13 or resolution of dichlorinated primary alcohols by peptide-catalyzed14 or enzymatic means were unsuccessful. Of course, highly effective examples of enzymatic resolution of chiral primary alcohols are few.15
Clearly, either resolution methods of later stage intermediates or Yoshimitsu’s elegant stereospecific dichlorodeoxygenation reactions of epoxides4a were the most promising ways to access enantioenriched intermediates. Owing to the single additional step involved in resolutions compared with the multiple steps involved in the epoxide-based strategy, we took the former approach to solve our problem.
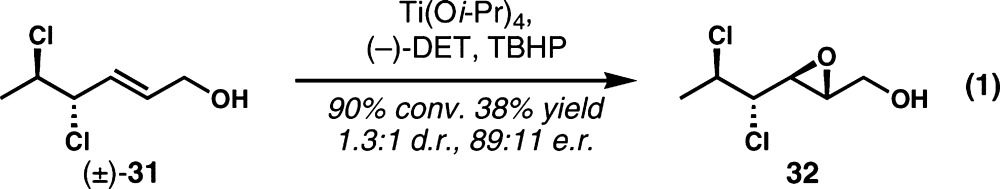
The Carreira group developed an asymmetric variant of their synthesis of mytilipin A (3) via (parallel) resolution of racemic dichloride 31 (eq 1).3d Sharpless asymmetric epoxidation of the allylic alcohol functional group afforded epoxide 32 in 1.3:1 dr; the enantiopurity of the desired diastereomer was moderate at 89:11 er.
 |
1 |
We undertook an extensive investigation into asymmetric carbonyl additions to α,β-dichloroaldehydes using chiral reagents or catalysts. The kinetic resolutions of racemic α,β-dichloroaldehyde (±)-27 via enantioselective haloallylation with chiral Oehlschlager/Brown haloallylborane reagents (33/34)10 proceeded with poor enantioselectivity (Scheme 3a). According to the enantioselective chloroallylation procedure of Kobayashi,16 the chiral zinc catalyst derived from the bipyridine ligand 38 afforded the product (−)-35 in moderate enantiopurity, and useful selectivity factors were achieved (Scheme 3b). However, the resolved starting material, which is always easier to obtain in higher enantiopurity via kinetic resolution,17 was unstable to the reaction conditions and could not be isolated, leaving only partially resolved product 35. Furthermore, as we showed in Scheme 2a, epoxide formation from α,α′-dichloroalcohols of type 35 was not selective, and the bromoallylation corresponding to that shown in Scheme 3b was never successfully implemented.
Scheme 3. a. Haloallylation of Dichloroaldehyde with a Chiral Boron Reagent. b. Chloroallylation of Dichloroaldehyde with Chiral Zinc Lewis Acid.

We next considered resolving racemic vinyl epoxide 30 derived from diastereoselective haloallylation/epoxide formation of the α,β-dichloroaldehyde (Scheme 2b). This type of vinyl epoxide was readily prepared on multigram scale and should be easily recovered after kinetic resolution. Furthermore, it has a strong bias for regiocontrol of ring opening; clearly the allylic terminus is activated while the other epoxide carbon is deactivated by the proximal chlorides. Therefore, we postulated that some of the many available enantioselective meso-epoxide desymmetrization protocols should be plausibly extended to kinetic resolution of substrates of type 30.
We began with Jacobsen’s epoxide opening chemistry, using highly reactive (R,R)-(oligosalen)Co catalysts.18 To the best of our knowledge, resolutions of internal epoxides with the Jacobsen system have not been reported; however, we felt that the cis-vinyl epoxide might be a close structural mimic of competent cyclic meso-epoxides that are frequently desymmetrized using Jacobsen chemistry. Surprisingly, substrate 30 proved unreactive toward nucleophiles such as water, phenol, or benzyl alcohol under published conditions for desymmetrization of meso-epoxides. For reasons that we do not understand, Denmark’s catalytic system for desymmetrization of meso-epoxides via ring-opening chlorinolysis,19a,19b using the “Lewis base activation of Lewis acids” concept,19c proved much more successful. In the original Denmark group study, meso-stilbene oxide (39) was effectively desymmetrized in the presence of a chiral phosphoramide Lewis base catalyst (R)-40 and SiCl4, a weak Lewis acid, to afford the syn-1,2-chlorohydrin (1S,2S)-42 in high enantiopurity (Scheme 4a).19a Later, it was found that the dimeric phosphoramide Lewis base (R,R)-41, which is typically more selective for other SiCl4-mediated enantioselective transformations, provided (1S,2S)-42 with notably diminished enantiopurity.19b The stereochemical outcome of desymmetrization of meso-epoxide suggested that the (R)-BINAM-derived phosphoramde Lewis base catalysts would enrich our cis-vinyl epoxide reactants in the desired enantiomer by selectively catalyzing ring-opening chlorinolysis of the undesired enantiomer.
Scheme 4. a. Denmark’s Desymmetrization of meso-Epoxides. b. Preliminary Study of Chiral Lewis Base-Catalyzed Kinetic Resolution of cis-Vinyl Epoxide (±)-30. Selectivity Factor, S = kfast/kslow = ln[(1 – conversion)(1 – ee)]/ln[(1 – conversion)(1 + ee)]. c. Chiral Lewis Bases Studied for Kinetic Resolution.

Under Denmark’s conditions, the cis-vinyl epoxide (±)-30 was found to be less reactive than meso-epoxides, probably because of the more sterically congested environment presented by the proximal chlorine bearing carbons. Consequently, the kinetic resolution with (R)-40 was carried out at slightly elevated temperature (−50 °C) with higher catalyst loading (20 mol%) (Scheme 4b). Unfortunately, the resolution with (R)-40 proceeded with poor selectivity (selectivity factor, S = 4). Surprisingly, in contrast to Denmark’s result, the dimeric chiral Lewis base (R,R)-41 was more selective (S = 14) for our kinetic resolution than the monomeric chiral Lewis base (R)-40. Interestingly and unexpectedly, the resolved vinyl epoxide from the kinetic resolutions with (R)-40 and (R,R)-41 were enriched in the opposite enantiomers. Other chiral Lewis bases such as trans-cyclohexanediamine-derived phosphoramide (R,R)-43 and (R)-BINAPO ((R)-44) were also tested. These Lewis bases were more reactive than (R)-40 or (R,R)-41 but virtually unselective (S < 3). Clearly, we had a good lead with catalyst (R,R)-41 at this point.
During the optimization of the kinetic resolution, it was found that the selectivity is highly dependent on the reaction temperature. When (±)-30 was resolved with 10 mol% of (R,R)-41 at −78 °C, a substantially improved selectivity factor of 33 was obtained (Table 1, entry 1). However, the reaction was even more sluggish and proceeded to only 24% conversion after 24 h. Even with higher catalyst loading (20 mol%) and extended reaction time (48 h), the conversion was improved to only 42% and the reaction became increasingly slower as the reaction progressed (entry 2). The amounts of SiCl4 and i-Pr2NEt seem to have little effect on the conversion and selectivity. Because it is reasonable to postulate that the rate of chlorinolysis would be increased at higher concentration of chloride nucleophile, the kinetic resolution was conducted in the presence of exogenous soluble chloride (entry 3). Although the conversion was improved to 61% in the presence of 1 equiv of Et4NCl, the selectivity factor diminished significantly. It was also possible to improve the conversion by adopting higher concentrations. The kinetic resolution could be efficiently carried out at 0.2 M with little attenuation of selectivity (entry 4). The reaction could be further accelerated by further increasing the concentration; however, the selectivity factor decreased substantially (entries 5 and 6). On the other hand, the selectivity factor could be improved by performing the reaction in more diluted condition. A selectivity factor of 61 was obtained at 0.05 M even though the reaction was too slow to be practical (entry 7). Unfortunately, the mechanistic origin for the drastic effects of the exogenous chloride and the reaction concentration on the selectivity was unclear. Eventually, an ideal 53% conversion was achieved with 20 mol% of (S,S)-41 after 24 h at 0.2 M, and the desired enantiomer of unreacted vinyl epoxide (−)-30 was isolated in 43% yield with 97.3:2.7 er on a preparative scale (entry 8 and eq 2). The catalyst could be fully recovered after reaction.
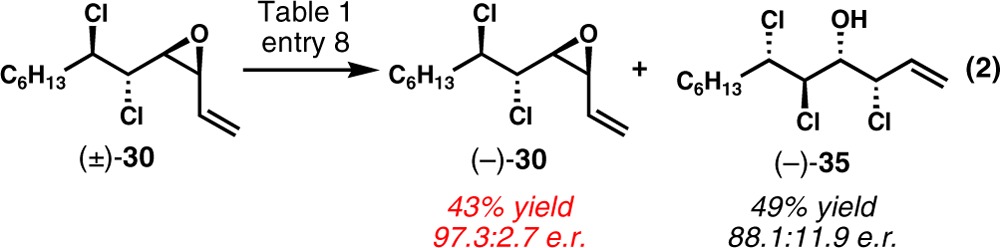 |
2 |
Table 1. Optimization of Kinetic Resolution of (±)-30a.
| entry | concentration (M) | time (h) | conversionb (%) | er (reactant)c | er (product)d | S |
|---|---|---|---|---|---|---|
| 1 | 0.1 | 24 | 24 | 66.0:34.0 | 3.9:96.1 | 33 |
| 2e | 0.1 | 48 | 42 | 84.5:15.5 | 5.3:94.7 | 37 |
| 3f | 0.1 | 48 | 61 | 93.1:6.9 | 22.2:77.8 | 9 |
| 4 | 0.2 | 48 | 41 | 83.9:16.1 | 5.8:94.2 | 33 |
| 5 | 0.4 | 24 | 44 | 83.7:16.3 | 10.0:90.0 | 18 |
| 6 | 1.0 | 24 | 66 | 88.7:11.3 | 31.9:68.1 | 5 |
| 7 | 0.05 | 72 | 32 | 72.4:27.6 | 2.5:97.5 | 61 |
| 8e,g | 0.2 | 24 | 53 | 2.7:97.3 | 88.1:11.9 | 27 |
All reactions employed 1.0 equiv of SiCl4 and 0.1 equiv of i-Pr2NEt on 0.1–2.0 mmol scale.
Determined by 1H NMR analysis.
Determined by CSP-GC.
Determined by CSP-SFC after 2,4-dinitrobenzoylation.
20 mol% of catalyst.
1.0 equiv of Et4NCl.
Preparative scale, using (S,S)-41, to preferentially recover (−)-30.
The optimized kinetic resolution conditions were next applied to a substrate that was destined for the enantioselective synthesis of mytilipin A. The cis-vinyl epoxide (±)-49 was prepared in a similar manner to that used to make (±)-30 (Scheme 5). (E)-Crotyl alcohol (45) was treated with molecular chlorine in the presence of Et4NCl to give the anti-1,2-dichloride (±)-46. Oxidation with the Dess–Martin periodinane followed by a careful workup afforded the sensitive and volatile α,β-dichloroaldehyde (±)-47 in crude form, which was immediately converted to volatile cis-vinyl epoxide (±)-49 via bromoallylalumination and epoxide formation, again with near perfect diastereocontrol. The moderate yield in this case can be attributed to volatility of the intermediate aldehyde and the vinyl epoxide product.
Scheme 5. Synthesis of Racemic cis-Vinyl Epoxide (±)-49.

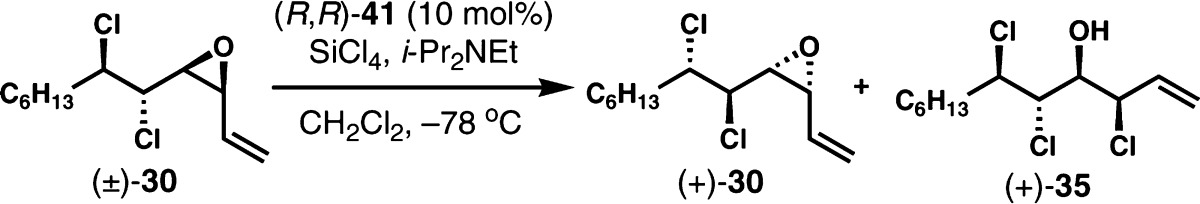
Surprisingly, the kinetic resolution of (±)-49, which differs only by alkyl chain length compared to (±)-30, was only moderately efficient with catalyst (R,R)-41. Under the optimized conditions developed for (±)-30, a selectivity factor of only 6 was obtained (Table 2, entry 1). Although it was possible to recover (+)-49 with an improved enantiopurity at higher conversion (entry 2), a more practical level of selectivity was desired. Similarly to the case of (±)-30, higher selectivity could be achieved at lower concentration. Consequently, the selectivity factor was improved to 8 at 0.15 M concentration (entry 3). Furthermore, a selectivity factor of 13 at 57% conversion was realized at 0.1 M concentration, and enantioenriched (+)-49 was isolated in 93.4:6.6 er and 43% yield (entry 4 and eq 3). The monomeric phosphoramide (R)-40 and (R)-BINAPO ((R)-44) were even less selective (S = ∼4, not shown), although (R)-BINAPO was more reactive than dimeric phosphoramide catalyst (R,R)-41. Curiously, it was difficult to reliably analyze the enantiopurity of the chlorohydrin product (+)-50 because of apparently facile selective sublimation of the major enantiomer under vacuum, which resulted in enantiodepletion of the sample (eq 4).20
 |
3 |
Table 2. Optimization of Kinetic Resolution of (±)-49a.
| entry | concentration (M) | time (h) | conversionb (%) | er (reactant)c | S |
|---|---|---|---|---|---|
| 1 | 0.2 | 24 | 56 | 84.6:16.4 | 6 |
| 2 | 0.2 | 36 | 66 | 92.4:7.6d | 6 |
| 3 | 0.15 | 72 | 65 | 94.8:5.2 | 8 |
| 4 | 0.1 | 72 | 57 | 93.4:6.6 | 13 |
All reactions employed 1.0 equiv of SiCl4 and 0.1 equiv of i-Pr2NEt and were conducted on 0.25–0.54 mmol scale.
Determined by 1H NMR analysis.
Determined by CSP-GC.
Determined by CSP-SFC after benzoylation.
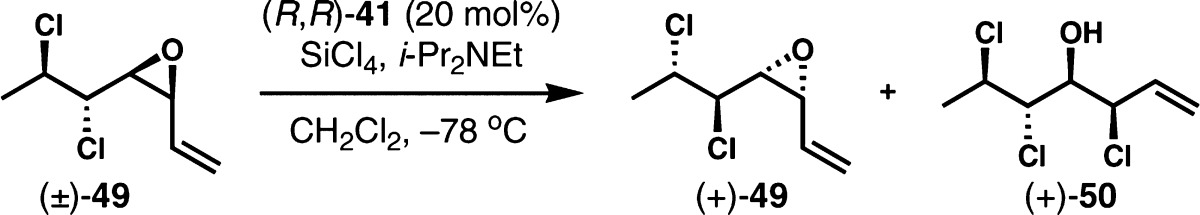
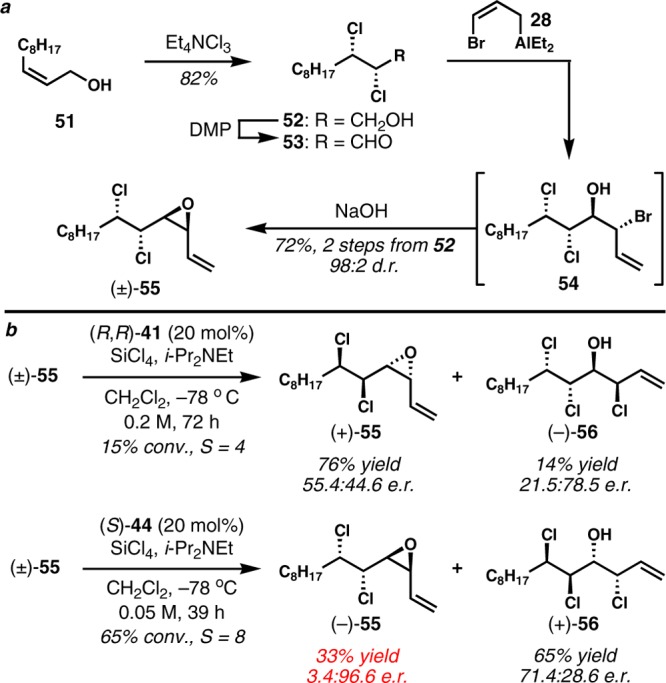
The same kinetic resolution strategy was also examined for the enantioselective synthesis of (+)-malhamensilipin A. The corresponding cis-vinyl epoxide (±)-55, which differs from substrates 30 and 49 by virtue of its syn-1,2-dichloride moiety, was prepared from (Z)-2-undecen-1-ol (51) via the same sequence used for previous substrates (Scheme 6a). Unfortunately, (±)-55 was considerably less reactive toward chlorinolysis than the other vinyl epoxide substrates, and the enantioselectivity was very poor (Scheme 6b). Under the general conditions with 20 mol% of (R,R)-41, the resolution proceeded to only 15% conversion even after 72 h and afforded a selectivity factor of only 4. Modified reaction conditions with higher concentrations, higher temperatures, or addition of exogenous chloride, while likely to accelerate the reaction, would equally likely attenuate the already very low enantioselectivity, as we had previously observed in the study of (±)-30. When (±)-55 was resolved with the typically more reactive (S)-BINAPO ((S)-44), a much higher reaction rate was indeed observed. Contrary to previous cases, the selectivity factor was also improved, although it was still moderate (S = 8–9). From a preparative scale reaction, the resolved vinyl epoxide (−)-55 was obtained in 3.4:96.6 er and 34% yield. Several related chiral bis(phosphine oxide)s such as (S)-Tol-BINAPO, (S)-H8-BINAPO, and (R)-SEGPHOS dioxide were also tested, but the selectivity was not improved.
Scheme 6. a. Synthesis of Racemic cis-Vinyl Epoxide (±)-55. b. Kinetic Resolution of (±)-55.
Convergent Z-Selective Alkene Cross Metathesis
Concurrent with the development of an effective kinetic resolution method, the key convergent metathesis step was investigated with (±)-30, a potential precursor to danicalipin A. At the time of conception of our metathesis-based second-generation approach, only the first hint that Z-selective alkene cross metathesis was a viable reaction had appeared in the literature.21 Moreover, whereas a Z-configured alkene is required en route to malhamensilipin A and mytilipin A so that stereospecific anti-dichlorination would afford the correct relative syn-configuration (at C11/C12 and C9/C10, respectively), it was not obviously a necessity for danicalipin A because of the unchlorinated carbon at C12. Therefore, the feasibility of the alkene cross metathesis approach was initially evaluated with normal alkene metathesis catalysts.
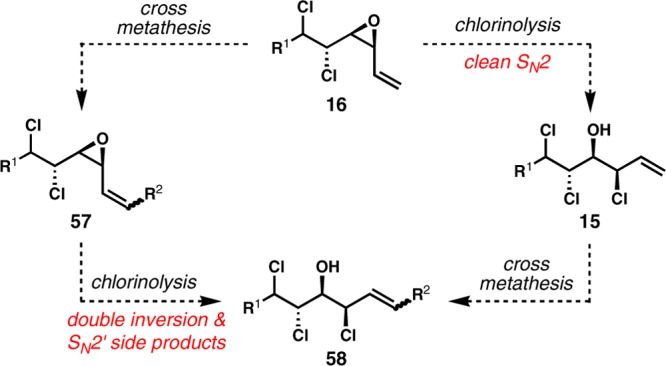
Two different orders of operations were considered for alkene cross metathesis and ring-opening chlorinolysis (Scheme 7). The left-hand sequence involves the alkene cross metathesis of a vinyl epoxide (16) followed by chlorinolysis of the resulting internal alkenyl epoxide, which might be plagued by double inversion at the allylic center and SN2′ side reactions, as seen in previous studies. On the other hand, the right-hand sequence is initiated with chlorinolysis of the terminal vinyl epoxide, which might proceed as a clean SN2 reaction under certain conditions, for example, the “racemic version” of the vinyl epoxide chlorinolysis resolution using SiCl4 and an achiral Lewis base catalyst such as HMPA. The resulting allylic chlorohydrin 15 would then be a potential substrate for subsequent alkene cross metathesis to deliver 58. With the latter sequence, the enantioenriched 1,2-chlorohydrin product from the kinetic resolution could also be conveniently utilized for the synthesis of enantiomeric chlorosulfolipids.
Scheme 7. Possible Orders of Operations for Alkene Cross Metathesis and Ring-Opening Chlorinolysis.
Electron-poor allylic chloride (±)-35 underwent alkene cross metathesis with 1-decene in the presence of 10 mol% of the Grubbs second-generation catalyst (G II) at room temperature to afford the (E)-alkene product 59 in 43% yield along with dimeric side products (Scheme 8); because of the relatively complex crude reaction mixture, it was difficult to determine the inherent E/Z selectivity of this reaction. The Hoveyda–Grubbs second-generation catalyst (HG II) promoted slower but cleaner alkene cross metathesis to give an 84:16 E:Z mixture of alkene isomers in 85% conversion and 60% yield of isomerically pure 59. Unfortunately, iodochlorination of 59 with ICl provided a complex crude mixture (not shown), in contrast to the case of the corresponding Z-isomer that had been iodochlorinated with high efficiency in our first-generation approach, although with low diastereoselectivity. Attempts to directly hydrochlorinate the unactivated alkene under iron-mediated radical hydrofunctionalization conditions recently reported by Boger22 was also unsuccessful, probably because of the low reactivity of the electron-deficient alkene.
Scheme 8. Alkene Cross Metathesis of (±)-35.

To generate the more desirable (Z)-alkene isomer, Z-selective alkene cross metathesis of allylic chloride 35 with various terminal alkene partners in the presence of recently developed Grubbs cycloadamantyl catalyst 60,23 which was generously provided first by the Grubbs group and later by Materia, was investigated. However, not only 35 but also hydroxy-protected substrates and the less chlorinated substrate 62 exhibited no reactivity (Scheme 9). Ruthenium metathesis catalysts are clearly able to execute cross metatheses of allylic chlorides; at this stage, we have no reasonable understanding of the apparent limitation of the Z-selective catalysts toward allylic chlorides, nor do we know if it is a truly general limitation.
Scheme 9. Alkene Cross Metathesis of 35 with Z-Selective Catalyst 60.

Alternatively, the corresponding cis-vinyl epoxide was examined as a substrate for alkene cross metathesis. Similarly to the corresponding allylic chloride, cis-vinyl epoxide (±)-30 underwent alkene cross metathesis with 1-decene in the presence of 10 mol% of G II to afford 64 with moderate 82:18 E:Z-selectivity (Scheme 10). Unlike the case of chlorohydrin substrates, it was difficult to separate the internal alkenyl epoxide product from the unreacted terminal vinyl epoxide reactant. These compounds were isolated as a mixture (estimated yields of the product and the recovered reactant: ∼57 and 10%, respectively based on NMR integration). In contrast, a complex mixture was obtained from the similar reaction with HG II. While the desired product was not detected, one of the major components in the crude mixture was identified as the unsaturated chloroaldehyde 65, which implies the formation of α,β-dichloroaldehyde 27 (Scheme 2b) under the reaction conditions. Additionally, the presence of chlorohydrin 66 as a minor component in the crude mixture further suggests the formation of 27 followed by elimination of HCl, which is presumably responsible for epoxide chlorinolysis of a small amount of desired alkene cross metathesis product. The formation of 65 was confirmed from the reactions between 30 and HG II (10 and 100 mol%) in the absence of other metathesis partners. At this stage, we cannot put forth a reasonable mechanism for this interesting three-carbon degradation of vinyl epoxides. We have not investigated the generality of this reaction type.
Scheme 10. Alkene Cross Metathesis between 30 and Simple Terminal Alkenes.

Gratifyingly, cis-vinyl epoxide 30 turned out to be a competent substrate for Z-selective alkene cross metathesis. In the presence of 1 mol% of catalyst 60, 30 underwent alkene cross metathesis with an excess of 1-hexene to 12% conversion at 35 °C in 1 h (Scheme 11). The Z-isomer of vinyl epoxide 67 was produced with exquisite selectivity. Such exceptionally high Z-selectivity had only been rarely observed with this catalyst.23 Other solvents such as toluene or dichloromethane had no significant impact on the conversion and selectivity. The catalytic activity was typically lost within a few hours, and the reactions would proceed no further. The conversion could be improved to about 50–60% (NMR estimate) with higher loading of catalyst (10 mol%), and the use of chlorinated solvents such as dichloromethane or 1,2-dichloroethane proved beneficial because of the poor solubility of 60 in other solvents. However, the decomposition of the starting vinyl epoxide was a serious side reaction, and significant amounts of an as yet unidentified decomposition product were formed.
Scheme 11. Z-Selective Alkene Cross Metathesis of 30 with 1-Hexene.

With this preliminary success in hand, we turned to the use of the relevant alkene 71 as the metathesis partner, which was made from known aldehyde 68(24) via a slight modification of Yoshimitsu’s procedure4c as shown in Scheme 12a. This high molecular weight compound could not be used in as large excess as the model alkenes owing to effects on reaction concentration; initial reactions suffered from very low efficiencies, and the decomposition of the starting vinyl epoxide remained problematic. The related alkene 70, with a free hydroxyl group and attendant lower molecular weight that could potentially be used in greater excess, was unreactive (Scheme 12b).
Scheme 12. a. Preparation of Potential Alkene Cross Metathesis Partners 70 and 71 for the Synthesis of Danicalipin A and Malhamensilipin A. b. Z-Selective Alkene Cross Metathesis of 30 for the Synthesis of Danicalipin A (all compounds shown are racemic).
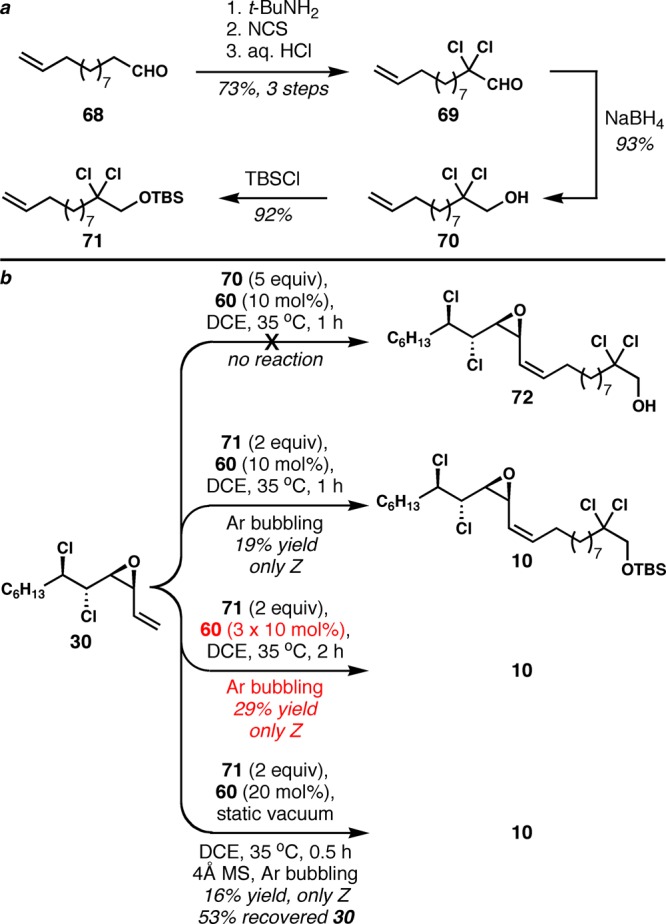
To achieve higher conversion and suppress the decomposition, an extensive optimization of the reaction conditions was conducted. A variety of reaction solvents including tetrahydrofuran, diethyl ether, t-butyl methyl ether, toluene, chlorobenzene, hexafluorobenzene,25 α,α,α-trifluorotoluene,25 and octafluorotoluene,25 as well as neat conditions were employed, but the reaction efficiency was not improved. The reaction was even slower at room temperature, and performing the reaction at higher temperature (60 °C) only resulted in greater decomposition.
A wide range of additives were also evaluated. Amine bases such as i-Pr2NEt and di-tert-butylpyridine promoted decomposition. 1,4-Benzoquinone, known to scavenge ruthenium hydride species26 that might be formed during reaction and cause decomposition, only attenuated the catalytic reactivity of 60. The reaction became slightly cleaner in the presence of 3 or 4 Å molecular sieves, but substrate decomposition could not be completely avoided. Ti(Oi-Pr)427 and hexachloroethane,28 which have been used to improve the reactivity of other alkene cross metathesis reactions, had no influence on the reaction.
Portionwise addition of catalyst and 1-hexene also provided no advantage. We hoped that removal of ethylene from the reaction mixture would shift the cross metathesis equilibrium and drive these reactions to higher conversion. Therefore, the reaction was carried out under static vacuum, continuous vacuum, and in an open vessel inside a glovebox, but to no avail. More rigorous removal of ethylene was attempted by vigorously bubbling argon through the reaction mixture, and gratifyingly, the formation of the unknown was finally prevented. Under optimized conditions, with 10 mol% of 60, (±)-10 was obtained in 19% yield along with 74% recovered starting material (Scheme 12b). It was more challenging to suppress the decomposition with higher catalyst loadings, and the mass balance was poorer. The decomposition could be minimized by slowing down the reaction rate via a portionwise addition of catalyst, giving the product 10 in 29% yield with 40% recovered starting material using 30 mol% of 60. Although we were unable to achieve more than the equivalent of a single turnover, this sequence still stands as a marked improvement over the previous Wittig-based route. Access to enantioenriched 10 now requires only five steps, compared with our previous eight-step approach that afforded racemic material. As a result, this moderate success completed a much shorter, enantioselective formal synthesis of danicalipin A because of the interception of intermediate 10 from our first-generation synthesis. However, more improvements in the end-game were still possible (see below).
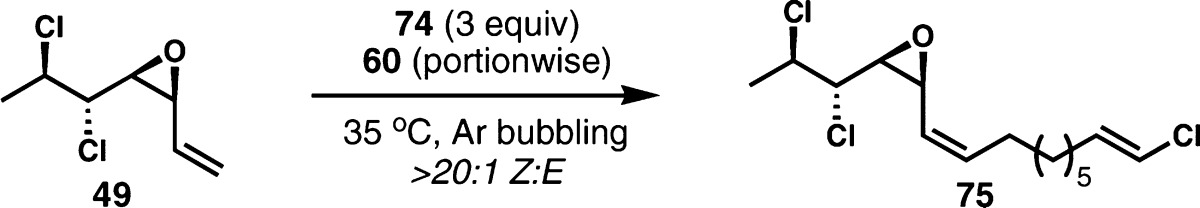
Convergent Z-selective alkene cross metathesis for mytilipin A with the corresponding cis-vinyl epoxide 49 proceeded similarly to the corresponding reaction for danicalipin A. Alkene metathesis partner 74 was obtained in two steps from 8-bromo-1-octene (73) via formylation of Grignard reagent followed by Takai-Utimoto chloroolefination (eq 5). The convergent metathesis reactions were carried out with vigorous bubbling of argon to prevent the decomposition of starting vinyl epoxide, and the desired alkene (±)-(Z)-75 was produced as a single geometrical isomer. Again, we were unable to achieve more than a single turnover with 10–30 mol% of catalyst 60 (Table 3, entries 1 and 2). The use of fluorinated solvents such as α,α,α-trifluorotoluene25 did not result in any improvement (entry 3). Unfortunately, higher loading of catalyst only resulted in significant loss of mass balance and the yield of product was only marginally improved (entries 4 and 5). Despite the low efficiency of the Z-selective alkene cross metathesis, the direct incorporation of the vinyl chloride is a marked improvement over previous syntheses because it eliminates at least three postconvergence steps. Cross metathesis partner 74 might appear upon cursory analysis to be poised for side reactivity because as a 1,9-diene cyclooctene formation could occur via ring-closing metathesis. However, vinyl chlorides are relatively slow to react in metathesis processes, and cyclo-octene formation can also be a sluggish reaction. Almost certainly, however, the high kinetic selectivity of catalyst 60 for (Z)-alkenes is presumably the most important factor that prevents reaction with the (E)-vinyl chloride in either RCM or cross metathesis events.
 |
5 |
Table 3. Z-Selective Alkene Cross Metathesis for the Synthesis of Mytilipin Aa.
| entry | 60 (mol%) | solvent | time (h) | yield (%) |
|---|---|---|---|---|
| 1 | 10 | DCE/CH2Cl2 | 2 | 10 |
| 2 | 30 | DCE/CH2Cl2 | 3 | 32 |
| 3 | 30 | PhCF3/CH2Cl2 | 3 | 33 |
| 4 | 50 | PhCF3/CH2Cl2 | 4 | 34 |
| 5b | 100 | DCE | 3 | 39 |
All reactions were carried out on 0.15–0.25 mmol scale.
5 equiv of 74 was employed. Catalyst 60 was added in one portion.
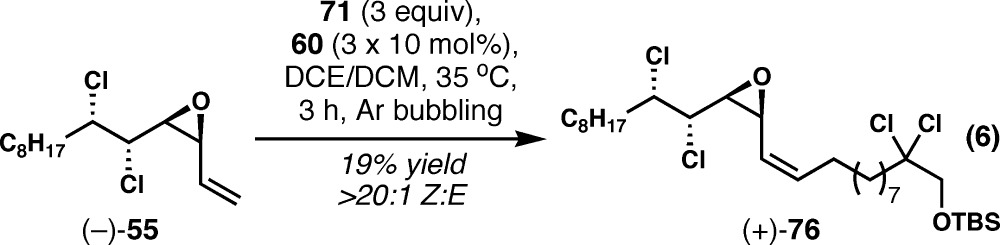
Unfortunately, the convergent Z-selective alkene cross metathesis was even less efficient for malhamensilipin A. The metathesis product (+)-76 was isolated only in 19% yield from the reaction of (−)-55 with 71 under the analogous conditions to those used for danicalipin A (eq 6). Cursory attempts to improve the efficiency of this reaction were unsuccessful. For reasons explained below, the improvement of this convergent step was not a priority.
 |
6 |
While admittedly not as efficient as desired, the convergent Z-selective alkene cross metathesis is noteworthy for its complete diastereoselectivity in all cases examined. To see if the extremely high selectivity we observed was general for cis-vinyl epoxides, as well as to investigate the low catalytic activity of 60 with respect to the specific chlorinated cases relevant to the chlorosulfolipids, we tested the reactivity of unchlorinated cis-vinyl epoxide 79 with 1-decene (Scheme 13). In the presence of 10 mol% of 60, complete conversion to (Z)-vinyl epoxide 80 was observed (83% isolated yield, >20:1 Z:E). Even with only 1 mol% of catalyst, the reaction proceeded to 46% conversion and the product was isolated in 43% yield with equal selectivity. Therefore, it appears that cis-vinyl epoxides are subject to highly Z-selective alkene cross metathesis with 60, and that the poor efficiency observed in the convergent steps for chlorosulfolipids is likely specific to chlorinated substrates. Recently, the Grubbs group also demonstrated that vinyl epoxides are excellent substrates for Z-selective metathesis using these catalysts.23o
Scheme 13. a. Synthesis of Simple Vinyl Epoxide 79. b. Z-Selective Alkene Cross Metathesis of (±)-79.

Postconvergent Manipulations and Completion of the Syntheses
Completion of the synthesis of (+)-danicalipin A took advantage of a similar reaction sequence to that previously developed in the context of our first-generation approach (Scheme 1a). Lewis acid-mediated chlorinolysis of the internal alkenyl epoxide 10 typically afforded a diastereomeric mixture of the desired SN2 product 11 and the double inversion3a,6b product 81 as well as the constitutional isomer 82 formed via SN2′ substitution (Scheme 14a). The extent of side product formation was highly dependent on the choice of Lewis acid and the concentration of chloride anion. Because exclusive SN2 reactivity was observed from the reaction of terminal cis-vinyl epoxide with SiCl4 in the presence of HMPA, a combination of SiCl4 and a number of Lewis base activators including pyridine, DMAP, pyridine N-oxide, HMPA, DMPU, DMI, and TMU was evaluated with or without Et4NCl. In all cases, a variable amount of side products were produced and a useful level of selectivity was not accomplished (90:10–31:69 dr, 2–50% SN2′). Both undesired pathways were reasonably attenuated when the epoxide was opened using dry HCl; however, high selectivity was desired specifically for the exclusion of double inversion product 81, which is more difficult to separate from the desired product. Double inversion could be completely overcome by employing BF3·OEt2 at −78 °C with a high concentration of Et4NCl. Despite the presence of a rather large amount of SN2′ product 82, the desired isomer 11 could be isolated in 73% yield as a single diastereomer. A major problem of our first-generation synthesis was the poorly diastereoselective iodochlorination reaction of 11 (∼1.8:1 dr), which was compounded further by the very painstaking separation of diastereomers at that stage or after deiodination. We found that transient introduction of a trimethylsilyl group on the C14 hydroxyl permitted high diastereocontrol (95:5 dr) in the iodochlorination, and because the silyl group could be introduced and removed in the same pot, this result had a significantly positive impact on the synthesis. Overall, the new approach facilitated a nine-step synthesis of enantioenriched (+)-danicalipin A (4.6% overall yield), which is a significant improvement over our 12-step racemic first-generation synthesis.
Scheme 14. a. Completion of the Synthesis of (+)-Danicalipin A. b. Completion of the Synthesis of (−)-Mytilipin A.

Completion of the synthesis of mytilipin A required only three postconvergence steps (Scheme 14b). BF3·OEt2-mediated vinyl epoxide chlorinolysis with inversion of configuration proceeded with exclusive diastereoselectivity and delivered diene 85. Dichlorination of the electron-deficient allylic chloride afforded hexachloride 86 in 86% yield with high diastereoselectivity (93:7 dr of crude product, purified to 97:3) and complete chemoselectivity with respect to the isolated vinyl chloride. Sulfation of the secondary alcohol according to Carreira’s conditions3a completed the synthesis of mytilipin A. In this way, racemic chlorosulfolipid could be accessed in 8.6% yield over the seven linear steps sequence, and enantioenriched mytilipin A is available via a longest linear sequence of eight steps (3.7% overall yield). These results compare favorably to the previously reported syntheses.
It is indeed fortuitous that we chose to first pursue danicalipin A with this new approach. The choice of malhamensilipin A as a first target could easily have discouraged us from pursuing this strategy. Although, as described above, this strategy led to much improved syntheses of mytilipin A and danicalipin A, there was ultimately little improvement in the synthesis of malhamensilipin A, for which we had already established an enantioselective synthesis, via the same number of steps, and for which the Wittig reaction was not improved upon with the metathesis option. Therefore, while we are pleased to claim a formal enantioselective synthesis of malhamensilipin A as part of this second-generation, general strategy for chlorosulfolipid synthesis, we would suggest that our first enantioselective synthesis of this single target would likely be the preferred method to access samples of this natural product. However, if new catalysts become available that can better effect these challenging Z-selective cross metatheses, and if a truly effective method for asymmetric dichlorination of allylic alcohols is discovered, the strategy described here would be hard to beat for any of these three chlorosulfolipid targets. Indeed, this approach has the distinct advantage that it can be rendered enantioselective without recourse to resolution once asymmetric catalysis technology is developed for allylic alcohol dichlorination.
Conclusions
We have developed a concise and general approach for the enantioselective synthesis of three chlorosulfolipid targets that takes strategic advantage of a common stereotriad. Diastereoselective carbonyl addition to sensitive α,β-dichloroaldehydes, Z-selective alkene cross metatheses, and kinetic resolution of chlorinated vinyl epoxides are key advances that permitted success in this second-generation approach. Enantioenriched danicalipin A, mytilipin A, and malhamensilipin A are accessed in nine, eight, and 11 steps, respectively.
Given the paucity of efforts toward this class of natural products until about five years ago, it is remarkable that so many effective solutions to these targets from multiple research groups have appeared in such short order. Certainly, polychlorinated natural products are not to be feared as objectives for chemical synthesis and are rather well-behaved in the contexts of many different reaction types. We look forward to extending our efforts toward other polyhalogenated natural products.
Experimental Section
General Experimental Protocols
All reactions were performed in oven-dried (140 °C) or flame-dried glassware under an atmosphere of dry argon unless otherwise noted. Reaction solvents including dichloromethane, toluene, N,N-dimethylformamide, and tetrahydrofuran were dried by percolation through a column packed with neutral alumina and a column packed with Q5 reactant, a supported copper catalyst for scavenging oxygen, under a positive pressure of argon. Dichloroethane (DCE) was heated to reflux over CaH2 for 3 h, distilled under argon, and stored over 3 Å molecular sieves prior to use. Column chromatography was performed using 60 Å (0.040–0.063 mm) mesh silica gel (SiO2). The following reagents were distilled from the indicated drying agents under argon prior to use: 2,2,6,6-tetramethylpiperidine (Na), allyl bromide (CaH2), triethylamine (CaH2), N,N-diisopropylethylamine (CaH2), trimethylsilyl chloride (TMSCl, CaH2), and ethylene diamine (CaH2). Silicon tetrachloride was heated at reflux for 2 h under a flow of argon and then distilled prior to use. Z-Selective Grubbs cycloadamantyl catalyst (60, Materia) was stored in the glovebox and used as received. Dimeric Denmark catalysts ((R,R)-41 and (S,S)-41, Obiter) were used as received and recovered by recrystallization from boiling benzene. (E)-2-Nonen-1-ol (25), boron trifluoride diethyl etherate, and tri-n-butyltin hydride were distilled prior to use. Tetraethylammonium chloride was heated to reflux in benzene with a Dean–Stark trap for 3 h and dried at 0.25 mmHg before use. Chlorine gas, Dess–Martin periodinane, diethylaluminum chloride, n-butyllithium, imidazole, iodine monochloride (1.0 M in CH2Cl2), camphorsulfonic acid (CSA), triethylborane (1.0 M in THF), chlorosulfonic acid, nickel(II) acetate tetrahydrate, sodium borohydride, magnesium (20–100 mesh), 1,2-dibromoethane, 1-bromo-10-undecene, N-chlorosuccinimide, t-butyldimethylsilyl chloride, and paraformaldehyde were used without further purification. Tetraethylammonium trichloride13 and (S)-BINAPO29 were prepared according to literature procedures.
1H and 13C spectra were referenced to residual solvent (CDCl3: 7.26 ppm, 1H, 77.00 ppm, 13C; CD3OD: 3.31 ppm, 1H, 49.00 ppm, 13C). Chemical shifts are reported in parts per million, and multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br s (broad singlet), and app (apparent). Coupling constants, J, are reported in Hertz. Infrared (IR) spectra were recorded on an FT-IR instrument on NaCl plates, and peaks are reported in cm–1. High-resolution mass spectra (HRMS) data are reported in the form of (m/z). Kugelrohr distillation temperatures reported are air bath temperatures (ABT). Visualization of analytical thin-layer chromatography was accomplished with UV(254) and potassium permanganate (KMnO4) or p-anisaldehyde staining solutions. Optical rotation data were obtained on a digital polarimeter and are reported as follows: concentration (c = g/100 mL) and solvent. Analytical gas chromatography (CSP-GC) was performed on a gas chromatograph equipped with a flame ionization detector and a dimethylated β-cyclodextrin (B-DM, 30 m) capillary column. The injector temperature and the detector temperature were 200 °C with a split ratio of approximately 100:1.
Synthesis of Danicalipin A
(±)-(2S,3R)-2,3-Dichloro-1-nonanol (26):(4c)
To a stirred solution of Et4NCl (6.63 g, 40.0 mmol)30 and (E)-2-nonen-1-ol (2.84 g, 20.0 mmol) in CH2Cl2 (60 mL) was bubbled Cl2 at 0 °C until the reaction mixture turned yellow (∼2 min). Ethylene was bubbled until the yellow color disappeared (∼2 min). The resulting colorless solution was diluted with CH2Cl2 (50 mL) and shaken with a mixture of saturated aqueous NaHCO3 solution (50 mL) and saturated aqueous Na2S2O3 solution (50 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (50 mL). The combined organic extracts were shaken with saturated aqueous NaCl solution (100 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by bulb-to-bulb distillation under reduced pressure (0.25 mmHg, ABT 123–126 °C) to afford (±)-26 (4.04 g, 95%, contained ∼1.5% of 1,3-dichloro-2-nonanol) as a colorless oil. Data for (±)-26: 1H NMR (600 MHz, CDCl3) δ 4.12 (app td, J = 8.7, 8.7, 2.7 Hz, 1H), 4.09–4.06 (m, 1H), 4.024 (d, J = 6.6 Hz, 1H), 4.017 (d, J = 6.6 Hz, 1H), 2.11–2.02 (m, 1H), 1.97 (app t, J = 6.9, 6.9 Hz, 1H), 1.82–1.75 (m, 1H), 1.64–1.54 (m, 1H), 1.48–1.39 (m, 1H), 1.39–1.23 (m, 6H), 0.89 (dd, J = 6.8, 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 66.4, 64.5, 61.8, 34.9, 31.6, 28.6, 25.5, 22.5, 14.0; IR (thin film) 3390, 2924, 2858, 1463, 1455, 1434, 1379, 1066, 725, 655 cm–1; HRMS (CI-TOF) m/z calcd for C9H1835Cl2ONH4 [M + NH4]+ 230.1078, found 230.1071.
(±)-(2S,3R)-2,3-Dichlorononanal (27):
To a stirred suspension of (±)-26 (2.13 g, 10.0 mmol) and NaHCO3 (2.52 g, 30.0 mmol) in CH2Cl2 (10 mL, saturated with H2O) was added Dess–Martin periodinane (6.36 g, 15.0 mmol) slowly over 1 min at 0 °C under air. After stirring for 10 min, the ice bath was removed and the reaction mixture was stirred at rt for 30 min prior to the addition of n-pentane (100 mL). The resulting mixture was filtered, washed with saturated aqueous NaHCO3 (50 mL), dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg) to give 27 (1.99 g) as a pale yellow oil. The crude material was used directly for the next reaction without further purification (∼3% 2-chloro-2-nonenal).31 Data for (±)-27: 1H NMR (600 MHz, CDCl3) δ 9.43 (d, J = 3.1 Hz, 1H), 4.25 (dd, J = 7.4, 3.1 Hz, 1H), 4.24–4.21 (m, 1H), 2.02–1.97 (m, 1H), 1.84–1.77 (m, 1H), 1.63–1.54 (m, 1H), 1.48–1.39 (m, 1H), 1.39–1.27 (m, 6H), 0.90 (dd, J = 6.9, 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 191.4, 64.9, 59.8, 34.0, 31.5, 28.5, 25.5, 22.5, 14.0; IR (thin film) 2926, 2858, 1734, 1458 cm–1; HRMS (CI-TOF) m/z calcd for C9H1535ClONH4 [M – HCl + NH4]+ 192.1155, found 192.1158.
(±)-(3S,4S,5S,6R)-5,6-Dichloro-3,4-epoxy-1-dodecene (30):
To a stirred solution of TMP (3.71 mL, 22.0 mmol) in THF (50 mL) was added n-BuLi (2.50 M in hexanes, 8.40 mL, 21.0 mmol) at −78 °C. After being stirred for 30 min, the LiTMP solution was cannulated into a solution of allyl bromide (1.82 mL, 21.0 mmol) and Et2AlCl (1.0 M in hexanes, 40.0 mL, 40.0 mmol) in THF (100 mL) at −78 °C over 5 min. The resulting solution was stored at −78 °C, while (±)-27 was prepared (see above). A solution of (±)-27 in THF (10 mL + rinsed with 5 mL × 2) was added dropwise over 15 min. After being stirred at −78 °C for 4 h, the reaction mixture was poured into an ice-cold 5 M aq NaOH solution (200 mL). Et4NCl (17 mg, 0.10 mmol) was added.32 The biphasic mixture was vigorously stirred at rt for 1 h prior to the dilution with n-pentane (100 mL) and filtration. The organic layer was separated, and the aqueous layer was extracted with n-pentane (100 mL × 2). The combined organic extracts were washed with saturated aqueous NH4Cl solution (200 mL × 2), dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 5.0 cm, l = 13.5 cm, n-pentane/CH2Cl2, 9/1, Rf = 0.29, p-anisaldehyde) and bulb-to-bulb distillation under reduced pressure (0.25 mmHg, ABT 123–127 °C) to give (±)-30 (1.89 g, 75% from (±)-26, 98:2 dr) as a colorless oil. Data for (±)-30: 1H NMR (600 MHz, CDCl3) δ 5.82 (ddd, J = 17.1, 10.6, 5.6 Hz, 1H), 5.52 (d, J = 17.1 Hz, 1H), 5.45 (d, J = 10.7 Hz, 1H), 4.21 (ddd, J = 9.4, 4.6, 4.1 Hz, 1H), 3.76 (dd, J = 9.0, 4.2 Hz, 1H), 3.57 (app t, J = 4.9, 4.9 Hz, 1H), 3.46 (dd, J = 9.0, 4.3 Hz, 1H), 1.98–1.87 (m, 2H), 1.65–1.59 (m, 1H), 1.47–1.39 (m, 1H), 1.36–1.26 (m, 6H), 0.89 (dd, J = 6.9, 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 130.4, 121.2, 65.2, 60.6, 57.7, 56.0, 34.5, 31.6, 28.6, 26.5, 22.5, 14.0; IR (thin film) 2956, 2928, 2858, 1463, 1455, 1250, 981, 934, 783, 668, 597 cm–1; HRMS (CI-TOF) m/z calcd for C12H2035Cl2ONH4 [M + NH4]+ 268.1235, found 268.1236.
(−)-(3S,4S,5S,6R)-5,6-Dichloro-3,4-epoxy-1-dodecene (30), (−)-(3S,4R,5R,6S)-3,5,6-trichloro-1-dodecen-4-ol (35):
To a stirred solution of (±)-30 (126 mg, 0.502 mmol) and (S,S)-41 (84 mg, 0.10 mmol) in CH2Cl2 (2.5 mL) were added i-Pr2NEt (9 μL, 0.05 mmol) and SiCl4 (57 μL, 0.50 mmol) at −78 °C. After 24 h, a solution of CH3OH/Et3N/CH2Cl2 (1/1/5, 4 mL) was added quickly at −78 °C. The resulting solution was vigorously stirred with a saturated aqueous NaHCO3 solution (20 mL) at rt for 2 h prior to filtration. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). (S,S)-41 was recovered from the residue by column chromatography (SiO2, ϕ = 2.2 cm, l = 7 cm, CH2Cl2/i-PrOH, 10/1, Rf = 0.37, UV). The fractions that contained 30 and 35 were combined and purified by column chromatography (SiO2, ϕ = 2.2 cm, l = 11 cm, n-pentane/CH2Cl2, 8/1 to 4/1, p-anisaldehyde) to give (−)-35 (70 mg, 49%, Rf = 0.12 in 8/1, 88.1:11.9 er) as a colorless oil and (−)-30 as a colorless oil, which was purified again by column chromatography (54 mg, 43%, Rf = 0.30 in 8/1, 2.7:97.3 er). Data for (−)-30: [α]D26 = −29.9 (c 1.00, CHCl3); GC (B-DM, 30 psi, 145 °C) tR 15.5 min (2.7%), 16.5 min (97.3%). Data for (−)-35: [α]D25 = −60.6 (c 1.00, CHCl3); GC (B-DM, 30 psi, 165 °C) tR 18.5 min (88.1%), 19.1 min (11.9%); 1H NMR (500 MHz, CDCl3) δ 6.03 (ddd, J = 16.9, 10.2, 7.7 Hz, 1H), 5.49 (d, J = 16.9 Hz, 1H), 5.35 (d, J = 10.2 Hz, 1H), 5.07 (d, J = 7.6 Hz, 1H), 4.51 (app dt, J = 10.5, 2.7, 2.7 Hz, 1H), 4.31 (dd, J = 9.4, 2.7 Hz, 1H), 3.89 (app td, J = 9.8, 9.8, 1.3 Hz, 1H), 2.23 (d, J = 9.9 Hz, 1H), 1.92–1.83 (m, 1H), 1.83–1.74 (m, 1H), 1.68–1.58 (m, 1H), 1.46–1.23 (m, 7H), 0.89 (app t, J = 6.6, 6.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 134.4, 119.6, 74.5, 66.5, 65.0, 62.5, 32.4, 31.6, 28.6, 26.5, 22.6, 14.0; IR (thin film) 3540, 2956, 2927, 2857, 1465, 1379, 1265, 1096, 1069, 987, 935 cm–1; HRMS (CI-TOF) m/z calcd for C12H2135Cl3ONH4 [M + NH4]+ 304.1002, found 304.1000.
(+)-(11Z,13S,14S,15S,16R)-1-tert-Butyldimethylsiloxy-2,2,15,16-tetrachloro-13,14-epoxy-11-docosene (10):(6b)
The solvents were bubbled with argon for 15 min before use. To a stirred solution of (−)-30 (52 mg, 0.21 mmol) and 71 (152 mg, 0.414 mmol) in DCE (210 μL) in a test tube (12 mm × 75 mm) was added a solution of 60 (39 mg, 0.062 mmol) in CH2Cl2 (210 μL) in three portions (0, 0.5, 1.0 h) at 35 °C while the reaction mixture was vigorously bubbled with argon (saturated with DCE).33 After being stirred at 35 °C with argon bubbling for an additional 2 h, the reaction mixture was cooled to rt, filtered through silica gel (ϕ = 2.2 cm, l = 9 cm, CH2Cl2, 40 mL), and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 3.8 cm, l = 15 cm, n-pentane/CH2Cl2, 8/1, Rf = 0.24, p-anisaldehyde) to give (+)-10 (35 mg, 29%, >20:1 = Z:E) as a colorless oil. Data for (+)-10: [α]D26 = +14.2 (c 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.86 (app dt, J = 10.9, 7.6 Hz, 7.6, 1H), 5.26–5.20 (m, 1H), 4.21 (ddd, J = 9.6, 4.4, 4.0 Hz, 1H), 3.92 (s, 2H), 3.76 (dd, J = 9.1, 4.0 Hz, 1H), 3.74 (dd, J = 7.9, 4.3 Hz, 1H), 3.44 (dd, J = 9.1, 4.2 Hz, 1H), 2.27–2.19 (m, 2H), 2.19–2.14 (m, 2H), 1.99–1.86 (m, 2H), 1.66–1.55 (m, 3H), 1.46–1.39 (m, 3H), 1.39–1.26 (m, 14H), 0.91 (s, 9H), 0.89 (app t, J = 6.9, 6.9 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 139.6, 121.5, 93.5, 72.1, 65.2, 61.5, 57.4, 52.5, 43.5, 34.3, 31.6, 29.29, 29.27, 29.26, 29.1, 29.0, 28.6, 28.1, 26.5, 25.7, 24.7, 22.5, 18.3, 14.0, −5.4.
(+)-(7R,8S,9S,10R,11Z)-22-tert-Butyldimethylsiloxy-7,8,10,21,21-pentachloro-11-docosen-9-ol (11):(6b)
To a stirred solution of (+)-10 (35 mg, 0.059 mmol) and Et4NCl (30 mg, 0.18 mmol) in CH2Cl2 (240 μL) was added BF3·OEt2 (15 μL, 0.12 mmol) at −78 °C. After being stirred for 1 h, the reaction mixture was poured into an ice-cold saturated aqueous NaHCO3 solution (10 mL). To the biphasic mixture were added CH2Cl2 (10 mL) and H2O (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 1.5 cm, l = 9 cm, n-pentane/CH2Cl2, 5/1 to 3/1 to 1/1, p-anisaldehyde) to give (+)-11 (27 mg, 73%, Rf = 0.25 in 3/1, >20:1 dr) as a colorless oil and SN2′ product 82 (9.6 mg, 26%, Rf = 0.33 and 0.24 in 1/1, 6:4 dr) as a colorless oil. Data for (+)-11: [α]D25 = +62.5 (c 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.73 (app t, J = 10.3, 10.3 Hz, 1H), 5.66 (app dt, J = 10.7, 7.4, 7.4 Hz, 1H), 5.38 (dd, J = 9.9, 1.7 Hz, 1H), 4.49 (app dt, J = 10.3, 2.9, 2.9 Hz, 1H), 4.29 (dd, J = 9.0, 3.1 Hz, 1H), 3.92 (s, 2H), 3.83–3.78 (m, 1H), 2.34 (d, J = 10.5 Hz, 1H), 2.21–2.09 (m, 4H), 1.91–1.83 (m, 1H), 1.83–1.76 (m, 1H), 1.67–1.62 (m, 1H), 1.61–1.56 (m, 2H), 1.46–1.36 (m, 3H), 1.36–1.25 (m, 14H), 0.91 (s, 9H), 0.89 (app t, J = 6.8, 6.8 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 135.7, 125.8, 93.5, 75.1, 66.8, 62.4, 60.1, 43.5, 32.5, 31.6, 29.3, 29.2, 29.1, 29.0 (2C), 28.6, 27.6, 26.5, 25.7, 24.7, 22.6, 18.3, 14.0, −5.4. Data for 82 (a 6:4 mixture of diastereomers): 1H NMR (600 MHz, CDCl3) δ 5.97–5.90 (m, 1H), 5.79 (ddd, J = 15.2, 13.9, 6.8, 1H), 4.77 (app td, J = 7.3, 7.3, 3.9 Hz, 0.4H), 4.72 (app td, J = 7.2, 7.2, 4.3 Hz, 0.6H), 4.39 (app quintet, J = 7.4, 7.4, 7.4, 7.4 Hz, 1H), 4.18 (ddd, J = 8.8, 6.7, 4.1 Hz, 1H), 3.92 (s, 2H), 3.91–3.88 (m, 1H), 2.20–2.14 (m, 2H), 2.13–2.04 (m, 2H), 1.89–1.73 (m, 3H), 1.65–1.51 (m, 3H), 1.50–1.37 (m, 3H), 1.37–1.23 (m, 14H), 0.91 (s, 9H), 0.89 (app t, J = 6.8, 6.8 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 136.0, 135.7, 128.22, 128.17, 93.5, 72.1, 72.0, 71.7, 69.4, 69.1, 61.9, 61.7, 61.6, 43.5, 38.4, 38.3, 34.7, 34.5, 31.6, 29.30, 29.26, 29.0, 28.9, 28.61, 28.60, 26.43, 26.35, 25.7, 25.3, 25.2, 24.7, 22.5, 18.3, 14.1, −5.4; IR (thin film) 3403, 2929, 2857, 1463, 1256, 1153, 1120, 970, 840, 780 cm–1.; HRMS (ESI-TOF) m/z calcd for C28H5335Cl5O2SiNa [M + Na]+ 647.2155, found 647.2143.
(−)-(11S,12R,13S,14R,15S,16R)-1-tert-Butyldimethylsiloxy-2,2,11,13,15,16-hexachloro-14-hydroxy-12-iododocosane (83):(6b)
To a stirred solution of (+)-11 (27 mg, 0.043 mmol) and imidazole (8.8 mg, 0.13 mmol) in CH2Cl2 (430 μL) was added TMSCl (11 μL, 0.086 mmol) at rt. After being stirred for 10 min, the reaction mixture was cooled to −78 °C and ICl (1.0 M in CH2Cl2, 215 μL, 0.215 mmol) was added. After being stirred for 20 min at −78 °C, a solution of CSA (100 mg, 0.43 mmol) in CH3OH (645 μL) was added and the cold bath was removed. After being stirred for 30 min, the brown solution was poured into a stirred mixture of saturated aqueous NaHCO3 solution (5 mL) and saturated aqueous Na2S2O3 solution (5 mL). The resulting colorless biphasic mixture was diluted with CH2Cl2 (10 mL) and H2O (10 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic extracts were washed with saturated aqueous NH4Cl solution (20 mL), and the aqueous layer was extracted with CH2Cl2 (10 mL × 2). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 1.1 cm, l = 5.5 cm, n-pentane/CH2Cl2, 5/1 to 3/1, Rf = 0.29 in 3/1, p-anisaldehyde) to give (−)-83 (28 mg, 82%, 95:5 dr) as a colorless oil. Data for (−)-83: [α]D26 = −7.6 (c 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.98 (d, J = 10.9 Hz, 1H), 4.72 (app t, J = 10.6, 10.6 Hz, 1H), 4.56 (dd, J = 10.9, 1.8 Hz, 1H), 4.48 (app dt, J = 10.5, 2.6, 2.6 Hz, 1H), 4.38 (dd, J = 9.8, 2.4 Hz, 1H), 3.92 (s, 2H), 3.75–3.71 (m, 1H), 2.19–2.16 (m, 2H), 2.14 (d, J = 11.3 Hz, 1H), 2.04–1.97 (m, 1H), 1.97–1.88 (m, 1H), 1.83–1.78 (m, 1H), 1.77–1.71 (m, 1H), 1.69–1.62 (m, 1H), 1.62–1.56 (m, 2H), 1.53–1.46 (m, 1H), 1.46–1.38 (m, 3H), 1.38–1.27 (m, 13H), 0.92 (s, 9H), 0.90 (app t, J = 6.8, 6.8 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 93.5, 74.6, 72.1, 66.3, 66.0, 62.9, 62.8, 43.5, 42.6, 40.6, 32.8, 31.6, 29.2 (2C), 29.0, 28.9, 28.6, 26.6, 26.2, 25.7, 24.7, 22.6, 18.3, 14.1, −5.3.
(+)-(11S,13S,14R,15S,16R)-1-tert-Butyldimethylsiloxy-2,2,11,13,15,16-hexachloro-14-hydroxydocosane (84):(6b)
Toluene was bubbled with argon for 20 min before use. To a stirred solution of (−)-83 (28 mg, 0.035 mmol) in toluene (355 μL) were added n-Bu3SnH (11 μL, 0.041 mmol, 99% pure by 1H NMR in C6D6)34 and Et3B (1.0 M in THF, 7 μL, 0.007 mmol) at −78 °C. After being stirred for 2 h at −78 °C, n-pentane (3.55 mL) was added and the resulting solution was concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 1.1 cm, l = 5.5 cm, n-pentane/CH2Cl2, 1/0 to 4/1, Rf = 0.25 in 4/1, p-anisaldehyde) to give (+)-84 (21.5 mg, 91%) as a colorless oil. Data for (+)-84: [α]D25 = +34.3 (c 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.96 (d, J = 10.3 Hz, 1H), 4.51 (app dt, J = 10.6, 2.4, 2.4 Hz, 1H), 4.30 (dd, J = 9.7, 2.4 Hz, 1H), 4.17–4.13 (m, 1H), 3.92 (s, 2H), 3.77 (app t, J = 10.7, 10.7 Hz, 1H), 2.35–2.28 (m, 1H), 2.20–2.16 (m, 2H), 2.16 (d, J = 11.6 Hz, 1H), 2.02–1.95 (m, 1H), 1.94–1.85 (m, 1H), 1.83–1.73 (m, 3H), 1.68–1.62 (m, 1H), 1.62–1.57 (m, 2H), 1.57–1.49 (m, 1H), 1.49–1.39 (m, 2H), 1.39–1.26 (m, 14H), 0.91 (s, 9H), 0.89 (app t, J = 6.8, 6.8 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 93.5, 75.1, 72.1, 66.5, 63.0, 62.7, 60.4, 44.3, 43.5, 38.7, 32.4, 31.6, 29.3, 29.2, 29.0 (2C), 28.6, 26.6, 26.2, 25.7, 24.7, 22.6, 18.3, 14.0, −5.3.
(+)-Danicalipin A Disodium Salt (1):(2k,5,4c,6b)
To a stirred solution of (+)-84 (21.5 mg, 0.0324 mmol) in CH2Cl2 (650 μL) was added ClSO3H (5 drops) via a Pasteur pipet at rt under air. After being stirred for 10 min, the reaction mixture was slowly poured into a vigorously stirred mixture of a saturated aqueous NaHCO3 solution (6.5 mL) and solid NaHCO3 (650 mg). The resulting heterogeneous mixture was diluted with EtOH (26 mL), filtered, and concentrated in vacuo (30 mmHg). The residue was suspended in THF (20 mL), filtered, and concentrated in vacuo (25 mmHg). The residue purified by column chromatography (SiO2, ϕ = 2.2 cm, l = 14.5 cm, CH2Cl2/CH3OH, 3/1, Rf = 0.38, p-anisaldehyde) to give (+)-1 (23.4 mg, 96%) as a colorless amorphous solid. Data for (+)-1: [α]D25 = +34.2 (c 2.34, CH3OH) (lit. [α]D26 +33.0 (c 0.40, CH3OH),4c [α]D28 +31.5 (c 0.25, CH3OH),5 [α]D25 +12.8 (c 0.2, CH3OH)2k); 1H NMR (600 MHz, CD3OD) δ 4.89 (d, J = 11.2 Hz, 1H), 4.75 (d, J = 10.7 Hz, 1H), 4.55 (d, J = 10.2 Hz, 1H), 4.45 (dd, J = 10.2, 1.5 Hz, 1H), 4.31 (s, 2H), 4.23–4.19 (m, 1H), 2.56–2.49 (m, 1H), 2.27–2.24 (m, 2H), 2.15–2.06 (m, 1H), 1.99–1.92 (m, 1H), 1.85–1.76 (m, 2H), 1.76–1.69 (m, 1H), 1.69–1.62 (m, 2H), 1.61–1.52 (m, 2H), 1.51–1.42 (m, 2H), 1.42–1.27 (m, 14H), 0.90 (app t, J = 6.9, 6.9 Hz, 3H); 13C NMR (126 MHz, CD3OD, 313 K) δ 91.3, 80.9, 75.6, 68.4, 63.3, 62.4, 62.2, 45.5, 45.1, 39.9, 33.5, 32.9, 30.4, 30.3, 30.07, 30.05, 30.0, 27.6, 27.4, 25.8, 23.6, 14.4. The analytical data for (+)-1 were in agreement with the data given in refs (2k), (4c), (5), and (6b).
Synthesis of Malhamensilipin A
(Z)-2-Undecen-1-ol (51):
To a stirred solution of Ni(OAc)2·4H2O (9.12 g, 36.7 mmol) in CH3OH (500 mL) was added NaBH4 (1.38 g, 36.7 mmol) portionwise over 5 min at 0 °C. The blue solution immediately turned black upon addition of NaBH4. After being stirred for an additional 5 min, the ice bath was removed and ethylene diamine (4.90 mL, 36.7 mmol) was added. After being stirred for 5 min, a solution of undec-2-yn-1-ol35 (24.7 g, 147 mmol) in CH3OH (230 mL) was added. The reaction mixture was quickly purged with H2 three times and stirred overnight under a balloon of H2 prior to the dilution with H2O (100 mL) and n-pentane (100 mL). After filtration through Celite, the organic layer was separated and the aqueous layer was extracted with n-pentane (100 mL × 3). The combined organic extracts were washed with H2O (50 mL) and saturated aqueous NaCl solution (50 mL), dried over MgSO4, filtered, and concentrated in vacuo (5 mmHg) to afford 51 (25.0 g, 98%) as a colorless oil. The crude material was used for the next reaction without any further purification. Data for 51: 1H NMR (600 MHz, CDCl3) δ 5.62–5.52 (m, 2H), 4.19 (d, J = 6.4 Hz, 2H), 2.07 (q, J = 7.2 Hz, 2H), 1.38–1.32 (m, 2H), 1.32–1.23 (m, 10H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 133.5, 128.4, 58.8, 32.0, 29.8, 29.6, 29.42, 29.38, 27.6, 22.8, 14.3; IR (thin film) 3347, 3938, 3857, 1015 cm–1; HRMS (CI-TOF) m/z calcd for C11H22ONH4 [M + NH4]+ 188.2014, found 188.2023.
(±)-(2S,3S)-2,3-Dichloro-1-undecanol (52):
To a stirred solution of (4.96 g, 29.1 mmol) in CH2Cl2 (70 mL) was added Et4NCl3 (13.8 g, 58.3 mmol) portionwise over 5 min at rt. After the yellow color disappeared over the course of 10 min, another portion of Et4NCl3 (6.89 g, 29.1 mmol) was added portionwise over 3 min. After being stirred for 30 min, the reaction mixture was poured into a mixture of saturated aqueous NaHCO3 solution (15 mL) and saturated aqueous Na2S2O3 solution (15 mL). The organic layer was separated, and the aqueous layer was extracted with hexanes (30 mL × 3). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo (5 mmHg). The residue was purified by column chromatography (150 mL of SiO2, 10% EtOAc/hexanes, Rf = 0.6 in 30% EtOAc/hexanes, KMnO4) to give (±)-52 (5.79 g, 82%) as a colorless oil. Data for (±)-52: 1H NMR (500 MHz, CDCl3) δ 4.22 (ddd, J = 8.1, 5.4, 2.6 Hz, 1H), 4.18 (m, 1H), 3.95 (ddd, J = 11.9, 7.7, 6.0 Hz, 1H), 3.89 (ddd, J = 12.1, 7.5, 5.6 Hz, 1H), 1.94 (dd, J = 7.7, 5.4 Hz, 1H), 1.90–1.84 (m, 2H), 1.57–1.51 (m, 1H), 1.44–1.22 (m, 11H), 0.88 (app t, J = 6.6, 6.6 Hz, 3H); 13C (126 MHz, CDCl3) δ 65.6, 64.7, 62.2, 35.3, 32.0, 29.5, 29.3, 29.1, 26.7, 22.8, 14.3; IR (thin film) 3363, 3923, 2855, 1455, 1041 cm–1; HRMS (CI-TOF) m/z calcd for C11H2235Cl2ONH4 [M + NH4]+ 258.1392, found 258.1401.
(±)-(2S,3S)-2,3-Dichloroundecanal (53):
To a stirred suspension of (±)-52 (2.20 g, 9.12 mmol) and NaHCO3 (2.30 g, 27.4 mmol) in CH2Cl2 (46 mL, saturated with H2O) was added Dess–Martin periodinane (5.80 g, 13.7 mmol) portionwise over 1 min at 0 °C under air. After being stirred for 5 min, the ice bath was removed and the reaction mixture was stirred at rt for 25 min prior to the addition of hexanes (20 mL) and saturated aqueous NaHCO3 solution (100 mL). The organic layer was separated, and the aqueous layer was extracted with hexanes (50 mL × 3). The combined organic extracts were filtered, dried over MgSO4, filtered, and concentrated in vacuo (5 mmHg) to give (±)-53 as a pale yellow oil. The crude material was generally used directly for the next reaction within 30 min and without further purification (it was often contaminated with up to 5% 2-chloro-2-undecenal). Data for (±)-53: 1H NMR (500 MHz, CDCl3) δ 9.55 (s, 1H), 4.40 (app s, 2H), 1.92–1.86 (m, 2H), 1.55–1.48 (m, 1H), 1.40–1.22 (m, 11H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 194.8, 67.0, 60.1, 35.1, 31.8, 29.3, 29.1, 28.8, 26.1, 22.6, 14.1; IR (thin film) 2927, 2856, 1736, 1465 cm–1; HRMS (ESI-TOF) m/z calcd for C11H1935ClONa [M – HCl + Na]+ 225.1022, found 225.1013.
(±)-(3S,4S,5S,6S)-5,6-Dichloro-3,4-epoxy-1-tetradecene (55):
To a stirred solution of TMP (3.39 mL, 20.1 mmol) in THF (46 mL) was added n-BuLi (2.47 M in hexanes, 7.75 mL, 19.2 mmol) at −78 °C. After being stirred for 15 min, the LiTMP solution was cannulated into a solution of allyl bromide (1.66 mL, 19.2 mmol) and Et2AlCl (1.0 M in hexanes, 36.5 mL, 36.5 mmol) in THF (46 mL) at −78 °C over 15 min. The resulting solution was stored at −78 °C, while (±)-53 was prepared (see above). A solution of (±)-53 in THF (10 mL + rinsed with 8 mL × 2) was added dropwise down the side of the flask. After being stirred at −78 °C for 5 h, the cooling bath was removed and a 6 M aq NaOH solution (100 mL) was added. After stirring vigorously for 1 h, the biphasic mixture was diluted with hexanes (100 mL) and shaken in a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with hexanes (100 mL × 3). The combined organic extracts were washed with saturated aqueous NaCl solution (50 mL × 3), filtered through silica gel (CH2Cl2, 300 mL), and concentrated in vacuo (5 mmHg). The residue was purified by column chromatography (500 mL of SiO2, 5% CH2Cl2/hexanes, Rf = 0.2, KMnO4) and bulb-to-bulb distillation under reduced pressure (0.1 mmHg, ABT 150 °C) to give (±)-55 as a colorless oil (1.83 g, 72% from (±)-52). Data for (±)-55: 1H NMR (500 MHz, CDCl3) δ 5.82 (ddd, J = 17.0, 10.6, 5.3 Hz, 1H), 5.49 (d, J = 17.2 Hz, 1H), 5.45 (d, J = 10.7 Hz, 1H), 4.26 (ddd, J = 8.3, 4.9, 2.7 Hz, 1H), 3.67 (dd, J = 8.9, 2.7 Hz, 1H), 3.64 (app t, J = 4.7 Hz, 1H), 3.54 (dd, J = 9.7, 4.7 Hz, 1H), 1.94 (app dtd, J = 14.2, 9.5, 4.8 Hz, 1H), 1.83, (app ddt, J = 14.0, 10.1, 5.4 Hz, 1H), 1.58–1.50, (m, 1H), 1.43–1.35 (m, 1H), 1.35–1.22 (m, 10H), 0.88 (app t, J = 6.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 130.5, 121.3, 63.5, 60.4, 58.6, 57.4, 35.8, 32.0, 29.5, 29.3, 29.1, 26.5, 22.8, 14.3; IR (thin film) 2926, 2855, 932 cm–1; HRMS (CI-TOF) m/z calcd for C14H2435Cl2ONH4 [M + NH4]+ 296.1548, found 296.1560.
(−)-(3S,4S,5S,6S)-5,6-Dichloro-3,4-epoxy-1-tetradecene (55), (+)-(3S,4R,5R,6R)-3,5,6-trichloro-1-tetradecen-4-ol (56):
To a stirred solution of (±)-55 (500 mg, 1.79 mmol) and (S)-BINAPO (234 mg, 0.358 mmol) in CH2Cl2 (36 mL) were added i-Pr2NEt (31.0 μL, 0.179 mmol) and SiCl4 (144 μL, 1.25 mmol) slowly at −78 °C. After 39 h, a solution of CH3OH/Et3N/CH2Cl2 (1/1/5, 5 mL) was added quickly at −78 °C. The resulting solution was vigorously stirred with a saturated aqueous NaHCO3 solution (20 mL) at rt for 2 h. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (20 mL × 3). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo (5 mmHg). The residue was purified by column chromatography (SiO2, 5% EtOAc/hexanes, KMnO4) to give (−)-55 (165 mg, 33%, 3.4:96.6 er, Rf = 0.7 in 10% EtOAc/hexanes) as a colorless oil and (+)-56 (367 mg, 65%, 71.4:28.6 er, Rf = 0.5 in 10% EtOAc/hexanes) as pale yellow crystals. The enantiopurity of the recovered reactant (−)-55 was measured after ring-opening chlorinolysis to form (−)-56. Data for (−)-55: [α]D25 = −23.5 (c 1.74, CHCl3); GC (B-DM, 30 psi, 180 °C) tR 23.9 min (2.6%), 25.1 min (97.4%). Data for (+)-56: mp 34.0–36.0 °C; [α]D24 = +1.6 (c 2.01, CHCl3); GC (B-DM, 30 psi, 180 °C) tR 23.7 min (71.4%), 25.3 min (28.6%); 1H NMR (600 MHz, CDCl3) δ 6.07 (ddd, J = 17.1, 10.3, 7.4 Hz, 1H), 5.49 (dd, J = 16.1, 1.0 Hz, 1H), 5.35 (dd, J = 10.3, 0.8 Hz, 1H), 5.10 (dd, J = 7.4, 1.0 Hz, 1H), 4.56 (ddd, J = 9.0, 5.2, 1.4 Hz, 1H), 4.10 (dd, J = 9.4, 1.5 Hz, 1H), 4.05 (dd, J = 9.1, 1.1 Hz, 1H), 2.18 (d, J = 8.8 Hz, 1H), 2.00 (app dtd, J = 14.0, 10.0, 4.7 Hz, 1H), 1.80 (app ddt, J = 15.5, 10.7, 5.5 Hz, 1H), 1.58–1.51 (m, 1H), 1.45–1.38 (m, 1H), 1.36–1.24 (m, 10H), 0.89 (app t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 134.9, 119.5, 74.5, 64.7, 64.5, 61.7, 36.5, 32.0, 29.5, 29.3, 29.2, 26.7, 22.8, 14.3; IR (thin film) 3390, 2925, 2855, 933 cm–1; HRMS (ESI-TOF) m/z calcd for C14H2535Cl4O [M + Cl]− 349.0659, found 349.0665.
(+)-(11Z,13S,14S,15S,16S)-1-tert-Butyldimethylsiloxy-2,2,15,16-tetrachloro-13,14-epoxy-11-tetracosene (76):
The solvents were bubbled with argon for 15 min before use. To a stirred solution of (−)-55 (134 mg, 0.481 mmol) and 71 (530 mg, 1.44 mmol) in DCE (480 μL) was added a solution of 60 (91.3 mg, 0.144 mmol) in CH2Cl2 (600 μL) in six portions (0, 15, 30, 45, 60, 75 min) at 35 °C while the reaction mixture was vigorously bubbled with argon (saturated with DCE).6 After being stirred at 35 °C with argon bubbling for an additional 105 min, the reaction mixture was cooled to rt, filtered through a plug of silica gel (CH2Cl2, 10 mL), and concentrated in vacuo (5 mmHg). The residue was purified via column chromatography (140 mL of SiO2, 5% CH2Cl2/hexanes, Rf = 0.23 in 10% CH2Cl2/hexanes) to give (+)-76 (57.1 mg, 19%, >20:1 = Z:E) as a colorless oil. Data for (+)-76: [α]D25 = +0.088 (c 2.65, CHCl3); 1H NMR (499 MHz, CDCl3) δ 5.86 (app td, J = 8.9, 8.4 Hz, 1H), 5.22 (app t, J = 8.7 Hz, 1H), 4.29–4.24 (m, 1H), 3.92 (s, 2H), 3.82–3.77 (m, 1H), 3.65 (d, J = 8.9 Hz, 1H), 3.52 (dd, J = 8.0, 2.2 Hz, 1H), 2.22 (app q, J = 7.2 Hz, 2H), 2.19–2.15 (m, 2H), 1.98–1.89 (m, 1H), 1.86–1.78 (m, 1H), 1.62–1.52 (m, 3H), 1.45–1.39 (m, 3H), 1.37–1.23 (m, 18H), 0.91 (s, 9H), 0.88 (app t, J = 6.3 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 139.8, 121.6, 93.7, 72.3, 63.5, 31.3, 58.3, 54.0, 43.7, 35.9, 32.0, 29.5, 29.45, 29.43 (2C), 29.32, 29.28, 29.18, 29.12, 28.4, 26.5, 25.9, 24.9, 22.8, 18.4, 14.2, −5.2; IR (thin film) 2927, 2855, 1119, 939, 779 cm–1; HRMS (ESI-TOF) m/z calcd for C30H56O235Cl4SiNa [M + Na]+ 639.2701, found 639.2719.
Preparation of Alkene Cross Metathesis Partner for Danicalipin A and Malhamensilipin A
11-Dodecenal (68):(36)
To a flask containing magnesium (3.43 g, 141 mmol) in THF (10 mL) was added 1,2-dibromoethane (275 μL, 3.19 mmol) slowly. The mixture was allowed to sit at rt until gray precipitate formed. After dilution with additional THF (70 mL), a solution of 1-bromo-10-undecene (10.0 mL, 45.6 mmol) in THF (20 mL) was added over 1 h via a syringe pump. After being stirred for 1 h, the mixture was cooled to 0 °C and allowed to settle. The liquid phase was transferred via a cannula to a rapidly stirred solution of DMF (53 mL, 684 mmol) and THF (53 mL) at 0 °C. After being stirred for 20 min at rt, the reaction mixture was diluted with hexanes (200 mL) and poured into 1 M aq HCl (200 mL). The organic layer was separated, and the aqueous layer was extracted with hexanes (200 mL × 3). The combined organic extracts were washed with brine (100 mL), dried over MgSO4, filtered, and concentrated in vacuo (5 mmHg). The residue was purified by column chromatography (300 mL of SiO2, 5% EtOAc in hexanes) to afford 68 (6.33 g, 76%) as a colorless oil. Data for 68: 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 5.81 (ddt, J = 17.0, 10.1, 6.7 Hz, 1H), 4.99 (dd, J = 17.0, 1.4 Hz, 1H), 4.92 (dd, J = 10.2, 0.8 Hz, 1H), 2.41 (td, J = 7.6, 1.7 Hz, 2H), 2.03 (app q, J = 7.1 Hz, 2H), 1.62 (tt, J = 7.3, 6.6 Hz, 2H), 1.39–1.35 (m, 2H), 1.33–1.25 (m, 10H); 13C NMR (126 MHz, CDCl3) δ 203.0, 139.2, 114.1, 43.9, 33.8, 29.4, 29.34, 29.31, 29.13, 29.07, 28.9, 22.1; IR (thin film) 2926, 2854, 2715, 1727 cm–1; HRMS (ESI-TOF) m/z calcd for C12H22ONa [M + Na]+ 205.1568, found 205.1561.
2,2-Dichloro-11-dodecenal (69):(4c)
To a flask containing t-butylamine (634 μL, 6.03 mmol) was added 11-dodecenal (68) (1.00 g, 5.49 mmol) dropwise at 0 °C. After being stirred at rt for 45 min, the cloudy reaction mixture was dried over K2CO3 (3.79 g, 27.4 mmol), filtered, and concentrated in vacuo (25 mmHg). The residue was purified by bulb-to-bulb distillation under reduced pressure (0.25 mmHg, ABT 128–135 °C) to give the corresponding t-butylimine4c (1.21 g, ∼92:8 imine:aldehyde) as a colorless oil. The t-butylimine was dissolved in CH2Cl2 (15 mL), and N-chlorosuccinimide (2.04 g, 15.3 mmol) was added at rt under air. After being stirred for 24 h, the reaction mixture was shaken with saturated aqueous Na2S2O3 solution. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with saturated aqueous NaCl solution, dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was diluted with hexanes, filtered, and concentrated in vacuo (25 mmHg) to give the corresponding α,α-dichloro-t-butylimine4c as a yellow oil (1.54 g, ∼94:6 dichloride:monochloride). The crude material was dissolved in THF (10 mL), and 6 M aq HCl (10 mL) was added at rt. The biphasic mixture was stirred for 2 h prior to dilution with Et2O. The organic layer was separated, and the aqueous layer was extracted with Et2O. The combined organic extracts were washed with saturated aqueous NaHCO3 solution, dried over MgSO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 2.2 cm, l = 7 cm, n-pentane/CH2Cl2, 2/1, Rf = ∼0.20, streaky, KMnO4) and bulb-to-bulb distillation under reduced pressure (0.25 mmHg, ABT 129–135 °C) to give 69 (1.01 g, 73% over three steps, ∼94% pure) as a colorless oil. Data for 69: 1H NMR (500 MHz, CDCl3) δ 9.25 (s, 1H), 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dd, J = 17.1, 1.9 Hz, 1H), 4.93 (dd, J = 10.2, 1.0 Hz, 1H), 2.31–2.23 (m, 2H), 2.04 (dd, J = 14.4, 6.9 Hz, 2H), 1.66–1.58 (m, 2H), 1.43–1.24 (m, 10H).
2,2-Dichloro-11-dodecen-1-ol (70):
To a stirred solution of 69 (1.00 g, 3.98 mmol) in ethanol (12 mL) was added NaBH4 (151 mg, 3.98 mmol) at 0 °C under air. After being stirred for 30 min at rt, 1 M aq HCl (12 mL) was added. The cloudy mixture was diluted with H2O and extracted with hexanes twice. The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo (25 mmHg). The residue was purified by column chromatography (SiO2, ϕ = 2.2 cm, l = 13 cm, n-pentane/CH2Cl2, 1/1, Rf = 0.29, p-anisaldehyde) to give 70 (934 mg, 93%) as a colorless oil. Data for 70: 1H NMR (500 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dd, J = 17.1, 1.5 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 3.90 (d, J = 7.6 Hz, 2H), 2.29 (t, J = 7.6 Hz, 1H), 2.24–2.18 (m, 2H), 2.04 (dd, J = 14.3, 6.9 Hz, 2H), 1.68–1.59 (m, 2H), 1.42–1.26 (m, 10H); 13C NMR (126 MHz, CDCl3) δ 139.1, 114.2, 94.7, 72.1, 43.5, 33.8, 29.28, 29.27, 29.02, 28.98, 28.8, 24.8; IR (thin film) 3484, 2926, 2854 cm–1; HRMS (CI-TOF) m/z calcd for C12H22O35Cl2NH4 [M + NH4]+ 270.1392, found 270.1390.
12-tert-Butyldimethylsiloxy-11,11-dichloro-1-dodecene (71):(4c)
To a stirred solution of 70 (348 mg, 1.37 mmol) and imidazole (187 mg, 2.75 mmol) in CH2Cl2 (2 mL) was added TBSCl (228 mg, 1.51 mmol) at rt. After being stirred for 48 h, the reaction mixture was diluted with CH2Cl2 (5 mL) and shaken with saturated aqueous NaHCO3 (5 mL). The organic layer was separated, and the aqueous layer was extracted with hexanes (10 mL × 3). The combined organic extracts were washed with saturated aqueous NaCl (3 mL), concentrated in vacuo (5 mmHg), and passed through a pad of silica gel (5% EtOAc in hexanes, 10 mL). The residue was purified by bulb-to-bulb distillation under reduced pressure (0.05 mmHg, ABT 170–180 °C) to afford 71 (453 mg, 90%) as a colorless oil. Data for 71: 1H NMR (500 MHz, CDCl3) δ 5.81 (ddt, J = 17.0, 10.1, 6.7 Hz, 1H), 4.99 (dd, J = 17.3, 1.7 Hz, 1H), 4.93 (dd, J = 10.2, 1.0 Hz, 1H), 3.92 (s, 2H), 2.15–2.19 (m, 2H), 2.04 (app q, J = 7.2 Hz, 2H), 1.55–1.62 (m, 2H), 1.27–1.40 (m, 10H), 0.91 (s, 9H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 139.2, 114.1, 93.5, 72.1, 43.5, 33.8, 29.30, 29.29, 29.1, 29.0, 28.9, 25.7, 24.7, 18.3, −5.4; IR (thin film) 2928, 2856, 1118, 838 cm–1; HRMS (CI-TOF) m/z calcd for C18H3635Cl2OSiNH4 [M + NH4]+ 384.2256, found 384.2257.
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM086483). J.S.C. is supported by a National Science Foundation Graduate Research Fellowship. We thank Materia for generous donation of metathesis catalyst 60, and Dr. Martin Schnermann, Dr. Paresma Patel, Dr. Vanessa Marx, and Prof. Robert Grubbs for assistance with the convergent metathesis step. We thank Prof. Scott Miller (Yale) and Dr. Matthew Tudge (Merck) for assistance with our attempts to resolved 2,3-dichloroalcohols. We acknowledge the intellectual contributions made to this project by Dr. D. Karl Bedke.
Supporting Information Available
Copies of NMR spectra for all new compounds and relevant chromatograms are available. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For reviews on the chlorosulfolipids, see:; a Bedke D. K.; Vanderwal C. D. Nat. Prod. Rep. 2011, 28, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nilewski C.; Carreira E. M. Eur. J. Org. Chem. 2012, 1685–1698. [Google Scholar]
- a Elovson J.; Vagelos P. R. Proc. Natl. Acad. Sci. U.S.A. 1969, 62, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Haines T. H.; Pousada M.; Stern B.; Mayers G. L. Biochem. J. 1969, 113, 565–566. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mayers G. L.; Pousada M.; Haines T. H. Biochemistry 1969, 8, 2981–2986. [DOI] [PubMed] [Google Scholar]; d Elovson J.; Vagelos P. R. Biochemistry 1970, 9, 3110–3126. [DOI] [PubMed] [Google Scholar]; e Haines T. H. Prog. Chem. Fats Lipids 1971, 11, 297–345. [Google Scholar]; f Haines T. H. Annu. Rev. Microbiol. 1973, 27, 403–412. [DOI] [PubMed] [Google Scholar]; g Chen J. L.; Proteau P. J.; Roberts M. A.; Gerwick W. H.; Slate D. L.; Lee R. H. J. Nat. Prod. 1994, 57, 524–527. [DOI] [PubMed] [Google Scholar]; h Ciminiello P.; Fattorusso E.; Forino M.; Magno S.; Di Rosa M.; Ianaro A.; Poletti R. J. Org. Chem. 2001, 66, 578–582. [DOI] [PubMed] [Google Scholar]; i Ciminiello P.; Dell’Aversano C.; Fattorusso E.; Forino M.; Di Rosa M.; Ianaro A.; Poletti R. J. Am. Chem. Soc. 2002, 124, 13114–13120. [DOI] [PubMed] [Google Scholar]; j Ciminiello P.; Dell’Aversano C.; Fattorusso E.; Forino M.; Magno S.; Di Meglio P.; Ianaro A.; Poletti R. Tetrahedron 2004, 60, 7093–7098. [Google Scholar]; k Kawahara T.; Kumaki Y.; Kamada T.; Ishii T.; Okino T. J. Org. Chem. 2009, 74, 6016–6024. [DOI] [PubMed] [Google Scholar]; l Chao C.-H.; Huang H.-C.; Wang G.-H.; Wen Z.-H.; Wang W.-H.; Chen I.-M.; Sheu J.-H. Chem. Pharm. Bull. 2010, 58, 944–946. [DOI] [PubMed] [Google Scholar]
- a Nilewski C.; Geisser R. W.; Carreira E. M. Nature 2009, 457, 573–576. [DOI] [PubMed] [Google Scholar]; b Nilewski C.; Geisser R. W.; Ebert M.-O.; Carreira E. M. J. Am. Chem. Soc. 2009, 131, 15866–15876. [DOI] [PubMed] [Google Scholar]; c Nilewski C.; Deprez N. R.; Fessard T. C.; Li D. B.; Geisser R. W.; Carreira E. M. Angew. Chem., Int. Ed. 2011, 50, 7940–7943. [DOI] [PubMed] [Google Scholar]; d Geisser R. W. D.Sc Dissertation, ETH Zürich, 2010. [Google Scholar]
- a Yoshimitsu T.; Fujumoto N.; Tanaka T. J. Org. Chem. 2009, 74, 696–702. [DOI] [PubMed] [Google Scholar]; b Yoshimitsu T.; Fujumoto N.; Nakatani R.; Kojima N.; Tanaka T. J. Org. Chem. 2010, 75, 5425–5437. [DOI] [PubMed] [Google Scholar]; c Yoshimitsu T.; Nakatani R.; Kobayashi A.; Tanaka T. Org. Lett. 2011, 13, 908–911. [DOI] [PubMed] [Google Scholar]
- Umezawa T.; Shibata M.; Kaneko K.; Okino T.; Matsuda F. Org. Lett. 2011, 13, 904–907. [DOI] [PubMed] [Google Scholar]
- a Shibuya G.; Kanady J.; Vanderwal C. D. J. Am. Chem. Soc. 2008, 130, 12514–12518. [DOI] [PubMed] [Google Scholar]; b Bedke D. K.; Shibuya G. M.; Pereira A.; Gerwick W. H.; Haines T. H.; Vanderwal C. D. J. Am. Chem. Soc. 2009, 131, 7570–7572. [DOI] [PubMed] [Google Scholar]; c Pereira A. R.; Byrum T.; Shibuya G. M.; Vanderwal C. D.; Gerwick W. H. J. Nat. Prod. 2010, 73, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bedke D. K.; Shibuya G. M.; Pereira A.; Gerwick W. H.; Vanderwal C. D. J. Am. Chem. Soc. 2010, 132, 2542–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chung W.-j.; Carlson J. S.; Bedke D. K.; Vanderwal C. D. Angew. Chem., Int. Ed. 2013, 52, 10052–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chung W.-j.; Vanderwal C. D.. Acc. Chem. Res. 2014, DOI: 10.1021/ar400246w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonyl additions to simple α-chloroaldehydes are often highly diastereoselective. See:Britton R.; Kang B. Nat. Prod. Rep. 2013, 30, 227–236. [DOI] [PubMed] [Google Scholar]
- The addition of organometallic nucleophiles to α-chloroaldehydes generally proceeds to afford the anti product, consistent with the polar Felkin–Anh model (hyperconjugative stabilization of the transition state/steric control), though the Cornforth model (dipole minimization/steric control) also accounts for the stereochemistry of the products. For an excellent discussion and lead reference, see:Cee V. J.; Cramer C. J.; Evans D. A. J. Am. Chem. Soc. 2006, 128, 2920–2930. [DOI] [PubMed] [Google Scholar]
- The corresponding chloroallylaluminum reagents have been previously reported:Hosomi A.; Kohra S.; Tominaga Y.; Ando M.; Sakurai H. Chem. Pharm. Bull. 1987, 35, 3058–3061. [Google Scholar]
- a Hu S.; Jayaraman S.; Oehlschlager A. C. J. Org. Chem. 1996, 61, 7513–7520. [DOI] [PubMed] [Google Scholar]; b Hu S.; Jayaraman S.; Oehlschlager A. C. J. Org. Chem. 1998, 63, 8843–8849. [Google Scholar]
- a Nicolaou K. C.; Simmons N. L.; Ying Y.; Heretsch P. M.; Chen J. S. J. Am. Chem. Soc. 2011, 133, 8134–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]; For enantioselective dibromination of alkenes, see:; b Hu D. X.; Shibuya G. M.; Burns N. Z. J. Am. Chem. Soc. 2013, 135, 12960–12963. [DOI] [PubMed] [Google Scholar]
- Halpern O. S. M.S. Dissertation, UC Irvine, 2008. [Google Scholar]
- Schlama T.; Gabriel K.; Gouverneur V.; Mioskowski C. Angew. Chem., Int. Ed. 1997, 36, 2341–2344. [Google Scholar]
- Copeland G. T.; Miller S. J. J. Am. Chem. Soc. 2001, 123, 6496–6502. [DOI] [PubMed] [Google Scholar]
- a Nordin O.; Nguyen B.-V.; Vörde C.; Hedenström E.; Högberg H.-E. J. Chem. Soc., Perkin Trans. 1 2000, 367–376and references therein. [Google Scholar]; For a review on lipase-catalyzed kinetic resolution, see:; b Ghanem A.; Aboul-Enein H. Y. Chirality 2004, 17, 1–15. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Endo T.; Ueno M. Angew. Chem., Int. Ed. 2011, 50, 12262–12265. [DOI] [PubMed] [Google Scholar]
- a Martin V. S.; Woodard S. S.; Katsuki T.; Yamada Y.; Ikeda M.; Sharpless K. B. J. Am. Chem. Soc. 1981, 103, 6237–6240. [Google Scholar]; b Kagan H. B.; Fiaud J. C. In Topics in Stereochemistry; Eliel E. L., Fiaud J. C., Eds.; Wiley: New York, 1988; Vol. 79, pp 249–330. [Google Scholar]; c Hoveyda A. H.; Didiuk M. T. Curr. Org. Chem. 1998, 2, 537–574. [Google Scholar]; d Cook G. R. Curr. Org. Chem. 2000, 4, 869–885. [Google Scholar]; e Keith J. M.; Larrow J. F.; Jacobsen E. N. Adv. Synth. Catal. 2001, 343, 5–26. [Google Scholar]; f Vedejs E.; Jure M. Angew. Chem., Int. Ed. 2005, 44, 3974–4001. [DOI] [PubMed] [Google Scholar]
- a Ready J. M.; Jacobsen E. N. J. Am. Chem. Soc. 2001, 123, 2687–2688. [DOI] [PubMed] [Google Scholar]; b Ready J. M.; Jacobsen E. N. Angew. Chem., Int. Ed. 2002, 41, 1374–1377. [DOI] [PubMed] [Google Scholar]; c White D. E.; Jacobsen E. N. Tetrahedron: Asymmetry 2003, 14, 3633–3638. [Google Scholar]; d Birrell J. A.; Jacobsen E. N. Org. Lett. 2013, 15, 2895–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Denmark S. E.; Barsanti P. A.; Wong K.-T.; Stavenger R. A. J. Org. Chem. 1998, 63, 2428–2429. [DOI] [PubMed] [Google Scholar]; b Denmark S. E.; Barsanti P. A.; Beutner G. L.; Wilson T. W. Adv. Synth. Catal. 2007, 349, 567–582. [Google Scholar]; c Denmark S. E.; Beutner G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. [DOI] [PubMed] [Google Scholar]
- Tarasevych A. V.; Sorochinsky A. E.; Kukhar V. P.; Guillemin J.-C. Origins Life Evol. Biospheres 2013, 43, 129–135and references therein. [DOI] [PubMed] [Google Scholar]
- For the first application of Z-selective alkene cross metathesis in natural product synthesis, see:Meek S. J.; O’Brien R. V.; Llaveria J.; Schrock R. R.; Hoveyda A. H. Nature 2011, 471, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Endo K.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Keitz B. K.; Endo K.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2011, 133, 9686–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Keitz B. K.; Endo K.; Patel P. R.; Herbert M. B.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464–1467. [DOI] [PubMed] [Google Scholar]; e Keitz B. K.; Fedorov A.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 2040–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Herbert M. B.; Lan Y.; Keitz B. K.; Liu P.; Endo K.; Day M. W.; Houk K. N.; Grubbs R. H. J. Am. Chem. Soc. 2012, 134, 7861–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Herbert M. B.; Marx V. M.; Pederson R. L.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Marx V. M.; Herbert M. B.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Rosebrugh L. E.; Herbert M. B.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Miyazaki H.; Herbert M. B.; Liu P.; Dong X.; Xu X.; Keitz B. K.; Ung T.; Mkrtumyan G.; Houk K. N.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 5848–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Cannon J. S.; Grubbs R. H. Angew. Chem., Int. Ed. 2013, 52, 9001–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Rosebrugh L. E.; Marx V. M.; Keitz B. K.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10032–10035. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Hartung J.; Grubbs R. H. J. Am. Chem. Soc. 2013, 135, 10183–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Endo K.; Herbert M. B.; Grubbs R. H. Organometallics 2013, 32, 5128–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Quigley B. L.; Grubbs R. H. Chem. Sci. 2014, 5, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odinokov V. N.; Ishmuratov G. Y.; Vakhidov R. R.; Berg A. A.; Muslukhov R. R.; Tolstikov G. A. Chem. Nat. Compd. 1992, 28, 237–240. [Google Scholar]
- Samojłowicz C.; Bieniek M.; Zarecki A.; Kadyrovb R.; Grela K. Chem. Commun. 2008, 44, 6282–6284. [DOI] [PubMed] [Google Scholar]
- Hong S. H.; Sander D. P.; Lee C. W.; Grubbs R. H. J. Am. Chem. Soc. 2005, 127, 17160–17161. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Langemann K. J. Am. Chem. Soc. 1997, 119, 9130–9136. [Google Scholar]
- Wdowik T.; Samojłowicz C.; Jawiczuk M.; Malińska M.; Woźniakb K.; Grela K. Chem. Commun. 2013, 49, 674–676. [DOI] [PubMed] [Google Scholar]
- Kotani S.; Hashimoto S.; Nakajima M. Tetrahedron 2007, 63, 3122–3132. [Google Scholar]
- At the initial stage of optimization, a catalytic amount of Et4NCl was employed as an indicator of the completion of the reaction. After all the alkene substrate is consumed, Et4NCl3 is formed with an excess of Cl2 and shows deep yellow color as a sign of completion. In the experiment described here, an excess of Et4NCl was employed to suppress the formation of oligomeric side products, which is usually problematic in the absence of an excess of exogenous Cl– source.
- An NMR sample of crude (±)-27 showed no noticeable further decomposition after standing at rt overnight.
- Although the epoxide formation can proceed without a phase transfer catalyst, the addition of a catalytic amount of Et4NCl facilitates the epoxide formation and ensures complete conversion.
- The vinyl epoxide decomposed significantly during reactions without argon bubbling.
- In the presence of an excess of n-Bu3SnH, dechlorination of one of the gem-dichlorides was often observed.
- Cabezas J. A.; Oehlschlager A. C. Synthesis 1999, 1, 107–111. [Google Scholar]
- Odinokov V. N.; Ishmuratov G. Yu.; Vakhidov R. R.; Berg A. A.; Muslukhov R. R.; Tolstikov G. A. Chem. Nat. Compd. 1992, 28, 237–240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.