

Abstract
Gene targeting provides a powerful tool to modify endogenous loci to contain specific mutations, insertions and deletions. Precise allele replacement, with no other chromosomal changes (e.g., insertion of selectable markers or heterologous promoters), maintains physiologically relevant context. Established methods for precise allele replacement in fission yeast employ two successive rounds of transformation and homologous recombination and require genotyping at each step. The relative efficiency of homologous recombination is low and a high rate of false positives during the second round of gene targeting further complicates matters. We report that pop-in, pop-out allele replacement circumvents these problems. We present data for 39 different allele replacements, involving simple and complex modifications at seven different target loci, that illustrate the power and utility of the approach. We also developed and validated a rapid, efficient process for precise allele replacement that requires only one round each of transformation and genotyping. We show that this process can be applied in population scale to an individual target locus, without genotyping, to identify clones with an altered phenotype (targeted forward genetics). It is therefore suitable for saturating, in situ, locus-specific mutation screens (e.g., of essential or non-essential genes and regulatory DNA elements) within normal chromosomal context.
Keywords: genetics, reverse genetics, mutagenesis, chromosome
Introduction
Gene targeting by homologous recombination allows scientists to modify chromosomal DNA sequences to engineer deletions, insertions and point mutations. Such reverse genetics provides powerful tools to dissect the structure and function of regulatory DNA elements, RNAs and proteins. The optimal approach involves precise allele replacement, with no other chromosomal changes (e.g., insertion of selectable markers or heterologous promoters), in order to maintain physiologically relevant context. Unfortunately, in most organisms precise allele replacement is difficult to achieve. We therefore developed a rapid, efficient process for precise allele replacement in fission yeast that can be used for reverse genetics —and remarkably —also for targeted forward genetics (saturating mutagenesis of discrete chromosomal elements in situ).
A widely used approach for precise allele replacement in fission yeast employs two sequential rounds of gene targeting [for recent examples see (Ahmed et al. 2010; Ivey et al. 2010; Singh et al. 2010; Steiner et al. 2011; Mudge et al. 2012)]. This approach takes advantage of the fact that one can select positively or negatively for the ura4+ gene, which encodes orotidine-5′-phosphate decarboxylase (Bach 1987). In the first round of gene targeting, a linear DNA fragment harboring a ura4+ cassette and, flanking each side of ura4+, regions of homology to the target locus is transformed into cells with a deletion at the endogenous ura4 locus (ura4-D18) (Grimm et al. 1988). Stable, uracil phototrophic (Ura+) transformants can arise by homologous recombination with the target locus (desired outcome) or by non-homologous integration of the ura4+ cassette elsewhere in the genome (Grimm and Kohli 1988; Grimm et al. 1988; Grallert et al. 1993; Tatebayashi et al. 1994; Davidson et al. 2004). Once ura4+ has been used to “mark” the target locus of interest, a second DNA fragment harboring the desired allelic changes (e.g., base pair substitutions) is transformed into the cells. Recombination with the chromosomal target simultaneously replaces the target allele and ejects the ura4+ cassette. Cells that have lost ura4+ can be selected for on media containing the pyrimidine analog 5-fluoroorotic acid (FOA), which kills cells that are ura4+.
Although this approach is used widely, it has two shortcomings. First, it requires two sequential rounds of gene targeting, which is inefficient in fission yeast due to a relatively high ratio of non-homologous to homologous integration events (Tatebayashi et al. 1994; Krawchuk and Wahls 1999; Davidson et al. 2004). Second, selection for recombinants in the second round of gene targeting is confounded further by FOAr colonies that arise from spontaneous mutations (in ura4+ or ura5+) whose frequency can be 60-fold higher than that of homologous targeting (Mudge et al. 2012). We describe how pop-in, pop-out allele replacement, originally developed for budding yeast [e.g., (Barton et al. 1990)], circumvents these problems. We show that this approach is more efficient and precise (hence, less expensive) than two-step methods applied routinely for fission yeast and that it can be used to engineer a wide variety of genomic modifications, such as untagged (clean) deletions, insertions encoding in-frame epitope fusions, and mutations that substitute specific amino acids in expressed proteins. We also developed and validated a streamlined approach for rapid and precise allele replacement that requires only one round each of transformation and genotyping (Fig. 1). This approach is suitable for additional applications, such as saturating mutation screens of essential and non-essential genes in situ.
Fig. 1.
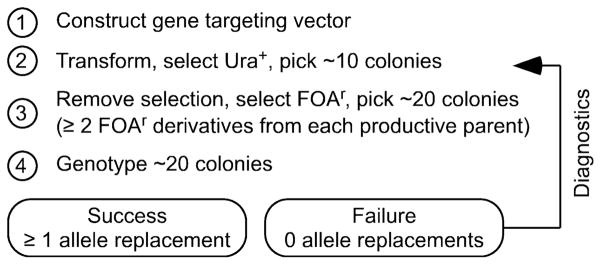
Streamlined approach for rapid and precise allele replacement. This overview of methodology and work flow also illustrates key advantages of the system: the process requires only one round each of transformation and genotyping. The streamlined approach has been validated using multiple different gene targeting vectors (nine successes and zero failures) with an observed allele replacement efficiency range of 24%–58%.
Materials and Methods
Yeast and molecular biology
Culture media, culture conditions and genotyping were as described previously (Gutz et al. 1974; Forsburg and Rhind 2006; Gao et al. 2008; Kan et al. 2011). Standard recombinant DNA methods were used to construct gene targeting vectors, which are described in the Results section and are available upon request. Methods for site-directed mutagenesis by inverse PCR and by recombinant PCR were as described (Krawchuk and Wahls 1999; Geiser et al. 2001; Sharif et al. 2002; Heckman and Pease 2007; Bryksin and Matsumura 2010; Kan et al. 2011). In some cases we used the QuikChange site directed mutagenesis kit from Stratagene or the GeneTailor kit from Invitrogen.
Pop-in homologous recombination
Cells containing a 1.8 kilobase pair deletion of the ura4 locus (ura4-D18) (Grimm et al. 1988) were transformed with 1–5 μg of linearized gene targeting vectors (ura4+) using lithium acetate protocols (Ito et al. 1983; Sabatinos and Forsburg 2010). Serial dilutions of transformed cells were plated on minimal nitrogen base agar (NBA; 0.67% yeast nitrogen base, 1% glucose, 2% agar) without uracil (supplemented as required with other growth factors) and were incubated at 32 °C for 4–5 days to select for Ura+ colonies. The Ura+ colonies were streaked or patched at least one additional time on NBA to ensure the fidelity of selection. Clones were then genotyped by RFLP and DNA sequence analyses of PCR products as described in the text.
Pop-out homologous recombination
Cells containing a ura4+ cassette, flanked by wild-type and modified alleles of the tandem target locus, were plated on NBA lacking uracil (supplemented as required with other growth factors) and were incubated at 32 °C for 3 days to form Ura+ (FOAs) colonies. Single colonies were each inoculated into 10 ml of YEL and incubated at 32 °C for 16–20 hours (to a final OD595 of 2–4). Aliquots containing about 25,000, 75,000 and 150,000 cells were plated on YEA containing 1 mg/ml of FOA and were incubated for 3 days at 32 °C to select for cells in which the ura4+ gene had been lost (pop-out) or inactivated (mutations). The FOAr (Ura−) colonies were streaked or patched at least one additional time on FOA-containing YEA to ensure the fidelity of selection. Clones were then genotyped by RFLP and DNA sequence analyses of PCR products as described in the text.
Results
We describe sequentially the rationale and approach for steps of pop-in, pop-out allele replacement mediated by homologous recombination. Key findings include development of a streamlined approach (Fig. 1) with multiple applications for reverse and forward genetics (described subsequently).
A. Construction of gene targeting vectors
The gene targeting vector contains one region that shares no significant homology to DNA sequences in the fission yeast genome (denoted by light lines in Figs. 2–3) and one region that shares DNA sequence homology to the endogenous location that is targeted for modification (bold lines in Figs. 2–3). There is flexibility in the design of the targeting vector with regard to functional elements and their relative positions.
Fig. 2.

Precise allele replacement by pop-in, pop-out gene targeting. Alleles are different shades for visual reference and differ by discrete modifications (open vs. filled circle). (a) The targeting vector contains a wild-type ura4+ gene and target DNA with desired changes (e.g., yfg1-M), but lacks an ARS (origin of replication). The target genome lacks the ura4 gene (ura4-D18). A restriction endonuclease is used to cut the vector within the region of homology (DSB) to promote homologous recombination (×) with the endogenous target. For efficient allele replacement, subregions of homology (a, b and c) should each be at least 250 bp in length. (b) Stable Ura+ transformants arise by homologous recombination with the target to produce tandem copies, one wild-type and one modified, or by non-homologous integration elsewhere (not shown). (c–d) Homologous recombination (×) between tandem repeats excises the plasmid from the genome. Because the plasmid lacks an ARS it cannot replicate and is lost during cell divisions. Removal of selection for uracil prototrophy allows pop-out cells to survive, and subsequent plating on media with FOA selects for cells that have lost the plasmid. Plasmid excision/loss from recombination to one side of the modification leaves that modification in the genome (c), whereas excision from recombination to the other side leaves the wild-type allele in the genome (d).
Fig. 3.
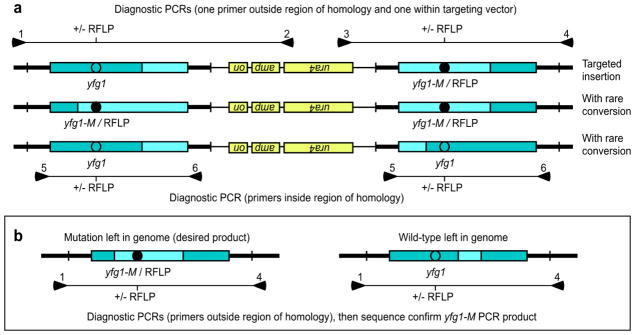
Confirmation of pop-in and pop-out genotypes. (a) Successful pop-in gene targeting produces a tandem integrant with one wild-type and one modified version of the target locus. These are readily identified by PCR with one primer outside of the region of homology used for targeting and one primer within non-homologous portions of the targeting vector (top, primer pairs 1–2, 3–4, or both). In rare cases, the locus can have two wild-type or two modified alleles (see text for details). These can be identified by the presence or absence of an RFLP in the diagnostic PCR products (top) or by RFLP analysis of PCR products amplified, simultaneously in one reaction, from both copies using primers inside the region of homology (bottom, primer pair 5–6). (b) Pop-out of the targeting vector leaves a single copy of the locus, which is confirmed by PCR using primers located outside the region of homology used for targeting (primer pair 1–4). The single copy can be either wild-type or modified and these can be distinguished by the presence or absence of the diagnostic RFLP.
The nonhomologous portion of each targeting vector contains an E. coli origin of replication and dominant selectable marker to support propagation in E. coli, plus the fission yeast gene ura4+. The ura4+ marker is portrayed to be in the nonhomologous portion of the targeting vector because the cells to be targeted for modification contain a deletion of the endogenous ura4 locus (ura4-D18). Any ura4+-containing plasmid can be used, provided that it lacks a fission yeast origin of replication (ARS).
The other portion of each targeting vector contains a modified DNA segment that is homologous to the endogenous DNA that is to be replaced (Fig. 2a). Non-replicating DNA transformed into fission yeast can integrate by non-homologous recombination, as well as by homologous recombination (desired outcome) (Grimm and Kohli 1988; Grallert et al. 1993; Davidson et al. 2004). In one study of 24 gene targeting constructs, 80 base pairs of homology flanking each side of the selectable marker produced an average targeting efficiency of 4% (96% non-homologous, off-target integrations), whereas constructs with at least 250 base pairs on each side yielded an average targeting efficiency of 58% (Krawchuk and Wahls 1999). During pop-in, pop-out allele replacement, the homologous recombination events can occur within one of three different subregions of homology (a, b and c in Fig. 2a), relative to the positions of the modification and a dsDNA break (DSB) used to promote recombination during pop-in. Correspondingly, to maximize efficient allele replacement, these three subregions of homology should each be at least 250 base pairs in length. Longer stretches of homology (recommended) might improve efficiency and shorter lengths can work, at reduced efficiency, but carry additional risks associated with the initiation and resolution of recombination events (examples provided below).
For the sake of illustration, our diagrams (Figs. 2–3) portray the open reading frame of “your favorite gene 1” (yfg1). However, precise allele replacement is applicable to other chromosomal features, such as loci that express non-coding RNAs or regulatory elements outside of coding regions. Data illustrating this flexibility of target choice and frequencies of pop-in, pop-out recombination events are presented below.
Our schematic diagrams portray genetic changes as allelic, simple point mutations (e.g., filled circles in Figs. 2–3) for the sake of simplicity and in order to depict most clearly their fates. However, other types of modifications, such as deletions and DNA insertions encoding in-frame epitope tags, can also be employed. The various types of potential changes are all functionally equivalent with regard to mechanisms and efficiencies of pop-in, pop-out allele replacement (data provided below) and are thus conceptually equivalent to the point mutation examples.
Standard recombinant DNA methods are used to clone the target (homologous) region into the targeting vector either before or after mutagenesis. Similarly, established methods are used for PCR-based, in vitro mutagenesis. The Electronic Supplementary Material file contains additional details for the following:
Approach to subclone chromosomal DNA fragments that lack convenient restriction endonuclease (RE) sites.
Approaches to create specific modifications by inverse PCR (Geiser et al. 2001).
Approaches to create specific modifications by recombinant PCR (Krawchuk and Wahls 1999; Heckman and Pease 2007; Bryksin and Matsumura 2010).
Approach to create additional changes, such as restriction fragment length polymorphisms (RFLPs), to facilitate genotyping (Kan et al. 2011).
Approaches to reduce the frequency of off-target mutations during in vitro mutagenesis.
We used PCR-based approaches to engineer 39 different gene targeting vectors directed to seven different loci (Table 1). In every case we obtained successfully PCR products and E. coli transformants with the desired changes, although for about half of the constructs we had to optimize conditions for PCR. We often included an engineered RFLP for diagnostic purposes (e.g., Fig. S1 of the Electronic Supplementary Material file) and DNA sequencing of RFPL-bearing clones revealed two things. First, when a clone harbors a diagnostic RFLP, the sequences within that region are almost always correct (i.e., they match each of the base pair substitutions in PCR primers, not just those affecting the RFLP). Second, off-target mutations were fairly frequent (occurred in about half of clones) when we used proofreading-deficient polymerases, but were rare when we used polymerases with proofreading activity. These findings illustrate the value of including diagnostic RFLPs and of using a DNA polymerase that has proofreading activity, each of which reduces the number of candidate clones that must be sequenced.
Table 1.
Efficiencies of pop-in, pop-out allele replacement in fission yeast.
| Allele | Modification | Pop-in homologous recombination a | Pop-out homologous recombination b | ||
|---|---|---|---|---|---|
| Homology L + R (bp) | Efficiency (H/T) | Homology L + R (bp) | Efficiency (M/T) | ||
| rec12-R76A | 2 bp substitutions | 937 + 676 | 7/8 (88%) | 225 + 1386 | 3/16(19%) |
| rec12-D79A | 2 bp substitutions | 937 + 676 | 7/8 (88%) | 231 + 1377 | 2/16 (13%) |
| rec12-E83A | 1 bp substitution | 937 + 676 | 7/8 (88%) | 247 + 1365 | 2/8 (25%) |
| rec12-R94A | 3 bp substitutions | 937 + 676 | 8/8 (100%) | 279 + 1331 | 3/16 (19%) |
| rec12-D95A | 3 bp substitutions | 937 + 676 | 7/8 (88%) | 337 + 1271 | 2/8 (25%) |
| rec12-Y97F | 2 bp substitutions | 937 + 676 | 3/3(100%) | 345 + 1265 | 3/8 (38%) |
| rec12-E179A | 2 bp substitutions | 209 + 1404 | 3/3 (100%) | 904 + 707 | 7/16 (44%) |
| rec12-K201A | 2 bp substitutions | 209 + 1404 | 3/3 (100%) | 770 + 841 | 9/12 (75%) |
| rec12-R209A | 4 bp substitutions | 209 + 1404 | 3/3 (100%) | 745 + 864 | 6/8 (75%) |
| rec12-K210A | 3 bp substitutions | 209 + 1404 | 3/3 (100%) | 743 + 865 | 6/8 (75%) |
| rec12-K214A | 4 bp substitutions | 209 + 1404 | 3/3 (100%) | 730 + 879 | 6/8 (75%) |
| rec12-D229A | 3 bp substitutions | 209 + 1404 | 2/2 (100%) | 685 + 924 | 6/16(38%) |
| rec12-D231A | 1 bp substitution | 209 + 1404 | 2/2 (100%) | 680 + 932 | 3/8 (38%) |
| rec12-K242A | 2 bp substitutions | 209 + 1404 | 2/2 (100%) | 647 + 964 | 5/8 (63%) |
| rec12-K282A | 4 bp substitutions | 937 + 676 | 2/2 (100%) | 526 + 1083 | 10/15 (67%) |
| rec12-R283A | 4 bp substitutions | 937 + 676 | 2/2 (100%) | 523 + 1086 | 5/12 (42%) |
| rec12-D284A | 3 bp substitutions | 937 + 676 | 2/2 (100%) | 521 + 1087 | 2/9 (22%) |
| rec12-R304A | 4 bp substitutions | 937 + 676 | 2/2 (100%) | 460 + 1149 | 3/8 (38%) |
| rec12-E305A | 1 bp substitution | 937 + 676 | 2/2 (100%) | 458 + 1154 | 4/26 (15%) |
| ade6-K87stop | 1 bp substitutions | 2012 + 854 | 8/10 c (80%) | 1134 + 1731 | 46/107 (43%) |
| ade6-Gal4BS | 7 bp substitutions | 2012 + 854 | 5/10 c(50%) | 1134 + 1705 | 25/67 (37%) |
| ade6- Gal4Control | 7 bp substitutions | 2012 + 854 | 8/10 c(80%) | 1134 + 1702 | 41/104 (39%) |
| ade6-rib+-M26 | 57 bp insertion | 1756 + 1110 | 5/6 (83%) | 899 + 1854 | 3/10 (30%) |
| ade6-ribm-M26 | 57 bp insertion | 1756 + 1110 | 4/6 (67%) | 899 + 1854 | 7/19 (37%) |
| ade6-rib+- M375 | 57 bp insertion | 1756 + 1110 | 3/6 (50%) | 899 + 1857 | 12/25 (48%) |
| ade6-ribm- M375 | 57 bp insertion | 1756 + 1110 | 2/3 (67%) | 899 + 1857 | 8/14 (57%) |
| ade6-M26-rib+ | 55 bp insertion | 2012 + 854 | 3/6 c (50%) | 1011 + 1720 | 27/94 (29%) |
| ade6-M26-ribm | 55 bp insertion | 2012 + 854 | 5/6 c (83%) | 1011 + 1720 | 33/100 (33%) |
| ade6-M375- rib+ | 55 bp insertion | 2012 + 854 | 4/6 c (67%) | 1008 + 1720 | 24/80 (30%) |
| ade6-M375- ribm | 55 bp insertion | 2012 + 854 | 5/6 c (83%) | 1008 + 1720 | 24/82 (29%) |
| ade6-M26- DP2 | 84 bp deletion | 1928 + 854 | 3/6 c (50%) | 441 + 1854 | 23/65 (35%) |
| ade6-M26- DP7 | 64 bp deletion | 1928 + 854 | 5/8 c (83%) | 750 + 1854 | 58/122 (48%) |
| prl10-rib+ | 57 bp insertion | 1557 + 383 | 6/7 (86%) | 1299 + 641 | 14/30 (47%) |
| prl10-ribm | 57 bp insertion | 1557 + 383 | 6/8 (75%) | 1299 + 641 | 14/30 (47%) |
| prl34-rib+ | 57 bp insertion | 1251 + 334 | 5/7 (71%) | 677 + 908 | 23/30 (77%) |
| prl34-ribm | 57 bp insertion | 1251 + 334 | 6/7 (86%) | 677 + 908 | 16/30 (53%) |
| fbp1- ΔM26 | 2 bp substitutions | 235 + 775 | 29/30 (97%) | 478 + 528 | 8/16 (50%) |
| ctt1- ΔM26 | 3 bp substitutions | 411 + 409 | 16/16 (100%) | 266 + 550 | 0/38 d (0%) |
| ctt1- ΔM26 | 3 bp substitutions | 411 + 409 | 5/20 (25%) | 266 + 550 | 3/16 (18%) |
| leu1-D1::prA- lexA | 1080 bp replacement | 939 + 1090 | 5/14 (36%) | 741 + 1288 | 3/12 (25%) |
Homology lengths are for regions to the left (L) and right (R) of the DSB used to promote recombination during transformation (Fig. 2a). The efficiency is the fraction of stable Ura+ transformants arising from pop-in homologous recombination (H) with the desired genomic target, as opposed to non-homologous integration elsewhere in the genome (T, total genotyped). For targeting constructs with insertions, the inserted DNA is not included in the lengths of homology.
Homology lengths flanking the desired modification are ordered relative to the schematic diagram in Fig. 2c–2d, where excision by recombination to the “left” (L) leaves the modification in the genome and excision to the “right” (R) leaves the wild-type allele in the genome. The efficiency is the fraction of pop-out recombinants that leave the modified allele (M) in the genome, as opposed to leaving the wild-type allele in the genome (T, total genotyped). Homology lengths do not include the region modified (e.g., where 3 base pairs were substituted within a 5 base pair stretch), so total lengths do not match precisely those reported for pop-in events.
In these cases we skipped genotyping of Ura+ transformants, picked 6–10 independent Ura+ colonies, sent them through pop-out protocols, and genotyped multiple FOAr derivatives of each clone.
In this unusual case, the Ura+ transformant had a tandem duplication of the target locus flanking the ura4+ cassette (desired structure), but after pop-out protocols never left a modified allele in the genome. We subsequently determined that both copies of the tandem integrant were wild-type. A different Ura+ transformant, with one wild-type and one modified allele in the tandem integrant, yielded the expected products (next line of table).
B. Modification of the genomic target by pop-in homologous recombination
In order to promote homologous recombination, the gene targeting vector is digested with a RE that introduces a DSB within the region of homology. The DSB can be to the “right” (Fig. 2a) or “left” (not shown) of the modified region. The linearized vector is transformed into cells of genotype ura4-D18 and cells are plated on media lacking uracil. Since the targeting vector lacks an origin of replication (ARS), the generation of Ura+ transformants requires integration of the targeting vector into the genome. In the absence of homology to the ura4 locus, this can occur by homologous recombination at the target locus of interest or by non-homologous recombination elsewhere (Grimm and Kohli 1988; Grallert et al. 1993; Davidson et al. 2004). Successful gene targeting will produce a tandem duplication of the homologous region in the target genome, one wild-type and one harboring the engineered modification, flanking the ura4+ cassette (Fig. 2b).
To have highest confidence in the likelihood of successful pop-in, pop-out allele replacement, one can genotype Ura+ transformants to identify those with successful gene targeting (pop-in). This is accomplished by PCR with appropriately configured primer pairs (Fig. 3a). However, we show below that the genotyping of Ura+ transformants is not required to achieve successful allele replacement (section entitled “Development of a streamlined approach for rapid and precise allele replacement”, see also Fig. 1). One might also wish to confirm that the tandem duplication contains one wild-type and one modified allele (Fig. 3a). This step of genotyping is also optional because it becomes relevant only in rare cases where the pop-out procedure fails to leave the desired modification in the genome (see below).
Data illustrating the efficiency of pop-in gene targeting are provided in Table 1 (which includes homology lengths in each vector). For 39 different gene targeting constructs directed to seven different loci we genotyped a total of 276 Ura+ transformants. We observed an average gene targeting efficiency of 83% using constructs whose average length of homology flanking each side of the DSB was 993 base pairs. Notably, we obtained a high targeting efficiency (97% mean) at the rec12 locus, which is known to be difficult to target using other approaches (Krawchuk and Wahls 1999). We conclude that pop-in gene targeting by homologous recombination in fission yeast is efficient, relative to non-homologous integration elsewhere, and that it can be used to introduce with precision a variety of specific changes (including point mutations, deletions and insertions) at genomic loci of interest.
C. Excision of the targeting vector by pop-out homologous recombination
Pop-in recombinants harbor a tandem duplication of the target locus, one wild-type and one modified, flanking the ura4+ cassette (Fig. 2b). Such tandem duplications are inherently unstable because recombination between the tandem repeats causes the vector to pop out of the genome (Fig. 2c–2d). Since the vector has no ARS it cannot replicate as a free episome and is lost during subsequent cell divisions. Thus, if one takes a Ura+ pop-in colony and grows it in the absence of selection for uracil prototrophy, then cells with pop-out recombination events can accumulate. Subsequent plating on media that contains FOA kills cells that are ura4+ and hence selects for cells that have lost the targeting vector due to pop-out recombination events. Spontaneous mutations in ura4+ and ura5+ can also confer resistance to FOA, which is a major problem for two-step allele replacement (Mudge et al. 2012), but the impact of spontaneous mutations on pop-in, pop-out allele replacement is negligible (data below).
Pop-out mediated by recombination to one side of the modified allele will leave the modified allele in the genome (Fig. 2c), whereas pop-out recombination events to the other side will leave the wild-type allele in the chromosome (Fig. 2d). Clones with the desired outcome (modified allele left in the chromosome) are identified easily by using PCR and diagnostic RFLPs to genotype the target locus (Fig. 3b). The PCR products are then sequenced to confirm the desired genotype and to identify potential off-target mutations.
Data illustrating the efficiency of pop-out excision of the targeting vector and frequencies of two alternative outcomes are provided in Table 1. In each case, removal of selection for uracil prototrophy for a short period of time (typically one passage through 10 ml of liquid culture medium that contains uracil) led to the appearance of FOAr colonies upon subsequent plating. The frequency of FOAr colonies (approximately 0.1% median value) was several orders of magnitude higher than that expected for spontaneous mutations. Furthermore, in 98% of cases genotyped, each FOAr colony harbored only a single copy of the target locus (each clone had lost the tandem duplication and the intervening ura4+ cassette). The remaining FOAr clones (2%) can be ascribed either to a failure of diagnostic PCR or to spontaneous mutations, so the frequency due to spontaneous mutations is ≤2%. We conclude that pop-out excision by homologous recombination occurs efficiently in fission yeast and, unlike the case for two-step allele replacement procedures (Mudge et al. 2012), is not confounded by FOAr colonies that arise from spontaneous mutations.
With one exception (described below), each of the 39 different tandem integrants produced pop-out clones of two distinct genotypes, with either a wild-type or modified allele left in the chromosome (Table 1). This was expected from the experimental design and the two regions of homology within which pop-out recombination can occur (Fig. 2c–2d). The fraction of pop-out recombinants leaving a modified allele in the chromosome increased as the fractional length of “productive” homology was increased (Fig. 4). This response was linear (slope = 0.95 ± 0.17) and the mean frequency with which the modified allele was left in the genome (41%) was close to the predicted value (36%) based on length ratios of homology. We conclude that pop-out excision of the gene targeting vector by homologous recombination occurs with precision and that the two alternative outcomes occur in proportion to lengths of homology within which recombination can occur (Fig. 4). Thus, the success rate can be predicted accurately from homology length ratios and, correspondingly, this information can be used to optimize the design of targeting vectors.
Fig. 4.

Success rates of precise allele replacement are proportional to length ratios of homology. For pop-out recombination events, the fraction of homology in which “productive” events occur [Fig. 2c, (length a)/(total length)] is predicted to influence the frequency with which the modified allele is left in the genome. Plot shows the Expected and Observed Efficiencies derived from data in Table 1. Linear regression analysis confirmed the predicted correlation (slope = 0.95 ± 0.17; intercept = 7.7 ± 6.5; R2 = 0.45; P < 0.0001). Scatter of data points is attributable, at least in part, to low n for individual measurements (Table 1, last column).
We were intrigued by the behavior of one targeted modification, ctt1-ΔM26. We had confirmed the genomic structure of a Ura+ pop-in recombinant (tandem duplication flanking the ura4+ cassette) by diagnostic PCR (as in Fig. 3a, top sets of primers). Unexpectedly, after pop-out protocols none of 38 FOAr colonies that we genotyped had a modified allele in the genome (Table 1, footnote “d”). Pop-out recombination had occurred in every case, because the tandem duplication collapsed to unit length and the ura4+ cassette was ejected, but at the target locus each clone was wild-type.
We backed up one step and genotyped each copy of the tandem integrant and discovered that both copies were wild-type (Fig. 3a, bottom genotype). Thus, at some point during transformation and pop-in homologous recombination, the modified ctt1-ΔM26 allele in the gene targeting vector had undergone gene conversion to wild-type. This makes sense, given that linear DNA transformed into fission yeast is often resected (Tatebayashi et al. 1994; Davidson et al. 2004) and that the position of the DSB used to linearize the targeting vector was close to (141 base pairs away from) the position of the ctt1-ΔM26 modification in that vector. Moreover, there is precedence for gene conversion of plasmid sequences during homologous recombination with chromosomal templates. For example, before it was superceded by PCR, gap repair was used to copy genomic DNA into replicating plasmid shuttle vectors in order to identify the DNA sequences of genomic mutations (Szankasi et al. 1988).
Subsequently, we repeated pop-out protocols using a clone known to harbor both wild-type and modified (ctt1-ΔM26) alleles in the tandem integrant. As expected, we obtained pop-out recombinants with two distinct genotypes (single copy, either wild-type or modified) (Table 1).
Together, these and other findings (above) indicate that lengths of homology in the gene targeting vector are important because they can influence the efficiency, fidelity and outcome of recombination events during both stages of pop-in, pop-out allele replacement. Within any given gene targeting fragment, three subregions of homology are delineated by the positions of the desired modification and the DSB used to promote recombination (a, b and c in Fig. 2a). Published findings for one-step gene targeting (Krawchuk and Wahls 1999) and our results for pop-in, pop-out (Table 1, a, b and cFig. 4) suggest that each subregion should be at least 250 base pairs in length to maximize the likelihood of success. We emphasize that we have not systematically varied lengths of homology for a single test allele/locus combination and that, in most of our gene targeting constructs, we used stretches of homology much longer than 250 base pairs (Table 1).
D. Development of a streamlined approach for rapid and precise allele replacement
With targeting vectors, lengths of homology and experimental approaches described in this study, the efficiency of successful pop-in gene targeting is high (83% mean) (Table 1). Pop-out recombination is easily accomplished and, among the selected FOAr colonies, its frequency and precision approach unity (≥98%). The frequency with which the desired modification is left in the genome is high (41% mean) (Table 1) and, for individual constructs, can be predicted from homology length ratios (Fig. 4). Based on these findings and probability theory, we reasoned that one could avoid the time and expense of genotyping primary transformants and still achieve precise allele replacement.
For example, if successful gene targeting (pop-in) at a given locus occurs with 50% efficiency and one picks seven independent Ura+ transformants, then there would be an approximately 99% probability [P = 1 - (0.5)7] that at least one clone harbors the desired tandem duplication. Such clones can immediately (without genotyping) be subjected to pop-out procedures and within about a week a few FOAr derivatives from each parental clone can be genotyped by PCR (or colony PCR) for unambiguous, diagnostic changes in the genome (Fig. 3b). Hypothetically, this lottery has a favorable rate of return.
We tested this hypothesis for nine distinct allele replacement constructs targeted to the ade6+ locus (Table 1, footnote “c”). Wild-type cells form white colonies on media with limiting amounts of adenine, whereas ade6 mutants have a red colony phenotype that is easily scored (Gutz et al. 1974). For each gene targeting construct we picked between six and ten Ura+ transformants (white on limiting adenine), sent them through pop-out protocols, and for each transformant we analyzed multiple FOAr derivatives. For each gene targeting construct, between 40% (4/10) and 83% (5/6) of the independent Ura+ isolates were capable of producing at high frequency FOAr colonies of alternative colors (red or white) on media with limiting adenine. Subsequently, DNA sequencing revealed that the red colony phenotype was due to the planned genetic modifications at ade6 (desired outcome).
We conclude that pop-in, pop-out allele replacement can be achieved rapidly and efficiently without any requisite genotyping of pop-in candidates. In short, we developed and validated (with nine independent experiments) a streamlined approach for rapid and precise allele replacement that involves only one round each of transformation and genotyping (Fig. 1). Success was achieved in all nine attempts (zero failures) and, among each pool of FOAr colonies, the frequency of precise allele replacement was high (range of 24%–58%) (Table 1). Obviating the need to genotype primary transformants correspondingly reduces the time, effort, and expense of precise allele replacement.
If one uses the streamlined approach and fails to obtain precise allele replacements, the loss is nominal and the solution is simple (Fig. 1, diagnostics). Back up one step and genotype the Ura+ transformants to determine if any of them (A) harbor a tandem duplication of the target flanking the ura4+ cassette, and (B) contain one wild-type and one modified allele (Figs. 2b, 3a). Such diagnostics will reveal if the problem lies in the pop-in step or the pop-out step and will, additionally, suggest potential solutions (e.g., to check the design and integrity of the gene targeting vector or to genotype more candidate colonies).
E. Development of a process for locus specific, saturating mutagenesis in situ (targeted forward genetics)
Classical, forward genetic screens are based on the assumptions that mutations (spontaneous or induced) are distributed in a stochastic or nearly stochastic fashion throughout the genome and that one can screen enough clones to ensure, statistically, a high probability of interrogating mutations that fall within the element(s) of interest. The number of mutants required to saturate a screen at the level of individual base pairs is several orders of magnitudes higher than the number required to saturate at the level of genes. Consequently, high resolution saturating mutagenesis of discrete elements is not practical and the best surrogate approach involves episomal (artificial) systems [e.g., plasmid shuffle (Paluh and Clayton 1996; Kiely et al. 2000)]. If it were possible to sprinkle mutations specifically within discrete loci and not to other sites in the genome, then saturating mutagenesis of specific elements in situ would become far more efficient (~10,000-fold, depending on size of target element relative to genome size). This would make it possible to carry out high resolution analyses of the structure and function of discrete chromosomal elements (and encoded factors) within their normal, physiologically relevant context.
Our results revealed that the pop-in, pop-out approach can be applied in population scale to an individual target locus, without genotyping, to identify clones with an altered phenotype (preceding section). It can therefore be used for saturating genetic screens targeted precisely to discrete chromosomal elements in situ. Details on efficiency and specificity are provided below and in the Electronic Supplementary Material file.
At the stage of transformation, Ura+ colonies can arise by pop-in homologous recombination at the correct target (observed mean of 83%) or by non-homologous integration elsewhere (Table 1). However, only the correctly targeted integrations contain tandem repeats that can undergo pop-out homologous recombination to produce at high frequency FOAr colonies (≥50-fold observed difference, relative to spontaneous mutations in the ura4+ cassette or elsewhere). Thus, starting with a population of Ura+ transformants that are mixed for homologous (pop-in) and nonhomologous integrations of the ura4+ cassette, the subsequent selection for resistance to FOA provides a powerful enrichment for clones derived from the pop-in (desired) class of transformants (see Table S1 and section “E” of the Electronic Supplementary Material file).
In the population approach, the vast majority of FOAr clones arise from pop-in, pop-out recombination at the correct target locus, even if the initial gene targeting (pop-in) efficiency is low (Table S1). Consequently, specific chromosomal elements can be efficiently targeted for saturating mutagenesis by pop-in, pop-out — with very low background of off-target events (high specificity).
In principle such precisely targeted, locus-specific, in situ mutation screens of essential or non-essential genes (or other chromosomal elements) can be carried to saturation. Implementation is, however, subject to probability theory for pop-in, pop-out allele replacement. Our results revealed that the positions of de novo mutations in the targeting vector and ratio lengths of homology (a, b and c in Fig. 2a) will influence the frequency with which a given mutation is left in the genome (Fig. 4). Gene conversion near DSBs during pop-in recombination also comes into play (Fig 3a and Table 1, footnote “d”). We therefore recommend that the transforming DNA be linearized in two batches, each with a RE cut site in a different location. Experimental evidence-based modeling revealed that this approach will produce a nearly uniform frequency distribution of mutations, spanning the chromosomal element of interest, in the population of pop-out (FOAr) colonies (Fig. 5; for additional details see section “E” of the Electronic Supplementary Material file).
Fig. 5.

Precisely targeted, saturating mutagenesis in situ. Diagram shows homology between targeting vector and genome (bold line), a hypothetical region of interest (box), and positions of DSBs used to promote pop-in recombination. Plot displays calculated frequency distribution of mutations left in the genome after pop-in, pop-out recombination (sum of mutations introduced using DSB1 and DSB2 in separate batches). Plot assumes that: (A) population size is infinite; (B) mutations are randomly distributed in the gene targeting vector (region of homology); and (C) all mutations at a DSB are lost to gene conversion, with the effect diminishing over distance from DSB in the “DSB zone” (shaded). Recombination events initiating from DSB2 compensate partially for conversion (loss) of mutations near DSB1 and vice versa. Thus, by linearizing the gene targeting vector in two batches (DSB1 and DSB2), one can generate and recover genomic mutations spanning the element of interest, including within the zones of gene conversion.
The ability to produce a nearly uniform frequency distribution of mutations within the chromosomal element of interest minimizes the number of clones that must be screened to achieve saturation.
Discussion
Precise allele replacement introduces the desired modifications, and only the desired modifications, at the target locus of interest. We developed and validated a streamlined pop-in, pop-out approach for rapid and precise allele replacement that requires only one round each of transformation and genotyping (Fig. 1). This approach has several advantages over widely used two-step methods. First, it cuts in half the number of targeting vectors that must be constructed for a given modification. Second, half as many transformations are required. Third, half as many successful gene targeting events are required 1. Fourth, one can dispense with genotyping of primary transformants. Fifth, far fewer FOAr colonies must be genotyped because pop-out is frequent and precise, which essentially eliminates false-positives due to spontaneous mutations in ura4+ and ura5+. Each of these advantages is accompanied by savings in time and cost.
We documented that the approach can be used to generate a wide variety of genomic modifications, such as clean deletions, insertions encoding in-frame epitope fusions, mutations that substitute specific amino acids in expressed proteins, and changes that affect the structure and function of RNAs (Table 1). It is therefore of broad utility for diverse applications of reverse genetics. It also provides a powerful, novel tool for precisely targeted forward genetics (below).
For broader perspective, recombinase mediated cassette exchange (RMCE) (Thomson and Ow 2006; Watson et al. 2008) and PCR vectorette-based (VB) gene targeting (Bähler et al. 1998; Krawchuk and Wahls 1999) were developed, in large part, to bypass the high effort and low efficiency of two-step allele replacement. However, RMCE and VB gene targeting do not support precise allele replacement because each involves off-target changes (e.g., ectopic placement of cassettes; or co-integration of dominant selectable markers and, in many cases, heterologous promoters or 3′ regulatory elements). Such off-target changes can have confounding effects (e.g., due to inappropriate expression of a modified protein under study). In addition, RMCE and VB methods are of limited (VB) or no (RCME) utility for recursive gene targeting to modify multiple loci in the same strain. In contrast, pop-in, pop-out allele replacement involves no off-target changes and can be carried out recursively in the same strain. In essence, streamlined pop-in, pop-out (Figs. 1, 2) matches the precision of two-step allele replacement and the efficiencies of RMCE and VB gene targeting, but without the shortcomings of those three other approaches.
We also report that pop-in, pop-out allele replacement can be carried out in population scale (Table 1, footnote “c”) to sprinkle mutations into the target element of interest (Fig. 5). It can therefore be used for precisely targeted, saturating mutation screens of discrete chromosomal elements in situ. This “targeted forward genetics” can be applied to diverse chromosomal elements, including protein coding genes and regulatory DNA elements.
Even essential genes can be targeted for pop-in using haploid cells because a wild-type copy in the tandem duplication will typically complement the modified version. The homologous region of the targeting vector can be sprinkled with mutations at an average density of a few (ideally, one) mutations per DNA molecule. The resulting population of targeting vector can be used for pop-in, pop-out protocols and then FOAr colonies can be screened for phenotypes such as conditional lethality. This approach is similar to plasmid shuffle approaches [e.g., (Paluh and Clayton 1996; Kiely et al. 2000)] and involves a similar amount of effort, but has the added benefit of physiologically relevant context. The mutations will be expressed from the endogenous chromosomal locus (single copy), under control of native regulatory sequences, without any heterologous sequences such as selectable markers.
Regulatory DNA sequence motifs function within a chromosomal context and some motifs, such as those that activate meiotic recombination hotspots, can only be studied in the chromosome (Schuchert et al. 1991; Steiner et al. 2009; Steiner et al. 2011). Population scale, precisely targeted, in situ mutagenesis using a stretch of randomized base pairs can be used to discover candidate regulatory motifs (gain of function approach). Such gain of function screens have been validated using the more laborious two-step allele replacement method and an episomal reporter construct (Steiner et al. 2009). Reciprocally, to test and define at single base pair resolution the functionality of a candidate motif in the genome, the population of targeting vector can be engineered to contain each single base pair substitution spanning the region of interest (loss of function approach). This essentially recapitulates, but with defined substitutions in a more narrow region, the targeted forward genetics described above for genes.
In conclusion, pop-in, pop-out homologous recombination provides a straightforward (Figs. 1, 2) and efficient (Table 1) method for precise allele replacement in fission yeast. It is a powerful approach for reverse genetic analyses of specific chromosomal features within their native, physiologically relevant context—unencumbered by any other off-target changes and heterologies. It is, to our knowledge, also the first practical approach for precisely targeted, saturating, forward genetic screens of specific genomic elements in situ (Fig. 5).
Supplementary Material
Acknowledgments
We thank Charlie Albright for helpful suggestions and Michelle Krawchuk and Wallace Sharif for assistance with method development. This work was supported by a grants from the National Institutes of Health (GM81766) and the UAMS Graduate Student Research Fund. Core facilities were supported in part by a National Institutes of Health Translational Research Institute grant (TR000039).
Footnotes
These three comparisons are based on a single allele replacement at the target locus. If multiple alleles are targeted to the same locus, and if the same ura4+ transgene can be used for each allele replacement in the two-step method, then the relative effort (E) of the pop-in, pop-out approach to the two-step approach is approximated by the equation [E = (n)/(n+1)], where n is number of allele replacements.
Electronic supplementary material: The manuscript is accompanied by supplementary material, which is provide in a single PDF file.
References
- Ahmed S, Brickner DG, Light WH, Cajigas I, McDonough M, Froyshteter AB, Volpe T, Brickner JH. DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nat Cell Biol. 2010;12:111–118. doi: 10.1038/ncb2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ML. Cloning and expression of the OMP decarboxylase gene URA4 from Schizosaccharomyces pombe. Curr Genet. 1987;12:527–534. doi: 10.1007/BF00419562. [DOI] [PubMed] [Google Scholar]
- Bähler J, Wu J-Q, Longtine MS, Shah NG, McKenzie A, III, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Barton MC, Hoekstra MF, Emerson BM. Site-directed, recombination-mediated mutagenesis of a complex gene locus. Nucleic Acids Res. 1990;18:7349–7355. doi: 10.1093/nar/18.24.7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MK, Young NP, Glick GG, Wahls WP. Meiotic chromosome segregation mutants identified by insertional mutagenesis of fission yeast Schizosaccharomyces pombe; tandem-repeat, single-site integrations. Nucleic Acids Res. 2004;32:4400–4410. doi: 10.1093/nar/gkh767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL, Rhind N. Basic methods for fission yeast. Yeast. 2006;23:173–183. doi: 10.1002/yea.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Davidson MK, Wahls WP. Distinct regions of ATF/CREB proteins Atf1 and Pcr1 control recombination hotspot ade6-M26 and the osmotic stress response. Nucleic Acids Res. 2008;36:2838–2851. doi: 10.1093/nar/gkn037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M, Cebe R, Drewello D, Schmitz R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88–90. 92. doi: 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- Grallert B, Nurse P, Patterson TE. A study of integrative transformation in Schizosaccharomyces pombe. Mol Gen Genet. 1993;238:26–32. doi: 10.1007/BF00279526. [DOI] [PubMed] [Google Scholar]
- Grimm C, Kohli J. Observations on integrative transformation in Schizosaccharomyces pombe. Mol Gen Genet. 1988;215:87–93. doi: 10.1007/BF00331308. [DOI] [PubMed] [Google Scholar]
- Grimm C, Kohli J, Murray J, Maundrell K. Genetic engineering of Schizosaccharomyces pombe: a system for gene disruption and replacement using the ura4 gene as a selectable marker. Mol Gen Genet. 1988;215:81–86. doi: 10.1007/BF00331307. [DOI] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Loprieno N. Schizosaccharomyces pombe. In: King RC, editor. Handbook of Genetics. Plenum Press; New York: 1974. pp. 395–446. [Google Scholar]
- Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey FD, Taglia FX, Yang F, Lander MM, Kelly DA, Hoffman CS. Activated alleles of the Schizosaccharomyces pombe gpa2+ Galpha gene identify residues involved in GDP-GTP exchange. Eukaryot Cell. 2010;9:626–633. doi: 10.1128/EC.00010-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan F, Davidson MK, Wahls WP. Meiotic recombination protein Rec12: functional conservation, crossover homeostasis and early crossover/non-crossover decision. Nucleic Acids Res. 2011;39:1460–1472. doi: 10.1093/nar/gkq993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely J, Haase SB, Russell P, Leatherwood J. Functions of fission yeast orp2 in DNA replication and checkpoint control. Genetics. 2000;154:599–607. doi: 10.1093/genetics/154.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinner U, Schafer B. Genetic aspects of targeted insertion mutagenesis in yeasts. FEMS Microbiol Rev. 2004;28:201–223. doi: 10.1016/j.femsre.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Krawchuk MD, Wahls WP. High-efficiency gene targeting in Schizosaccharomyces pombe using a modular, PCR-based approach with long tracts of flanking homology. Yeast. 1999;14:1419–1427. doi: 10.1002/(SICI)1097-0061(19990930)15:13<1419::AID-YEA466>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge DK, Hofman CA, Lubinski TJ, Hofman CS. Use of a ura5+-lys7+ cassette to construct unmarked gene knock-ins in Schizosaccharomyces pombe. Curr Genet. 2012;58:59–64. doi: 10.1007/s00294-011-0360-4. [DOI] [PubMed] [Google Scholar]
- Paluh JL, Clayton DA. Mutational analysis of the gene for Schizosaccharomyces pombe RNase MRP RNA, mrp1, using plasmid shuffle by counterselection on canavanine. Yeast. 1996;12:1393–1405. doi: 10.1002/(SICI)1097-0061(199611)12:14%3C1393::AID-YEA29%3E3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Sabatinos SA, Forsburg SL. Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 2010;470:759–795. doi: 10.1016/S0076-6879(10)70032-X. [DOI] [PubMed] [Google Scholar]
- Schuchert P, Langsford M, Kaslin E, Kohli J. A specific DNA sequence is required for high frequency of recombination in the ade6 gene of fission yeast. EMBO J. 1991;10:2157–2163. doi: 10.1002/j.1460-2075.1991.tb07750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif WD, Glick GG, Davidson MK, Wahls WP. Distinct functions of S. pombe Rec12 (Spo11) protein and Rec12-dependent crossover recombination (chiasmata) in meiosis I; and a requirement for Rec12 in meiosis II. Cell Chromosome. 2002;1:1. doi: 10.1186/1475-9268-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Sabatinos S, Forsburg S, Bastia D. Regulation of replication termination by Reb1 protein-mediated action at a distance. Cell. 2010;142:868–878. doi: 10.1016/j.cell.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Davidow PA, Bagshaw AT. Important characteristics of sequence-specific recombination hotspots in Schizosaccharomyces pombe. Genetics. 2011;187:385–396. doi: 10.1534/genetics.110.124636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Steiner EM, Girvin AR, Plewik LE. Novel nucleotide sequence motifs that produce hotspots of meiotic recombination in Schizosaccharomyces pombe. Genetics. 2009;182:459–469. doi: 10.1534/genetics.109.101253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szankasi P, Heyer WD, Schuchert P, Kohli J. DNA sequence analysis of the ade6 gene of Schizosaccharomyces pombe. Wild-type and mutant alleles including the recombination hot spot allele ade6-M26. J Mol Biol. 1988;204:917–925. doi: 10.1016/0022-2836(88)90051-4. [DOI] [PubMed] [Google Scholar]
- Tatebayashi K, Kato J, Ikeda H. Structural analyses of DNA fragments integrated by illegitimate recombination in Schizosaccharomyces pombe. Mol Gen Genet. 1994;244:111–119. doi: 10.1007/BF00283511. [DOI] [PubMed] [Google Scholar]
- Thomson JG, Ow DW. Site-specific recombination systems for the genetic manipulation of eukaryotic genomes. Genesis. 2006;44:465–476. doi: 10.1002/dvg.20237. [DOI] [PubMed] [Google Scholar]
- Watson AT, Garcia V, Bone N, Carr AM, Armstrong J. Gene tagging and gene replacement using recombinase-mediated cassette exchange in Schizosaccharomyces pombe. Gene. 2008;407:63–74. doi: 10.1016/j.gene.2007.09.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.