

Abstract
Detailed computational and experimental studies reveal the crucial role that hydrophobic interactions play in the self-assembly of small molecule-DNA hybrids (SMDHs) into cyclic nanostructures. In aqueous environments, the distribution of the cyclic structures (dimers or higher-order structures) greatly depends on how well the hydrophobic surfaces of the organic cores in these nanostructures are minimized. Specifically, when the cores are attached to the 3′-ends of the DNA component strands, they can insert into the minor groove of the duplex that forms upon self-assembly, favoring the formation of cyclic dimers. However, when the cores are attached to the 5′-ends of the DNA component strands, such insertion is hindered, leading to the formation of higher-order cyclic structures. These computational insights are supported by experimental results that show clear differences in product distributions and stabilities for a broad range of organic core-linked DNA hybrids with different linkage directions and flexibilities.
Introduction
Recently, a new class of hybrid materials has been generated by attaching DNA to organic molecules,1−11 polymers,12−14 metal complexes,15−21 and nanoparticles.22,23 Some of the resulting 2D24−26 and 3D27−30 nanostructures have been used in DNA detection31−33 and electronic applications;34−38 other applications such as drug delivery and therapeutics are emerging. A key factor that enables these applications is the degree of structural control available when DNA are linked to other molecules. Types of linkers and linkage direction are crucial for providing the desired control on product distributions of the assemblies and their stability.39−41
Recent investigations into the self-assembly of small molecule-DNA hybrids in aqueous media have demonstrated that the structures of these assemblies are dictated by several factors, including the number of single-strand (ss) DNAs attached to the organic cores, the specific geometry and concentration of the DNA strands, and the type (Na+, K+, Ca2+, Mg2+, etc.) and concentration of ions used in aqueous media. We additionally showed that the hydrophobic properties of organic cores is an important parameter to be considered in the self-assembly of the small-molecule-DNA hybrids (SMDHs).39 Indeed, Richert and co-workers have utilized the hydrophobic properties of stilbenes and anthraquinones linked to DNA duplexes to stabilize small-duplex DNA-detection probes.42,43 In a related study, Bergstrom and co-workers end-capped small DNA hairpins with rigid, hydrophobic organic molecules to afford enhanced stability.44 Moreover, perylenedimide (PDI)-DNA hybrids have been found to form stable hairpin dimers45 and larger supramolecular oligomers due to the hydrophobicity of the PDI cores, which leads to PDI–PDI stacking via π–π interactions.2,4
Despite emerging evidences concerning the critical importance of the hydrophobic interactions between organic cores and DNA duplexes in organic-DNA hybrids, the factors that give rise to these interactions are not well understood. Specifically, the effects of linking hydrophobic organic cores to DNA strands through either 3′- or 5′-ends are still not known although it is clear that such different linkages will lead to different types of hydrophobic interactions—such as insertion into the minor groove, intercalation, and π–π stacking—in the self-assembly of the small molecule-DNA building blocks into DNA-hybrid nanostructures. Probing these interactions experimentally is a challenging task, so we decided to combine experimental studies with molecular dynamics (MD) simulations at the atomistic level with the goal of unraveling the assembly of two complementary SMDHs into a cyclic dimer, the simplest DNA-hybrid nanostructure possible. With the continuous improvements in computer technology, utilization of computational methods and tools to describe self-assembly is gaining popularity. While there are different methods available to describe the properties of nucleic acid systems such as melting, mechanical properties, and conformations46−49 using coarse-grain simulations,46,47 the atomistic details of these systems at long time scales can best be described using atom-based force fields, such as AMBER50−52 and CHARMM.53,54
Previously, we reported the remarkable effects of hydrophobic organic cores in the assembly of small-molecule DNA hybrids (SMDHs) into caged structures.39 Our experimental results and computational simulations showed that the final nanostructures assemble in aqueous environments in a manner that minimizes exposure of the hydrophobic surfaces of the organic cores to solvent. These hydrophobic interactions are greatly influenced by the incorporation of multiple noncomplementary deoxythymidine (T) spacers between the core and the DNA duplex, as the solvent-accessible surface area (SASA) of the hydrophobic cores is greatly reduced when these spacers are wrapped around the cores. Soon after our report, Sleiman and co-workers published a broad study on the self-assembly of cyclic nanostructures made from SMDH2 building blocks.40,41 They showed that while the small molecule-DNA hybrid derived from the flexible organic core 1 (fSMDH2) assembled exclusively into cyclic dimers, that derived from the rigid organic core 2 (SMDH2) assembled into a mixture of dimers and higher-order (tetramer, hexamer, etc.) cyclic nanostructures (Figure 1). They proposed that the presence of rigid cores at either the 3′- or 5′-ends of DNA duplexes constrains the ways in which these duplexes can assemble and affects the distributions of the final products (dimer, tetramer, hexamer, etc.).
Figure 1.

Schematic descriptions of the assembly of cyclic nanostructures (dimer, tetramer, hexamer) from SMDH2 building blocks containing organic cores described by the Sleiman group40 (1, flexible; 2, rigid) and us (3, partially rigid).
To explain why cyclic dimers are preferred in the case of core 1, Sleiman and co-workers invoked a “strand-end alignment” model that focuses only on the importance of duplex alignment; once the DNA duplexes are ideally aligned by the first rigid core, dimers can form if the other ends of these duplexes are properly aligned to accommodate the second rigid core.40,41 The orientation of the ends of the DNA duplexes was defined as either convergent (i.e., can accommodate the second rigid core) or divergent (i.e., cannot accommodate the second rigid core). While this assumption appears to be a reasonable one, it was based on two cores that are quite different: one (1) that is flexible and moderately hydrophilic and the other (2) that is quite rigid and very hydrophobic. We suspect that a model built upon two such different cores may not accurately reflect the assembly process of SMDH2, particularly when the hydrophobic nature of the organic cores can force them to interact very strongly with the bases of the DNA component strands as well as with stacked base-pairs of the assembled duplexes in aqueous environments. Furthermore, the magnitude of this hydrophobic interaction would be strongly modulated by the flexibility of the connection points between the cores and the DNA component strands. Indeed, we have observed for the SMDH3 system39 that when there is not enough flexibility at the connection points, the drive to minimize exposure of the hydrophobic organic cores to water can sometime become so overwhelming that the system will destabilize some of the DNA base pairings in the assembled duplex arms. These observations prompted us to explore the assembly of SMDH2 further using the core 3, which is hydrophobic but much more flexible than 2.
Herein, we present a detailed computational study, supported by experimental results, which suggests that both the hydrophobic nature and the flexibility of the organic core play very important roles in the self-assembly of SMDH2s into cyclic dimers and higher-order nanostructures. While duplex alignment may be important, the interactions between the cores and the DNA duplexes to minimize their SASA comprise a dominant force that cannot be ignored, especially when there is restricted flexibility. Indeed for cyclic dimers involving core 3, when the cores are attached to the 3′-ends of the DNA component strands, they prefer to insert into the minor groove of the DNA duplexes in the product dimers to minimize their SASA. In contrast when cores such as 3 are attached to the 5′-ends of the DNA component strands, they can only partially insert themselves into the minor groove of the DNA duplexes in the product dimers, resulting in much less stable cyclic dimers. Consistent with these insights, SMDH2 building blocks containing cores linked to 3′-ends of the DNA duplexes were found to yield higher percentages of cyclic dimers compared to that obtained from 5′-linked SMDH2s. The important roles that such hydrophobic interactions play in SMDH2 assembly is further supported by the enhanced thermodynamic stability of face-to-face (ff) dimers over analogous cyclic dimers; the former benefits from strong interactions between two overlapping hydrophobic cores.
Methods
Experimental Details
Synthesis of unsymmetric SMDH2s was achieved by adding the phosphoramidite core55 (Scheme S1 in the Supporting Information) to the initial DNA arm grown from the surface of a controlled porosity glass bead (CPG) from either the 3′- or 5′-end of the DNA strand, followed by synthesis of the second arm via either 3′-normal or 5′-reverse phosphoramidite chemistry (Scheme S2 in the Supporting Information).8 The final DMT-protected products were then cleaved from the solid support, purified by reverse-phase (RP) high-performance liquid chromatography (HPLC), and subjected to DMT deprotection to yield the desired SMDH2s (Table 1). The purities of all SMDH2s were ascertained via analytical RP-HPLC and their length and base compositions were confirmed via MALDI-ToF mass spectrometry (see Figures S1–S14 in the Supporting Information). Building blocks (SMDH2s, Table 1) containing two different strands (one of the strands is the reverse sequence of the other to maintain similar thermal properties) were designed to control the formation of cyclic (Figure 1) versus ff SMDH2 (Figure S15 in the Supporting Information) nanostructures, depending on the orientation of the linkage between the core and the DNA strands. The SMDH2 assemblies and their controls (Table 2) were prepared by combining equimolar amounts of two complementary components (Table 1) in TAMg buffer (40 mM Tris base, 20 mM acetic acid, 7.5 mM MgCl2·6H2O) and annealing the resulting mixtures using both normal and slow-cooling methods (see Section S3 in the Supporting Information for more details).
Table 1. List of the SMDH2 Oligonucleotides Used in This Work.
| # | full name | short name |
|---|---|---|
| 1 | 3′-X-5′-C-3′-Y-5′ | [5′-C-3′] |
| 2 | 3′-X′-5′-C-3′-Y′-5′ | [5′-C-3′]′ |
| 3 | 3′-X-5′-C-5′-Y-3′ | [5′-C-5′] |
| 4 | 3′-X′-5′-C-5′-Y′-3′ | [5′-C-5′]′ |
| 5 | 5′-X-3′-C-3′-Y-5′ | [3′-C-3′] |
| 6 | 5′-X′-3′-C-3′-Y′-5′ | [3′-C-3′]′ |
| 7 | 3′-X-5′-T3CT3-3′-Y-5′ | [5′-T3CT3-3′] |
| 8 | 3′-X′-5′-T3CT3-3′-Y′-5′ | [5′-T3CT3-3′]′ |
| 9 | 3′-X-5′-T6-3′-Y-5′ | [5′-T6-3′] |
| 10 | 3′-X′-5′-T6-3′-Y′-5′ | [5′-T6-3′]′ |
| 11 | 3′-X-5′-T6-5′-Y-3′ | [5′-T6-5′] |
| 12 | 3′-X′-5′-T6-5′-Y′-3′ | [5′-T6-5′]′ |
| 13 | 3′-X-5′-T3-3′-Y-5′ | [5′-T3-3′] |
| 14 | 3′-X′-5′-T3-3′-Y′-5′ | [5′-T3-3′]′ |
| Sequence X: 3′-ATC CTT ATC AAT ATT-5′ | ||
| Sequence X′: 5′-TAG GAA TAG TTA TAA-3′ | ||
| Sequence Y: 3′-TTA TAA CTA TTC CTA-5′ | ||
| Sequence Y′: 5′-AAT ATT GAT AAG GAT-3′ | ||
| To maintain similar thermal properties, sequence X is the reverse of Y and sequence X′ is the reverse of Y′. | ||
Table 2. List of SMDH2 Assemblies Used in This Work, Including Cyclic and ff Structures and the Appropriate Controls.
| # | componentsa | short name |
|---|---|---|
| 1 | 3′-X-5′-C-3′-Y-5′ | cyclic-[5′-C-3′]:[5′-C-3′]′ |
| 3′-X′-5′-C-3′-Y′-5′ | ||
| 2 | 3′-X-5′-C-5′-Y-3′ | cyclic-[5′-C-5′]:[5′-C-5′]′ |
| 3′-X′-5′-C-5′-Y′-3′ | ||
| 3 | 5′-X-3′-C-3′-Y-5′ | cyclic-[3′-C-3′]:[3′-C-3′]′ |
| 5′-X′-3′-C-3′-Y′-5′ | ||
| 4 | 3′-X-5′-T3CT3-3′-Y-5′ | cyclic-[5′-T3CT3-3′]:[5′-T3CT3-3′]′ |
| 3′-X′-5′-T3CT3-3′-Y′-5′ | ||
| 5 | 3′-X-5′-T6-3′-Y-5′ | cyclic-[5′-T6-3′]:[5′-T6-3′]′ |
| 3′-X′-5′-T6-3′-Y′-5′ | ||
| 6 | 3′-X-5′-T6-5′-Y-3′ | cyclic-[5′-T6-5′]:[5′-T6-5′]′ |
| 3′-X′-5′-T6-5′-Y′-3′ | ||
| 7 | 3′-X-5′-T3-3′-Y-5′ | cyclic-[5′-T3-3′]:[5′-T3-3′]′ |
| 3′-X′-5′-T3-3′-Y′-5′ | ||
| 8 | 3′-X-5′-C-3′-Y-5′ | cyclic-[5′-C-3′]:[5′-T6-3′]′ |
| 3′-X′-5′-T6-3′-Y′-5′ | ||
| 9 | 3′-X-5′-C-5′-Y-3′ | cyclic-[5′-C-5′]:[5′-T6-5′]′ |
| 3′-X′-5′-T6-5′-Y′-3′ | ||
| 10 | 3′-X-5′-C-3′-Y-5′ | cyclic-[5′-C-3′]:[5′-T3CT3-3′]′ |
| 3′-X′-5′-T3CT3-3′-Y′-5′ | ||
| 11 | 5′-X-3′-C-3′-Y-5′ | ff-[3′-C-3′]:[5′-C-5′]′ |
| 3′-X′-5′-C-5′-Y′-3′ | ||
| 12 | 5′-X-3′-C-3′-Y-5′ | ff-[3′-C-3′]:[5′-T6-5′]′ |
| 3′-X′-5′-T6-5′-Y′-3′ | ||
| 13 | 3′-X′-5′, 3′-Y′-5′ | control-[5′-C-3′] |
| 3′-X-5′-C-3′-Y-5′ | ||
| 14 | 3′-X′-5′, 5′-Y′-3′ | control-[5′-C-5′] |
| 3′-X-5′-C-5′-Y-3′ | ||
| 15 | 5′-X′-3′, 3′-Y′-5′ | control-[3′-C-3′] |
| 5′-X-3′-C-3′-Y-5′ | ||
| 16 | 3′-X′-5′, 3′-Y′-5′ | control-[5′-T3CT3-3′] |
| 3′-X-5′-T3CT3-3′-Y-5′ |
Total DNA concentration = 5 μM in TAMg buffer.
Computational Details
AMBER force field parameters for the organic core were calculated as previously described (Section S4 in the Supporting Information).39 The AMBER99 force field56,57 with revised χ58 and α/γ59 torsional parameter sets was used to define the DNA parameters. All the model DNA systems (with sequences described in Table 1) were created in B-form using the nucgen module of AMBER 9.50 The SMDH2 systems (Table 2, entries 1–5 and 13) were then prepared, where the organic cores were attached at either 3′- or 5′-ends of the DNA sequences (Section S5 in the Supporting Information). Most MD simulations were run in a Generalized Born implicit-solvent model (GBHCT)60,61 with 0.3 M salt concentrations. Unrestrained GB MD simulations were carried for our three different cyclic dimers (Table 2, entries 1–3) and the cyclic tetramer and hexamer for one SMDH2 system (Table 2, entry 1, see Section S6 in the Supporting Information for details). Restrained GB MD simulations (both normal and annealed) were carried out for cyclic dimers (Table 2, entries 1–3) with varying DNA duplex lengths (11, 13, 15, 17, 19, 21, and 23 base pairs) and a model system (used for explicit solvent calculations that were used to calibrate the GB model) consisting of an 11 bp DNA duplex with a single organic core attached at either 3′- or 5′-end of the DNA strands (see Section S7 in the Supporting Information). SASA and root-mean-square deviation (rmsd) analysis for all the MD simulations were performed according to our previously published work (see Sections S5 in the Supporting Information for details).39
The implicit-solvent model used in this study is preferred over explicit-solvent ones due to the large size of our systems, whose computation would be quite long and expensive if explicit-solvent models were used. Compared to explicit-solvent MD simulations, the viscosity in implicit-solvent MD simulations is low and this accelerates the sampling of MD space. Yet, implicit-solvent models are less realistic because they employ empirical parameters to calculate the solvation free energies. To date, implicit-solvent simulations of proteins are well developed62,63 while the corresponding simulations of nucleic acids still require improvements. While GB implicit-solvent simulations of regular DNA oligomers produce average structures that are in line with explicit-solvent MD simulations,64 the formers can distort the DNA backbone and promote fraying effects at terminal base pairs that might not be physical. This is one of the shortcomings of implicit-solvent models and can cause DNA to be more flexible than it actually is. Indeed, Harris and co-workers showed that the use of implicit solvents in DNA minicircle topoisomers with varying lengths display structures that are different from those seen in explicit-solvent simulation, which can be attributed to DNA flexibility.65 One of the reasons for this outcome is the neglect of specific interactions of water molecules with DNA in implicit-solvent models (such as the “spine of hydration” observed in the minor grooves of A-tracks). Thus, the improper description of DNA flexibility will show its effects in exotic systems such as in DNA minicircles65 and SMDH molecules. As a result, to keep the DNA structures in their known native B-form, we had to use Watson–Crick and torsional restraints so that the MD simulations have a physical meaning. Additionally, explicit-solvent calculations were carried out on our smallest model system (see Section S7 in the Supporting Information for more details) to verify the accuracy of the GB calculations with restraints.
Results and Discussion
Self-Assembly and Characterization of Cyclic Nanostructures
Combining equimolar amounts of [5′-C-3′], [5′-C-5′], [3′-C-3′], [5′-T3CT3-3′], [5′-T6-3′], [5′-T6-5′], [5′-T3-3′] with their complements, [5′-C-3′]′, [5′-C-5′]′, [3′-C-3′]′, [5′-T3CT3-3′]′, [5′-T6-3′]′, [5′-T6-5′]′, and [5′-T3-3′]′, respectively, was expected to result in cyclic structures (Figure 1; see also Table 2, entries 1–7) as previously reported.1,8,40 Our earlier study showed that systems with core 3 with noncomplementary T spacers (Table 2, entry 4) exclusively formed cyclic dimers at low concentrations of ss-DNA (up to 5 μM total ss-DNA concentration in which each arm’s concentration in the SMDH2 was calculated separately).8 However, Sleiman and co-workers recently reported that the rigidity of the core and the linkage between the core and the DNA strands (3′3′, 5′5′, and 3′5′ orientations; see Figure 1, inset B) directly influence the product distributions:40 systems with flexible core 1 (Figure 1, inset A) attached to 3′- or 5′-end of DNA strands mostly formed cyclic dimers, while those with rigid core 2 (Figure 1, inset A) afforded a mixture of all cyclic products. Interestingly, PAGE-gel analysis of the [5′-C-3′]:[5′-C-3′]′, [5′-C-5′]:[5′-C-5′]′, and [3′-C-3′]:[3′-C-3′]′ combinations in our present work revealed a mixture of cyclic structures (Figure 2) instead of solely cyclic dimers even though our core 3 can be classified as being overall flexible with highly flexible (−CH2OCH2CH2O−) arms flanking a rigid 1,3-bis(ethynylphenyl)phenyl component (Figure 1, inset A).
Figure 2.

Nondenaturing PAGE-gel image (6%) of DNA assemblies with 5 μM total ss-DNA concentration (gel was prepared in 1× TAMg buffer (40 mM Tris base, 20 mM acetic acid, 7.5 mM MgCl2·6H2O), and run at 4 °C for 2 h at a 200 V potential). From left to right: lane 1 = HL5 DNA ladder, lane 2 = cyclic-[5′-C-3′]:[5′-C-3′]′ (normal annealing), lane 3 = cyclic-[5′-C-3′]:[5′-C-3′]′ (slow annealing), lane 4 = control-[5′-C-3′], lane 5 = cyclic-[5′-C-5′]:[5′-C-5′]′ (normal annealing), lane 6 = cyclic-[5′-C-5′]:[5′-C-5′]′ (slow annealing), lane 7 = control-[5′-C-5′], lane 8 = cyclic-[3′-C-3′]:[3′-C-3′]′ (normal annealing), lane 9 = cyclic-[3′-C-3′]:[3′-C-3′]′ (slow annealing), and lane 10 = control-[3′-C-3′].
According to the strand-end alignment model,40,41 the flexible arms of our core 3 should be long enough to accommodate any strain put on the DNA duplexes by the rigid 1,3-bis(ethynylphenyl)phenyl component, favoring the formation of cyclic dimers. However, this was not experimentally observed. Rather, exclusive formation of cyclic dimers is observed when three noncomplementary deoxythymidine spacers (T3) were added to each side of our core 3 ([5′-T3CT3-3′]:[5′-T3CT3-3′]′), according to the nondenaturing PAGE-gel analysis (see Figure S17 in the Supporting Information, lane 9). These results, however, are in agreement with our previous report, which concluded that dimer formation is favored when T3 spacers are available to shield the hydrophobic surfaces of the cores from the aqueous media.39
The nondenaturing PAGE-gel analysis also revealed that there is a higher percentage of dimer formed for [3′-C-3′]:[3′-C-3′]′ compared to the [5′-C-5′]:[5′-C-5′]′ and [5′-C-3′]:[5′-C-3′]′ combination (Figure 2, lanes 8, 5, and 2, respectively). To further probe this observation and eliminate the possibility that the formation of cyclic dimers may be affected by the length of the annealing time, we annealed our SMDH2 systems (Table 2, entries 1–3) with a cooling rate of 0.01 °C/minute in a PCR instrument in the range of 60–25 °C, which is 34 times slower than our normal annealing protocol (0.34 °C/minute in the 60–40 °C range (Figure S16 in the Supporting Information), where the complete melting of a mixture of cyclic structures occur). Analysis of the nondenaturing PAGE-gel image (Figure 2) showed a clear increase in dimer formation for all cases during slow annealing. Moreover, the cyclic dimer formed in high yield (79%) for [3′-C-3′]:[3′-C-3′]′ in slow annealing, much higher than the [5′-C-3′]:[5′-C-3′]′ and [5′-C-5′]:[5′-C-5′]′ combinations (43 and 34%, respectively). The latter afforded the highest percentage of large cyclic structures (31%, Figure 2, lane 6) in the slow annealing method compared to the former two (11 and 5%, respectively). Similar observations were also reported by Sleiman and co-workers for systems derived from core 2.40 Cyclic dimers were formed exclusively in the nondenaturing PAGE-gel analysis when deoxythymidine linkers (T3 and T6, Table 2, entries 5–7) were used in place of organic core 3 (see Figure S18 in the Supporting Information, lanes 3–5).
Molecular Dynamics Simulations
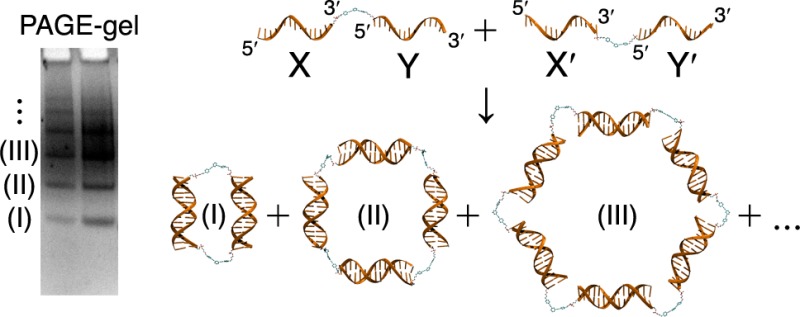
Our previous computational work showed that hydrophobic interactions between the organic cores and noncomplementary T spacers in SMDH3 building blocks play a crucial role in the self-assembly process and the final properties of SMDH3-based nanostructures.39 In such structures, the cores “strive” to minimize their SASA values through interaction with both the T spacer and the base pairs of the duplexes. When the SASA values were too high for dimers, other structures with lower SASA become dominant. Nevertheless, we were surprised to observe that the directionality of the linkage (3′- or 5′-) between the core and the duplex dramatically affected the product distributions in the assembly of SMDH2-based nanostructures in the absence of noncomplementary T spacers (see the discussion of experimental results). As such, we utilized MD simulations to elucidate the details of the self-assembly process. Computationally, we began by examining the simpler DNA-hybrid components that constitute the larger cyclic nanostructures (dimers, tetramers, hexamers, etc.): (1) organic core 3 attached to either 3′- or 5′-ends of a DNA duplex (Figure 3a); (2) single organic core attached at 3′- and 5′-ends of two DNA duplexes, control-[5′-C-3′] (Figure 3b); (3) cyclic-[5′-C-3′]:[5′-C-3′]′ structures (dimer structure is shown in Figure 3c; tetramer and hexamer structures are shown in Figure 1).
Figure 3.
Initial modeling of (a) organic core 3 attached to a DNA duplex, (b) control-[5′-C-3′], and (c) cyclic-[5′-C-3′]:[5′-C-3′]′ dimer.
MD Simulations of Organic Core 3 Attached to the 3′- or 5′-End of a DNA Duplex
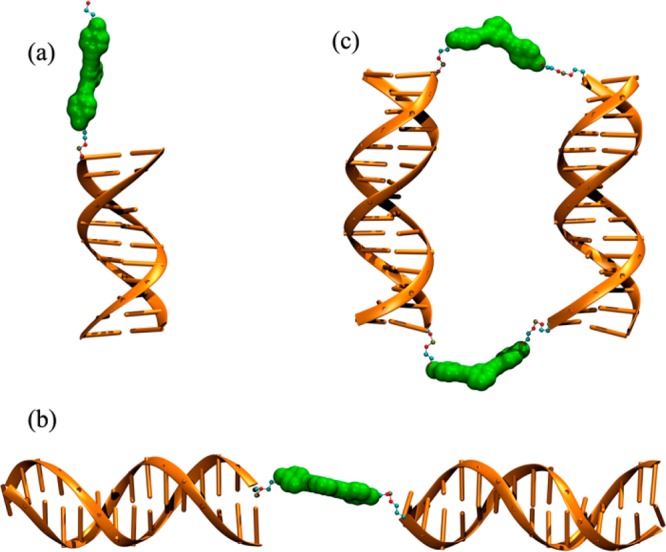
The simplest component system, consisting of the organic core 3 attached to an 11 bp DNA duplex, allows us to investigate how the organic cores attached to either 3′- or 5′-ends of DNA duplexes (see Section S7 in the Supporting Information) interact with these duplexes in an aqueous environment. An initial set of MD simulations utilized GB implicit solvent models while Watson–Crick base pairing and torsional restraints were applied to DNA duplexes to keep them in their native B-form since GB implicit solvent MD simulations can distort the DNA backbone and cause fraying at terminal base pairs (see the discussions below concerning unrestrained and restrained MD simulations and Section S7 in the Supporting Information).65 After 10 ns of MD simulations, the 3′-linked organic core is already inserted fully into the DNA minor groove (Figure 4b) while the 5′-linked core is only partially inserted (Figure 4c). Average SASA values for the 3′- and 5′-linked organic cores are 153.7 ± 19.6 and 250.8 ± 18.4 Å2, respectively, and remain constant over the course of the simulation time (55 ns, Figure 4a). Because the 3′-ends of DNA duplexes are spatially closer to the minor grooves compared to 5′-ends, it is easier for a 3′-linked organic core to be fully inserted into the DNA minor groove. On the other hand, the 5′-linked organic core needs to stretch over the terminal base pairs before it can reach DNA minor grooves; so it is only partially inserted, has a much larger SASA value, and is less stable in aqueous solution. These results suggest that the differences in the distribution of cyclic nanostructures, as observed in the PAGE-gel experiments (Figure 2, lanes 6 versus 9), can be rationalized by how well organic cores are inserted in DNA minor grooves to minimize exposure to the aqueous outside environment.
Figure 4.
MD simulation results for 3′- and 5′-linked core-DNA duplex systems. (a) SASA values of hydrophobic cores attached at 3′- (black) and 5′-ends (red) of DNA duplexes over the course of the simulation. Final structures of 3′- and 5′-linked 3-DNA hybrids are shown in (b,c), respectively. The 3′-linked core is fully inserted into DNA minor groove compared to the 5′-linked core (see Figure S22 in the Supporting Information).
To calibrate the stabilities of these restrained GB-based structures, we also performed explicit-solvent calculations for the same model. In these calculations, each final conformation from the GB calculations was solvated with Na+/Cl– ions and water molecules and the imposed restraints were removed on the DNA duplexes (see Section S7 in the Supporting Information for more details). The simulations show that the 3′-linked organic core stays in the DNA minor groove while the global DNA conformation remains in B-form (Figure S22 in the Supporting Information). It is also found that the organic core in the 5′-linked core-DNA system is dynamic and does not stay in the DNA minor groove for a long time. The terminal base pair is broken, and the organic core stacks within the bases of the distorted terminal side (Figure S22 in the Supporting Information). This clearly shows that DNA minor groove insertion is a viable option for 3′-linked organic cores to lower their SASA without distorting the DNA. This is not the case in 5′-linked core-DNA systems, where the only option for organic cores to lower their SASA is through stacking within terminal bases after disrupting the canonical base pairing. These results indicate that the restrained implicit-solvent MD simulations exhibit similar behavior for the hydrophobic organic cores to the explicit-solvent MD simulations without restraints.
MD Simulations of Organic Core 3 Attached at 3′- and 5′-Ends of Two DNA Duplexes
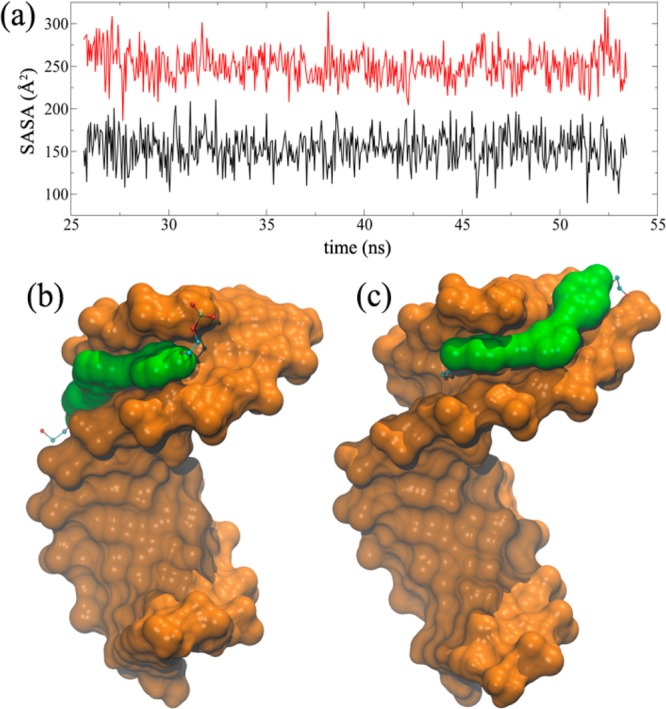
We next examined the control-[5′-C-3′] system (Figure 3b) to evaluate potential interactions between organic core 3 and the two DNA duplex arms that are linked to it. After 96 ns, restrained implicit-solvent MD simulations showed that the organic core already minimized its SASA by inserting itself within the minor groove of one DNA duplex and “stacking” with the terminal DNA base pairs of the other duplex (Figure 5; see also Movie S1 in the Supporting Information). As a result, the control-[5′-C-3′] system forms an almost continuous 30 bp DNA duplex with a single core stacked in the middle.
Figure 5.
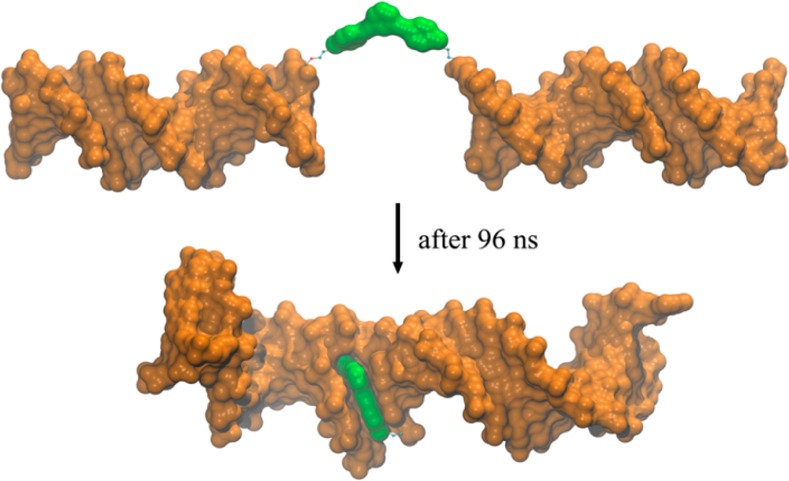
Molecular surface representations of initial (top) and final (below, after 96 ns) structures of the control-[5′-C-3′] system obtained from MD simulations. The organic core 3 is shown in green color. The two 15 bp DNA duplexes are brought together by the hydrophobic nature of 3. Two MD simulations were carried out; see Movie S1 in the Supporting Information for further details.
Unrestrained MD Simulations of Cyclic Nanostructures
As we have shown in previous MD simulations,39 the hydrophobic nature of core 3 forces it to minimize its exposed surfaces in an aqueous environment through interactions with the DNA duplexes. To simulate the cyclic nanostructures that may form in an experiment, we carried out implicit-solvent MD simulations (this is a more feasible option compared to expensive explicit-solvent MD simulations for these large systems; see detailed discussion in Section S7 in the Supporting Information) for all possible combinations of cyclic dimers (Figure 6) as well as tetramer and hexamer structures (Figures 7a-b) for the [5′-C-3′]:[5′-C-3′]′ system without any restraints on DNA duplexes (see Section S6 in the Supporting Information for more details). A SASA analysis of the cyclic-[5′-C-3′]:[5′-C-3′]′ dimer, tetramer, and hexamer systems shows that the organic cores 3 in these structures can lower their SASA better in higher-order structures (Table 3, entries 1–3). While the average SASA value of the cyclic-[5′-C-3′]:[5′-C-3′]′ dimer is 200 Å2, it is reduced to 134 Å2 in the tetramer and 122 Å2 in the hexamer (Table 3). In the cyclic dimer, the organic cores cannot lower their SASA as much due to strain. As shown above for the 3′-linked core model system, the organic cores in cyclic dimers can minimize their SASAs through interactions with the DNA minor grooves, which severely distort the two DNA arms (Figure 6). On the other hand, the organic cores in the tetramer and hexamer have additional means to minimize their SASAs (Figures 7c–f), such as “stacking” with the terminal base pairs of the adjacent DNA duplex, as shown for the control-[5′-C-3′] system (Figure 5).
Figure 6.
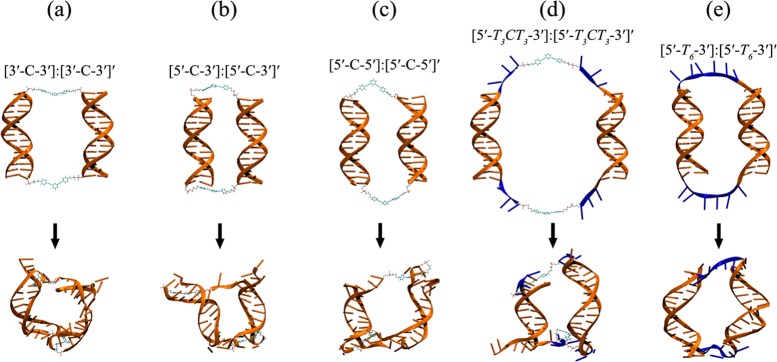
Schematic representations of initial (top) and lowest SASA (below) cyclic-dimer structures after 91 ns MD simulations. Residues colored with blue are deoxythymidine (T) linkers. Note that due to the flexible T linkers, DNA deformations in (d,e) are not severe compared to (a–c) (see Table S2 in the Supporting Information).
Figure 7.
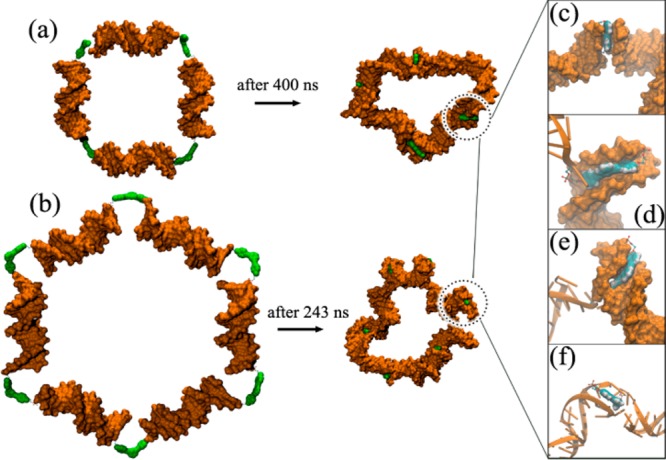
Molecular surface representations of the initial (left) and final (middle) structures of the cyclic tetramer (a) and hexamer (b) formed from the [5′-C-3′]:[5′-C-3′]′ system (see Figures S20 and S21 in the Supporting Information). The organic cores 3 are represented in green color and are shown interacting with neighboring DNA duplexes in different manners: (c) being sandwiched between neighboring DNA duplexes via π-stacking, (d) fully inserting into the minor groove, (e) inserting into the minor groove in a distorted manner, and (f) π-stacking with distorted terminal base pairs of neighboring DNA duplexes (the DNA distortions are not as severe in comparison to that observed in the cyclic-dimer structures shown in Figure 6).
Table 3. RMSD, SASA, and H-Bond Analysis of Unrestrained MD Simulations for Cyclic-Dimer, -Tetramer, and -Hexamer Systemsa.
| entry | components | structure | duplex rmsd (Å) | SASA (Å2 per core) | % loss of H-bonds |
|---|---|---|---|---|---|
| 1 | [5′-C-3′]:[5′-C-3′]′ | hexamer | 6.63 | 122.21 | % 17 |
| 2 | [5′-C-3′]:[5′-C-3′]′ | tetramer | 6.50 | 133.79 | % 22 |
| 3 | [5′-C-3′]:[5′-C-3′]′ | dimer | 6.82 | 200.39 | % 24 |
| 4 | [3′-C-3′]:[3′-C-3′]′ | dimer | 6.64 | 163.56 | % 22 |
| 5 | [5′-C-5′]:[5′-C-5′]′ | dimer | 5.88 | 140.37 | % 31 |
| 6 | [5′-T3CT3-3′]:[5′-T3CT3-3′]′ | dimer | 5.37 | 118.90 | % 5 |
| 7 | [5′-T6-3′]:[5′-T6-3′]′ | dimer | 5.89 | % 9 |
One MD simulation was carried out for each of the hexamer and tetramer; four independent MD simulations were carried out for cyclic-dimer systems but only the lowest rmsd results are shown (see Section S6 in the Supporting Information for more details).
Interestingly, MD simulations of the cyclic-[5′-T3CT3-3′]:[5′-T3CT3-3′]′ dimer structure show average SASA values around ∼119 Å2 (Table 3). The rmsd’s of the DNA duplexes are lower than those for the other cyclic-dimer systems that have only organic cores 3, implying that introducing T3 spacers provides additional flexibility that helps to minimize the SASAs of the cores without distorting the DNA duplexes (Table 3 and Figure 6d). Consistently, the percentage of hydrogen bond loss in the cyclic-[5′-T3CT3-3′]:[5′-T3CT3-3′]′ dimer is only 5% while those for the cyclic-[5′-C-3′]:[5′-C-3′]′, cyclic-[3′-C-3′]:[3′-C-3′]′, and cyclic-[5′-C-5′]:[5′-C-5′]′ dimers are over 20% (Table 3). These results are in line with our previous work39 and PAGE-gel experiments, which show exclusive formation of dimer structures in cyclic-[5′-T3CT3-3′]:[5′-T3CT3-3′]′ (see Figure S17 in the Supporting Information, lane 9). In the absence of the organic cores 3, exclusive cyclic dimer formation was observed for [5′-T6-3′]:[5′-T6-3′]′, [5′-T6-5′]:[5′-T6-5′]′, and [5′-T3-3′]:[5′-T3-3′]′ (see Figure S17 in the Supporting Information, lanes 3–5). Similarly, the MD simulations show no major DNA distortions in [5′-T6-3′]:[5′-T6-3′]′, with ∼9% hydrogen bond loss (Table 3 and Figure 6e).
Restrained MD Simulations of Cyclic Dimer Nanostructures
In the unrestrained MD simulations of the cyclic dimers discussed above, the DNA duplexes can be easily distorted due to the conformational stress put on by the organic cores while trying to minimize their SASAs (Figures 6a–c). One consequence of this is excessive fraying of the terminal base pairs in the DNA duplex arms of the dimers (Figures 6a–c), making it difficult to evaluate their relative stabilities. Thus, restrained MD simulations (see detailed discussion in Section S7 of the Supporting Information) were carried out where the DNA duplex arms of the dimers were constrained in B-form while their lengths were varied over one helix turn (11, 13, 15, 17, 19, 21, and 23 bp, see Section S7 in the Supporting Information for more details). On average, the total SASA values of the organic cores in the [3′-C-3′]:[3′-C-3′]′ systems are lower than those for the [5′-C-3′]:[5′-C-3′]′ and [5′-C-5′]:[5′-C-5′]′ systems (see Figure S23 and Table S3 in the Supporting Information), suggesting that the manner in which the cores are attached to the DNA duplexes does matter.
Simulated Annealing of Cyclic Dimer Nanostructures
While constraining the DNA duplex arms gave us a starting point to compare the three different dimer systems via MD simulations, these structures may be trapped in local minima and do not accurately reflect the energy landscape of the system. Thus, we performed simulated annealing on all of the 21 SMDH2 dimers shown in Table S4 in the Supporting Information (see also Section S8 in the Supporting Information). While the DNA structures were kept in B-form conformations with Watson–Crick, torsional, and chirality restraints, the temperature of each system was increased to 3000 K and gradually cooled down to 100 K. For each system, 301 simulated annealing MD simulations were run sequentially where the starting structure for each run was taken from the final structure of the previous run. This way, each system was allowed to move away from any potential minimum state and sample other regions in phase space. For each dimer, SASA values of the organic cores were calculated from the simulated annealing MD simulations for structures having final restraint energies and duplex rmsd values less than 10 kcal/mol and 10 Å, respectively. Within these structures, SASA values that are uniformly lower than those obtained for the restrained MD simulations shown earlier can be obtained for each cyclic dimer (see data in Table S3 in Supporting Information and Figure 8; see also Figures S23 and S24 in the Supporting Information), suggesting that simulated annealing offers a more self-consistent basis for comparing the dimers.
Figure 8.
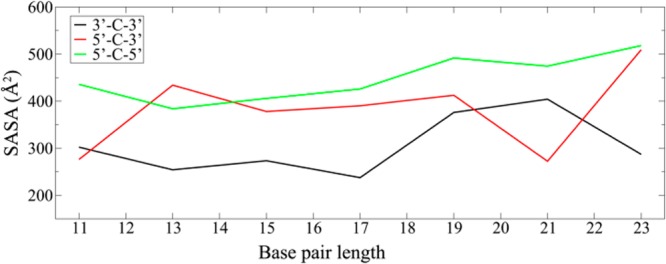
SASA values of the organic cores in SMDH2 cyclic dimers as a function of DNA length. The black, red, and green trends represent the results for [3′-C-3′]:[3′-C-3′]′, [5′-C-3′]:[5′-C-3′]′, and [5′-C-5′]:[5′-C-5′]′, respectively, where the lowest SASA values for the organic cores were extracted from simulated annealing MD simulations with final restraint energies and duplex rmsd values less than 10 kcal/mol and 10 Å, respectively.
As discussed above, implicit-solvent simulations of SMDH2 can yield unphysical structures if no restraints are imposed on DNA to keep them in their native B-form conformation. To see the effects of removal of restraints imposed on DNA duplexes in these cyclic dimers, the lowest SASA structures were simulated for over 125 ns without restraints. As expected and similar to the results shown in Figure 6, the DNA duplexes are distorted from the B-form conformation (Table S5 in the Supporting Information). In some cases, one of the DNA duplexes in these cyclic dimers is fully unfolded (see Table S5 in the Supporting Information, bp_17 [5′-C-5′]:[5′-C-5′]′). Furthermore, the % of hydrogen bonds lost increased with the size of the duplex while the SASA of the organic cores decreased suggesting that DNA duplexes are more flexible than they actually are (Table S5 in the Supporting Information). Therefore, to study these types of large DNA-hybrid systems with the current implicit-solvent models one is required to impose restraints on DNA structures to keep them in B-form conformation so that results have physical meaning.
As shown in Figure 8, there is a clear overall trend, which shows that the dimers with organic cores linked at the 3′-ends of the DNA strands (cyclic-[3′-C-3′]:[3′-C-3′]′) are much better at minimizing the SASAs of their organic cores than the other two dimer systems. There is a higher chance for the organic cores 3 to be inserted into the minor grooves of the flanking DNA duplex in cyclic-[3′-C-3′]:[3′-C-3′]′. As a result, organic cores can lower their SASAs more significantly in these systems than would be possible for the cores in the cyclic-[5′-C-5′]:[5′-C-5′]′ systems, which prevent full minor groove insertion of organic cores. For the [5′-C-3′]:[5′-C-3′]′ cyclic dimer, where the organic cores are linked at both 5′- and 3′-ends of the DNA strands, the cores can be inserted into the minor groove only if the DNA orientations are favorable. Not surprisingly, the SASA values for this system lie in the middle of the other two. (For a more detailed discussion of this system, please see Section S8 in the Supporting Information)
One interesting result shown in Figure 8 is the nonmonotonic behavior observed around base pair lengths 11 and 21. This phenomenon could be due to the DNA helical turn, which is approximately 10 base pairs, suggesting a real connection to the physical system. The SASA of organic cores in the 11 base paired cyclic-[5′-C-3′]:[5′-C-3′]′ system is lower than the cyclic-[3′-C-3′]:[3′-C-3′]′ system. After adding 10 base pairs to the DNA sequences, which results in DNA duplexes with 21 base pairs, a similar result was observed (Figure S8 and Table S24 in the Supporting Information). In cyclic-[5′-C-3′]:[5′-C-3′]′ systems with 11 and 21 DNA base pairs the organic cores almost fully insert themselves into the DNA minor grooves, which provides an explanation for the nonmonotonic behavior observed in these DNA sequences (Figure S23 in the Supporting Information). Note that no such behavior has been observed in cyclic-[5′-C-5′]:[5′-C-5′]′ systems (green curve in Figure 8).
Comparison of Theory and Experiment
The idea that the hydrophobic organic cores 3 in SMDH2 cyclic dimers can be stabilized in aqueous solutions by inserting into the minor groove can be used to explain the results that we reported above for the slow-cooling experiments of 15 bp SMDH2 systems (Figure 2). In that experiment, the percentage of cyclic dimers formed in [3′-C-3′]:[3′-C-3′]′ is 79%, much higher than those observed for [5′-C-3′]:[5′-C-3′]′ and [5′-C-5′]:[5′-C-5′]′ (43 and 34%, respectively; see Figure 2, lanes 3, 6, and 9). This trend closely follows the simulated annealing MD simulation results described in Figure 8: a higher proportion of dimers can form when the cores are linked to both 3′-ends of the DNAs because the cores can be better stabilized by inserting into the minor grooves of the two DNA arms. In other SMDH2 systems where the cores are linked to 5′-ends of DNAs, other higher-order structures are formed because there is no predominant stabilization mechanism for the core in the dimer structures.
Synthesis and Characterization of Cyclic Structures with Mixed Cores
To further test the insights obtained from our MD calculations, we designed an SMDH system where one side of the cyclic dimer had the organic core 3 and the other side had flexible linkers such as T3-C-T3 and T6 (Table 2, entries 8–10). These systems would also allow us to assess the relative importance of minimizing hydrophobicity versus strand-end alignment. If strand-end alignment is of primary importance, cyclic dimers would form predominantly if one of the linkage sites is flexible (i.e., with T3-C-T3 or T6 linkers) enough to alleviate the strain resulting from the rigid core on the other side. However, as shown in Figure 9, the nondenaturing PAGE-gel analyses for [5′-C-3′]:[5′-T6-3′]′, [5′-C-5′]:[5′-T6-5′]′, and [5′-C-3′]:[5′-T3CT3-3′]′ systems, both normal and slow-annealing, all afforded a mixture of dimers, tetramers, and hexamers.
Figure 9.

Nondenaturing PAGE-gel image (6%) of DNA assemblies from an SMDH component possessing the organic core 3 and the complementary SMDH component possessing a flexible linker (either T3-C-T3 or T6) with 5 μM total ss-DNA concentration. (Gel was prepared in 1× TAMg buffer (40 mM Tris base, 20 mM acetic acid, 7.5 mM MgCl2·6H2O), and run at 4 °C for 2 h under a 200 V field). From left to right: lane 1 = HL5 DNA ladder, lane 2 = cyclic-[5′-C-3′]:[5′-T6-3′]′ (normal annealing), lane 3 = cyclic-[5′-C-3′]:[5′-T6-3′]′ (slow annealing), lane 4 = cyclic-[5′-C-5′]:[5′-T6-5′]′ (normal annealing), lane 5 = cyclic-[5′-C-5′]:[5′-T6-5′]′ (slow annealing), lane 6 = cyclic-[5′-C-3′]:[5′-T3CT3-3′]′ (normal annealing), lane 7 = cyclic-[5′-C-3′]:[5′-T3CT3-3′]′ (slow annealing).
That all six experiments shown in Figure 9 formed a mixture of products instead of only cyclic dimers points to the importance of the organic core not having the freedom to isolate its hydrophobic surface. Moreover, imageJ analysis of the gel image showed that [5′-C-3′]:[5′-T6-3′]′ gave higher percentages of cyclic dimer (82% dimer, 14% tetramer, slow cooling, Figure 9, lanes 2 and 3) compared to [5′-C-5′]:[5′-T6-5′]′ (62% dimer, 29% tetramer, slow cooling, Figure 9, lanes 4 and 5), which is consistent with the 3′-linkage providing better shielding for the hydrophobic organic cores via minor-groove insertion. In effect, the better that the organic cores insert into the minor groove, the larger is the fraction of cyclic dimers formed. This argument is further supported by the results for [5′-C-3′]:[5′-T3CT3-3′]′ (Figure 9, lanes 6 and 7), both of which show higher cyclic dimer formation than [5′-C-5′]:[5′-T6-5′]′.
Melting Properties of Cyclic and Face-to-Face (ff) Dimers
The strong hydrophobic interactions between cores can also be invoked to explain the exclusive formation of ff dimers in the [3′-C-3′]:[5′-C-5′]′ and [3′-C-3′]:[5′-T6-5′]′ systems (Table 2, entries 11 and 12; see also Figure S18 in the Supporting Information, lanes 9 and 10).66 We attribute this behavior to the combination of reduced configurational entropy and increased ion-cloud sharing that occurs when the organic linkages bring the DNA duplexes into close proximity. Notably, the [3′-C-3′]:[5′-C-5′]′ ff dimer has a much higher thermal stability, as illustrated by its higher Tm, compared to the cyclic-[5′-T3-3′]:[5′-T3-3′]′ dimer, whose T3 linker is comparable in size to the core C (Figure 10). Indeed, the [3′-C-3′]:[5′-C-5′]′ ff dimer is consistently more stable than all the cyclic dimers that we examined (Tm = 52.4–53 °C, Table 4, cf entry 10 and entries 7–9). However, when the linker for one side of the ff dimer was changed to a flexible T6 ([3′-C-3′]:[5′-T6-5′]′), the Tm was decreased by 4.7 °C (Table 4, cf entries 10 and 11).
Figure 10.
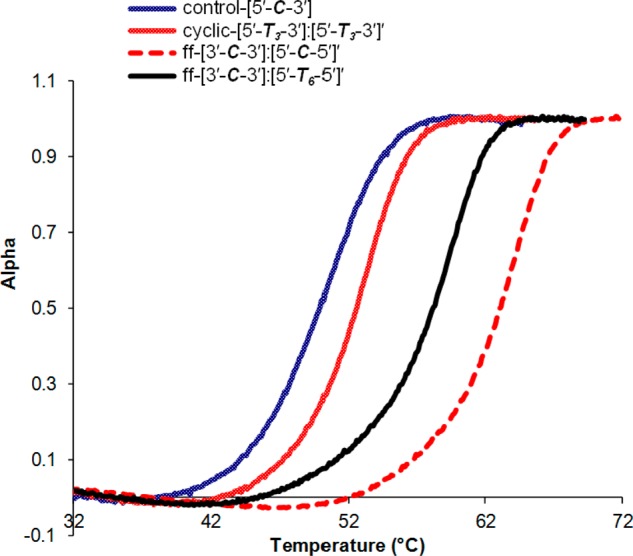
Melting profiles for control, cyclic-, and ff-dimer SMDH2 assemblies (5 μM) in TAMg buffer (40 mM Tris base, 20 mM acetic acid, 7.5 mM MgCl2·6H2O).
Table 4. Melting Data for Cyclic Dimers, ff Dimers, and the Controls with 5 μM total DNA Concentration in TAMg Buffer (40 mM Tris Base, 20 mM Acetic Acid, 7.5 mM MgCl2·6H2O)a.
| entry | short nameb | Tm (°C) | fwhm (°C) |
|---|---|---|---|
| 1 | X:X′ | 49.7 ± 0.1 | 10.7 ± 0.1 |
| 2 | control-[5′-C-3′]c | 49.9 ± 0.2 | 8.9 ± 0.1 |
| 3 | control-[5′-C-5′] | 50.4 ± 0.1 | 8.7 ± 0.1 |
| 4 | control-[3′-C-3′] | 49.6 ± 0.1 | 8.9 ± 0.1 |
| 5 | control-[3′-T3CT3-5′] | 48.6 ± 0.2 | 10.0 ± 0.1 |
| 6 | cyclic-[5′-T3CT3-3′]:[5′-T3CT3-3′]′c | 52.6 ± 0.2 | 7.4 ± 0.1 |
| 7 | cyclic-[5′-T6-3′]:[5′-T6-3′]′ | 52.8 ± 0.2 | 6.7 ± 0.1 |
| 8 | cyclic-[5′-T6-5′]:[5′-T6-5′]′ | 53.0 ± 0.1 | 7.1 ± 0.1 |
| 9 | cyclic-[5′-T3-3′]:[5′-T3-3′]′ | 52.4 ± 0.1 | 7.6 ± 0.1 |
| 10 | ff-[3′-C-3′]:[5′-C-5′]′c | 62.9 ± 0.2 | 7.3 ± 0.1 |
| 11 | ff-[3′-C-3′]:[5′-T6-5′]′c | 58.2 ± 0.1 | 7.2 ± 0.1 |
The aforementioned results are consistent with our previous work,39 indicating that hydrophobic interactions between the two organic cores (and that between cores and the DNA duplexes) can play a major role in determining the thermal properties of the final hybridized systems. Thus, such hydrophobic interactions must be taken into account in the consideration of product distributions in the assembly of organic-linked DNA materials. We note that Sleiman and co-workers have also observed similar thermal stability enhancements for ff dimers that are analogous to the one reported herein.40 However, these Tm increases were only attributed to allosteric and chelate cooperativities of the DNAs; contributions by hydrophobic interactions were not discussed. Comparing to the cyclic dimers, the increases in thermal stabilities for our ff-[3′-C-3′]:[5′-C-5′]′ and ff-[3′-C-3′]:[5′-T6-5′]′ dimer systems can be attributed to two factors: (1) hydrophobic interactions between the cores, and (2) effective extension of the DNA helix through the organic core/linkers,67 which was referred by Sleiman and co-workers as chelate cooperativity.40 The first effect is strongly supported by fluorescent studies in similar ff diphenyl acetylene-68 and 1,3,5-tris(p-ethynylphenyl)benzene-linked dimer systems.39 The second factor has been ascribed by Leumann and co-workers as due to close structural communications between the two linked helical domains.67 While chelate cooperativity may be important, our thermal data clearly point to hydrophobic interactions between the cores as a major effect.
Conclusions
In summary, we have elucidated the effect of linking hydrophobic organic cores to the 3′- and 5′- ends of the DNA components used in the assembly of SMDH2 materials. MD simulations39 show that the organic cores of these building blocks minimize their hydrophobic surfaces by choosing the best stacking pattern possible when they self-assemble in aqueous media. Computational results indicate a high correlation between the linkage type (3′ or 5′) and the final SASA of organic cores that can be attributed to the extent that the cores can insert into the DNA minor grooves in the duplex arms of the resulting SMDH2. While 3′-linked organic cores can insert almost perfectly into the minor groove, 5′-linked cores can only insert partially, resulting in less-stable dimers (i.e., with higher SASA). These results can be used to explain why higher percentages of cyclic dimers form in SMDH2 materials with 3′-linked organic cores in comparison to those with 5′-linked cores, as observed experimentally for a broad range of structures: SMDH2 materials with 5′-linked cores are simply less stable in aqueous media due to the inability of the hydrophobic cores to insert completely into the minor groove. Inadequate shielding of the hydrophobic cores then forces these systems to choose other assembly patterns such as higher-order structures (tetramer, hexamer, etc.), as shown by nondenaturing PAGE gel analysis.
Notably, the important role that hydrophobic organic cores play in the assembly of SMDH2 hybrids is strongly supported by comparing the thermodynamic stability of several ff and cyclic dimers: ff-dimer systems consistently have Tm’s that are several degrees higher than analogous cyclic dimers, suggesting that hydrophobic interactions between the cores as a major factor contributing to stability. Together with the strong interactions observed between 3′-linked cores and the minor groove in SMDH2, this result opens up the possibility of controlling the product distribution in SMDH assembly using the hydrophobic nature of the organic cores in conjunction with the different linking modes (3′ and 5′) to DNA strands. Incorporating these design parameters to the synthesis of small molecule-DNA hybrids should expand the range of future applications for DNA-based hybrid materials.
Acknowledgments
All computations were carried out using the Advanced Research Computing (QUEST) cluster at the Northwestern University. This work was supported by the Air Force Office of Scientific Research (under agreement FA-9550-11-1-0275) (S.T.N. and G.C.S.) and the PS-OC Center of the NIH/NCI Grant 1U54CA143869-01 (G.C.S.). We thank Dr. Andrea Greschner (McGill University) for helpful discussions regarding the details of the PAGE-gel experiments.
Supporting Information Available
(Experimental) General procedures, materials, and instrumentations; analytical HPLC and MALDI-ToF analysis of SMDH2s; hybridization procedures and PAGE-Gel and optical melting experiments of the SMDH2 assemblies. (Computational) Parameterization of organic core and preparation of the DNA systems for MD simulations; unrestrained and restrained MD simulations; data analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ I.Y. and I.E. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Shi J.; Bergstrom D. E. Assembly of Novel DNA Cycles with Rigid Tetrahedral Linkers. Angew. Chem., Int. Ed. 1997, 36, 111–113. [Google Scholar]
- Wang W.; Wan W.; Zhou H.-H.; Niu S.; Li A. D. Q. Alternating DNA and Pi-Conjugated Sequences. Thermophilic Foldable Polymers. J. Am. Chem. Soc. 2003, 125, 5248–5249. [DOI] [PubMed] [Google Scholar]
- Abdalla M. A.; Bayer J.; Rädler J. O.; Müllen K. Synthesis and Self-Assembly of Perylenediimide–Oligonucleotide Conjugates. Angew. Chem., Int. Ed. 2004, 43, 3967–3970. [DOI] [PubMed] [Google Scholar]
- Neelakandan P. P.; Pan Z.; Hariharan M.; Zheng Y.; Weissman H.; Rybtchinski B.; Lewis F. D. Hydrophobic Self-Assembly of a Perylenediimide-Linked DNA Dumbbell into Supramolecular Polymers. J. Am. Chem. Soc. 2010, 132, 15808–15813. [DOI] [PubMed] [Google Scholar]
- Kuroda T.; Sakurai Y.; Suzuki Y.; Nakamura A. O.; Kuwahara M.; Ozaki H.; Sawai H. Assembly of DNA Nanostructures with Branched Tris-DNA. Chem. Asian J. 2006, 1, 575–580. [DOI] [PubMed] [Google Scholar]
- Aldaye F. A.; Sleiman H. F. Guest-Mediated Access to a Single DNA Nanostructure from a Library of Multiple Assemblies. J. Am. Chem. Soc. 2007, 129, 10070–10071. [DOI] [PubMed] [Google Scholar]
- Meng M.; Ahlborn C.; Bauer M.; Plietzsch O.; Soomro S. A.; Singh A.; Muller T.; Wenzel W.; Brase S.; Richert C. Two Base Pair Duplexes Suffice to Build a Novel Material. ChemBioChem 2009, 10, 1335–1339. [DOI] [PubMed] [Google Scholar]
- Eryazici I.; Prytkova T. R.; Schatz G. C.; Nguyen S. T. Cooperative Melting in Caged Dimers with Only Two DNA Duplexes. J. Am. Chem. Soc. 2010, 132, 17068–17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.; Tolev M.; Meng M.; Klenin K.; Plietzsch O.; Schilling C. I.; Muller T.; Nieger M.; Bräse S.; Wenzel W.; et al. Branched DNA That Forms a Solid at 95°C. Angew. Chem., Int. Ed. 2011, 50, 3227–3231. [DOI] [PubMed] [Google Scholar]
- Scheffler M.; Dorenbeck A.; Jordan S.; Wüstefeld M.; von Kiedrowski G. Self-Assembly of Trisoligonucleotidyls: The Case for Nano-Acetylene and Nano-Cyclobutadiene. Angew. Chem., Int. Ed. 1999, 38, 3311–3315. [PubMed] [Google Scholar]
- Shchepinov M. S.; Mir K. U.; Elder J. K.; Frank-Kamenetskii M. D.; Southern E. M. Oligonucleotide Dendrimers: Stable Nano-Structures. Nucleic Acids Res. 1999, 27, 3035–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs-Davis J. M.; Schatz G. C.; Nguyen S. T. Sharp Melting Transitions in DNA Hybrids without Aggregate Dissolution: Proof of Neighboring-Duplex Cooperativity. J. Am. Chem. Soc. 2007, 129, 15535–15540. [DOI] [PubMed] [Google Scholar]
- Alemdaroglu F. E.; Ding K.; Berger R.; Herrmann A. DNA-Templated Synthesis in Three Dimensions: Introducing a Micellar Scaffold for Organic Reactions. Angew. Chem., Int. Ed. 2006, 45, 4206–4210. [DOI] [PubMed] [Google Scholar]
- Ding K.; Alemdaroglu F. E.; Börsch M.; Berger R.; Herrmann A. Engineering the Structural Properties of DNA Block Copolymer Micelles by Molecular Recognition. Angew. Chem., Int. Ed. 2007, 46, 1172–1175. [DOI] [PubMed] [Google Scholar]
- Waybright S. M.; Singleton C. P.; Tour J. M.; Murphy C. J.; Bunz U. H. F. Synthesis and Self-Assembly of an Oligonucleotide-Modified Cyclobutadiene Complex. Organometallics 2000, 19, 368–370. [Google Scholar]
- Waybright S. M.; Singleton C. P.; Wachter K.; Murphy C. J.; Bunz U. H. F. Oligonucleotide-Directed Assembly of Materials: Defined Oligomers. J. Am. Chem. Soc. 2001, 123, 1828–1833. [DOI] [PubMed] [Google Scholar]
- Vargas-Baca I.; Mitra D.; Zulyniak H. J.; Banerjee J.; Sleiman H. F. Solid-Phase Synthesis of Transition Metal Linked, Branched Oligonucleotides. Angew. Chem., Int. Ed. 2001, 40, 4629–4632. [DOI] [PubMed] [Google Scholar]
- Stewart K. M.; McLaughlin L. W. Design and Synthesis of DNA-Tethered Ruthenium Complexes That Self-Assemble into Linear Arrays. Chem. Commun. 2003, 2934–2935. [DOI] [PubMed] [Google Scholar]
- Mitra D.; Di Cesare N.; Sleiman H. F. Self-Assembly of Cyclic Metal–DNA Nanostructures Using Ruthenium Tris(Bipyridine)-Branched Oligonucleotides. Angew. Chem., Int. Ed. 2004, 43, 5804–5808. [DOI] [PubMed] [Google Scholar]
- Stewart K. M.; McLaughlin L. W. Four-Arm Oligonucleotide Ni(II)-Cyclam-Centered Complexes as Precursors for the Generation of Supramolecular Periodic Assemblies. J. Am. Chem. Soc. 2004, 126, 2050–2057. [DOI] [PubMed] [Google Scholar]
- Stewart K. M.; Rojo J.; McLaughlin L. W. Ru(Ii) Tris(Bipyridyl) Complexes with Six Oligonucleotide Arms as Precursors for the Generation of Supramolecular Assemblies. Angew. Chem., Int. Ed. 2004, 43, 5808–5811. [DOI] [PubMed] [Google Scholar]
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. A DNA-Based Method for Rationally Assembling Nanoparticles into Macroscopic Materials. Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- Alivisatos A. P.; Johnsson K. P.; Peng X.; Wilson T. E.; Loweth C. J.; Bruchez M. P.; Schultz P. G. Organization of ’Nanocrystal Molecules’ Using DNA. Nature 1996, 382, 609–611. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. DNA in a Material World. Nature 2003, 421, 427–431. [DOI] [PubMed] [Google Scholar]
- Lin C.; Liu Y.; Yan H. Designer DNA Nanoarchitectures. Biochemistry 2009, 48, 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- Zhang C.; He Y.; Su M.; Ko S. H.; Ye T.; Leng Y.; Sun X.; Ribbe A. E.; Jiang W.; Mao C. DNA Self-Assembly: From 2D to 3D. Faraday Discuss. 2009, 143, 221–233. [DOI] [PubMed] [Google Scholar]
- Lo P. K.; Metera K. L.; Sleiman H. F. Self-Assembly of Three-Dimensional DNA Nanostructures and Potential Biological Applications. Curr. Opin. Chem. Biol. 2010, 14, 597–607. [DOI] [PubMed] [Google Scholar]
- He Y.; Ye T.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. Hierarchical Self-Assembly of DNA into Symmetric Supramolecular Polyhedra. Nature 2008, 452, 198–201. [DOI] [PubMed] [Google Scholar]
- Andersen E. S.; Dong M.; Nielsen M. M.; Jahn K.; Subramani R.; Mamdouh W.; Golas M. M.; Sander B.; Stark H.; Oliveira C. L. P.; et al. Self-Assembly of a Nanoscale DNA Box with a Controllable Lid. Nature 2009, 459, 73–76. [DOI] [PubMed] [Google Scholar]
- Elghanian R.; Storhoff J. J.; Mucic R. C.; Letsinger R. L.; Mirkin C. A. Selective Colorimetric Detection of Polynucleotides Based on the Distance-Dependent Optical Properties of Gold Nanoparticles. Science 1997, 277, 1078–1081. [DOI] [PubMed] [Google Scholar]
- Taton T. A.; Mirkin C. A.; Letsinger R. L. Scanometric DNA Array Detection with Nanoparticle Probes. Science 2000, 289, 1757–1760. [DOI] [PubMed] [Google Scholar]
- Gibbs J. M.; Park S.-J.; Anderson D. R.; Watson K. J.; Mirkin C. A.; Nguyen S. T. Polymer-DNA Hybrids as Electrochemical Probes for the Detection of DNA. J. Am. Chem. Soc. 2005, 127, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Yang H.; McLaughlin C. K.; Aldaye F. A.; Hamblin G. D.; Rys A. Z.; Rouiller I.; Sleiman H. F. Metal-Nucleic Acid Cages. Nature. Chem. 2009, 1, 390–396. [DOI] [PubMed] [Google Scholar]
- Park S. Y.; Lytton-Jean A. K. R.; Lee B.; Weigand S.; Schatz G. C.; Mirkin C. A. DNA-Programmable Nanoparticle Crystallization. Nature 2008, 451, 553–556. [DOI] [PubMed] [Google Scholar]
- Aldaye F. A.; Sleiman H. F. Dynamic DNA Templates for Discrete Gold Nanoparticle Assemblies: Control of Geometry, Modularity, Write/Wrase and Structural Switching. J. Am. Chem. Soc. 2007, 129, 4130–4131. [DOI] [PubMed] [Google Scholar]
- Lo P. K.; Karam P.; Aldaye F. A.; McLaughlin C. K.; Hamblin G. D.; Cosa G.; Sleiman H. F. Loading and Selective Release of Cargo in DNA Nanotubes with Longitudinal Variation. Nature. Chem. 2010, 2, 319–328. [DOI] [PubMed] [Google Scholar]
- Macfarlane R. J.; Lee B.; Jones M. R.; Harris N.; Schatz G. C.; Mirkin C. A. Nanoparticle Superlattice Engineering with DNA. Science 2011, 334, 204–208. [DOI] [PubMed] [Google Scholar]
- Eryazici I.; Yildirim I.; Schatz G. C.; Nguyen S. T. Enhancing the Melting Properties of Small Molecule-DNA Hybrids through Designed Hydrophobic Interactions: An Experimental-Computational Study. J. Am. Chem. Soc. 2012, 134, 7450–7458. [DOI] [PubMed] [Google Scholar]
- Greschner A. A.; Toader V.; Sleiman H. F. The Role of Organic Linkers in Directing DNA Self-Assembly and Significantly Stabilizing DNA Duplexes. J. Am. Chem. Soc. 2012, 134, 14382–14389. [DOI] [PubMed] [Google Scholar]
- Greschner A. A.; Bujold K. E.; Sleiman H. F. Intercalators as Molecular Chaperones in DNA Self-Assembly. J. Am. Chem. Soc. 2013, 135, 11283–11288. [DOI] [PubMed] [Google Scholar]
- Dogan Z.; Paulini R.; Stütz J. A. R.; Narayanan S.; Richert C. 5′-Tethered Stilbene Derivatives as Fidelity- and Affinity-Enhancing Modulators of DNA Duplex Stability. J. Am. Chem. Soc. 2004, 126, 4762–4763. [DOI] [PubMed] [Google Scholar]
- Patra A.; Richert C. High Fidelity Base Pairing at the 3′-Terminus. J. Am. Chem. Soc. 2009, 131, 12671–12681. [DOI] [PubMed] [Google Scholar]
- Ng P.-S.; Laing B. M.; Balasundarum G.; Pingle M.; Friedman A.; Bergstrom D. E. Synthesis and Evaluation of New Spacers for Use as dsDNA End-Caps. Bioconjugate Chem. 2010, 21, 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan M.; Zheng Y.; Long H.; Zeidan T. A.; Schatz G. C.; Vura-Weis J.; Wasielewski M. R.; Zuo X.; Tiede D. M.; Lewis F. D. Hydrophobic Dimerization and Thermal Dissociation of Perylenediimide-Linked DNA Hairpins. J. Am. Chem. Soc. 2009, 131, 5920–5929. [DOI] [PubMed] [Google Scholar]
- Knotts T. A.; Rathore N.; Schwartz D. C.; de Pablo J. J. A Coarse Grain Model for DNA. J. Chem. Phys. 2007, 126, 084901. [DOI] [PubMed] [Google Scholar]
- Drukker K.; Schatz G. C. A Model for Simulating Dynamics of DNA Denaturation. J. Phys. Chem. B 2000, 104, 6108–6111. [Google Scholar]
- Prytkova T. R.; Eryazici I.; Stepp B. R.; Nguyen S. T.; Schatz G. C. DNA Melting in Small-Molecule-DNA-Hybrid Dimer Structures: Experimental Characterization and Coarse-Grained Molecular Dynamics Simulations. J. Phys. Chem. B 2010, 114, 2627–2634. [DOI] [PubMed] [Google Scholar]
- Sayar M.; Avşaroğlu B.; Kabakçıoğlu A. Twist-Writhe Partitioning in a Coarse-Grained DNA Minicircle Model. Phys. Rev. E 2010, 81, 041916. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Merz K. M.; Pearlman D. A.; Crowley M.. et al. AMBER 9; University of California: San Francisco, 2006.
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Crowley M.; Walker R. C.; Zhang W.. et al. AMBER 10; University of California: San Francisco, 2008.
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.. et al. AMBER 11; University of California: San Francisco, 2010.
- MacKerell A. D. Jr.; Banavali N.; Foloppe N. Development and Current Status of the CHARMM Force Field for Nucleic Acids. Biopolymers 2000, 56, 257–265. [DOI] [PubMed] [Google Scholar]
- Hart K.; Foloppe N.; Baker C. M.; Denning E. J.; Nilsson L.; MacKerell A. D. Jr. Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput. 2011, 8, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen C. H.; Pedersen E. B. Design of an Intercalating Linker Leading to the First Efficiently 5′,5′-Linked Alternate-Strand Hoogsteen Triplex with High Stability and Specificity. Helv. Chim. Acta 2004, 87, 2465–2471. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Wang J.; Cieplak P.; Kollman P. A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules?. J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar]
- Yildirim I.; Stern H. A.; Kennedy S. D.; Tubbs J. D.; Turner D. H. Reparameterization of RNA χ Torsion Parameters for the AMBER Force Field and Comparison to NMR Spectra for Cytidine and Uridine. J. Chem. Theory Comput. 2010, 6, 1520–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez A.; Marchán I.; Svozil D.; Sponer J.; Cheatham T. E. III; Laughton C. A.; Orozco M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of A/Γ Conformers. Biophys. J. 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins G. D.; Cramer C. J.; Truhlar D. G. Pairwise Solute Descreening of Solute Charges from a Dielectric Medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar]
- Hawkins G. D.; Cramer C. J.; Truhlar D. G. Parametrized Models of Aqueous Free Energies of Solvation Based on Pairwise Descreening of Solute Atomic Charges from a Dielectric Medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar]
- Nguyen H.; Roe D. R.; Simmerling C. Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput. 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H.; Mark A. E.; Zhu J.; Honig B. Comparative Study of Generalized Born Models: Protein Dynamics. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6760–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chocholoušová J.; Feig M. Implicit Solvent Simulations of DNA and DNA–Protein Complexes: Agreement with Explicit Solvent vs Experiment. J. Phys. Chem. B 2006, 110, 17240–17251. [DOI] [PubMed] [Google Scholar]
- Mitchell J.; Harris S. Testing the Use of Implicit Solvent in the Molecular Dynamics Modelling of DNA Flexibility. Prog. Theor. Phys. Supplement 2011, 191, 96–108. [Google Scholar]
- We previously reported cooperative melting in cage- and ff-dimer structures with two and three duplexes in the presence of Na+ ions (see refs (8) and (39)) The same cooperative behavior can be observed in the cyclic dimers used in this study in presence of Mg2+ ions. As shown in Table 4, higher melting temperatures, Tm, and sharper transitions (fwhm = 6.7–7.6 °C) are observed for these dimers compared to the corresponding free-DNA duplex.
- Becaud J.; Pompizi I.; Leumann C. J. Propagation of Melting Cooperativity Along the Phosphodiester Backbone of DNA. J. Am. Chem. Soc. 2003, 125, 15338–15342. [DOI] [PubMed] [Google Scholar]
- Letsinger R. L.; Wu T.; Yang J.-S.; Lewis F. D. DNA-Templated Formation and Luminescence of Diphenylacetylene Dimeric and Trimeric Complexes. Photochem. Photobiol. Sci. 2008, 7, 854–859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.