

Abstract
The insulin sensitizing thiazolidinedione drugs, rosiglitazone and pioglitazone are specific peroxisome proliferator-activated receptor-gamma (PPARγ) agonists and reduce pro-inflammatory responses in patients with type 2 diabetes and coronary artery disease and may be beneficial in sepsis. Sepsis was induced in 8–10 wk old C57BL/6 mice by cecal ligation and puncture (CLP) with a 22g double puncture technique. Mice received intraperitoneal injection of vehicle (DMSO:PBS) or pioglitazone (20mg/kg) at 1h and 6h after CLP and were sacrificed at various timepoints. In sepsis, vehicle-treated mice had hypoglycemia, increased lung injury and increased lung neutrophil infiltration. Pro-inflammatory plasma cytokines were increased but the plasma adipokine, adiponectin, was decreased in vehicle-treated septic mice. This corresponded with inhibitor κB (IκBα) protein degradation and an increase in NF-κB activity in lung. Pioglitazone treatment improved plasma glucose and adiponectin levels and decreased pro-inflammatory cytokines. Lung IκBα protein expression increased and corresponded with a decrease in nuclear factor kappa-B (NF-κB) activity in the lung from pioglitazone treated mice. Pioglitazone reduces the inflammatory response in polymicrobial sepsis in part through inhibition of NF-κB and may be a novel therapy in sepsis.
Keywords: sepsis, pioglitazone, inflammation, nuclear factor kappa-b
Introduction
Sepsis is a potentially life-threatening disorder causing death in an estimated 215,000 patients per year in the United States.1 The mortality rate increases with age from 10% in children to 38% in the elderly population.1, 2 Treating patients with severe sepsis costs nearly 17 billion dollars.3–5 Unfortunately, few clinical studies show a difference in outcomes in patients with sepsis.6–9
The nuclear receptor peroxisome proliferator-activated receptor-gamma (PPARγ) is involved in the regulation of sepsis-induced inflammation. Data from our laboratory and others demonstrate that in animal models of sepsis PPARγ expression is markedly downregulated in the lung, liver, and peripheral blood cells.10–14 This downregulation is associated with severe inflammation and multiple organ failure. On the contrary, treatment with activators of PPARγ reduces inflammation and improves survival in experimental animals. In a pilot clinical trial in pediatric patients with sepsis, we found that PPARγ expression is decreased in peripheral blood mononuclear cells from children with septic shock compared with controls.10
The thiazolidinediones (TZDs), pioglitazone and rosiglitazone, are Food and Drug Administration (FDA)-approved medications that activate PPARγ. TZDs are used therapeutically as insulin-sensitizers for patients with type 2 diabetes. In patients with type 2 diabetes TZDs have favorable effects on vascular function and inflammation.15–17 Pioglitazone also decreases inflammatory mediators in patients with type 2 diabetes and coronary artery disease.15 PPAR gamma agonists are beneficial in experimental models of infection. In influenza pneumonia, PPAR gamma agonists improve survival.18–20 Additionally, in mice subjected to cecal ligation and puncture (CLP) pioglitazone administration decreased inflammation and improved survival in apolipoprotein E knockout mice.21 As a result, TZDs may modulate inflammation and be beneficial in infection-related inflammatory conditions.
Therefore, pioglitazone, through its activation of PPARγ and subsequent anti-inflammatory response, may prove as a novel therapeutic in sepsis. However the mechanisms involved in the anti-inflammatory responses of pioglitazone treatment in sepsis remain unknown. We hypothesized that pioglitazone treatment will decrease the inflammatory response in mice subjected to polymicrobial sepsis.
Materials and Methods or Experimental Procedures
Materials
The primary antibody for PPARγ was obtained from Thermo Fisher Scientific (Rockford, IL) and antibodies for inhibitor kB (IκBα), intercellular adhesion molecule (ICAM)-1 and actin were obtained from Santa Cruz Biotechnology (Dallas, TX). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Pioglitazone was obtained from Enzo Life Sciences (Farmingdale, NY).
Mice
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health and was approved by the institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center.22 The experimental groups consisted of male C57BL/6 mice at 8–10 weeks of age supplied from Charles River Laboratories International, Inc. (Wilmington, MA). The mice were housed in the animal facility at the Cincinnati Children’s Research Foundation (CCRF). Food and water were provided ad libitum. Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) and mice were sacrificed at various time points thereafter. Plasma and lung samples were collected for biochemical studies as described below.
Mouse model of cecal ligation and puncture
CLP was performed as previously described.23 Mice were anesthetized with isoflurane via nose cone throughout the surgical procedure. After opening the abdomen, the cecum was exteriorized and ligated by a 6.0 silk ligature at its base without obstructing intestinal continuity. The cecum was punctured twice with a 22-gauge needle and returned to the peritoneal cavity. The abdominal incision was closed with silk running sutures and liquid topical adhesive. After the procedure, animals were fluid resuscitated with sterile saline (0.6 ml) injected subcutaneously but did not receive antibiotics. Sham mice underwent an abdominal incision and exteriorization of the cecum but did not undergo CLP. Mice received intraperitoneal injection of vehicle (DMSO:PBS) or pioglitazone (20 mg/kg, 1:1 DMSO:PBS) at 1 and 6h after CLP. The dose of pioglitazone was chosen based on published reports and modified as a result of results from our laboratory.24, 25
Histology
Lungs were fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin.
Measurement of myeloperoxidase activity
Myeloperoxidase (MPO) activity was determined as an index of neutrophil accumulation in lung as previously described.23 Tissues were homogenized in a solution containing 0.5% hexa-decyl-trimethyl-ammonium bromide dissolved in 10mM potassium phosphate buffer (pH 7) and were centrifuged for 30 min at 20,000 × g at 4°C. An aliquot of the supernatant was allowed to react with a solution of tetra-methyl-benzidine (1.6mM) and 0.1 mM H2O2. The rate of change in absorbance was measured by spectrophotometry at 650 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol hydrogen peroxide/min at 37°C and was expressed in units per 100 mg of tissue.
Plasma levels of adipokines and cytokines
Plasma levels of tumor necrosis factor-α (TNFα), interleukin (IL)-6, and adiponectin (high molecular weight adiponectin hexamers and trimers) were measured by use of the multiplex assay kit (Millipore, Billerica, MA) using the protocol recommended by the manufacturer.
Blood glucose levels
Glucose levels were determined by i-STAT measurement at time of tissue harvest.
Plasma levels of 15d-PGJ2
Plasma samples of 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) were measured by enzyme immunoassay kit (Enzo Life Sciences, Farmingdale, NY) using the protocol recommended by the manufacturer.
Subcellular fractionation and nuclear protein extraction
Tissue samples were homogenized in a buffer containing 0.32 M sucrose, 10 mM Tris-HCl, 1 mM ethylene glycol tetraacetic acid (EGTA), 2 mM ethylenediaminetetraacetic acid (EDTA), 5 mM NaN3, 10 mM β-mercaptoethanol, 50 mM NaF, 20 μM leupeptin, 0.15 μM pepstatin A, and 0.2 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM sodium orthovanadate, 0.4 nM microcystin.11 The homogenates were centrifuged (1,000 × g at 4°C, 10 min). The supernatant (cytosol + membrane extract) was collected and stored. The pellets were solubilized in Triton buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH7.4), 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovanadate, 20 μM leupeptin A, and 0.2 mM PMSF). The lysates were centrifuged (15,000 × g, at 4°C, 30 min) and the supernatant (nuclear extract) collected and stored at −80°C. The amount of protein was quantified by Bradford assay.
Western blot analysis
The nuclear or cytosol content of PPARγ, IκBα, ICAM-1 and actin in the lung were determined by immunoblot analyses. Extracts were boiled in equal volumes of loading buffer (125 mmol/L Tris-HCL, ph 6.8, 4% SDS, 20% glycerol, and 10% 2-mercaptoethanol) and 50 μg of protein was loaded per lane on 8%–16% Tris-glycine gradient gel. Proteins were separated electrophoretically and transferred to nitrocellulose membranes. For immunoblotting, membranes were blocked with 5% nonfat dried milk in Tris-buffered saline for 1 hour and then incubated with primary antibodies against PPARγ, IκBα, and ICAM-1. The membranes were washed in Tris-buffered saline with 0.1% Tween 20 and incubated with secondary peroxidase-conjugated antibody. Membranes were reprobed with primary antibody against actin to ensure equal loading. Detection was enhanced by chemiluminescence and exposed to photographic film. Densitometric analysis of blots was performed using ImageQuant (Molecular Dynamics, Sunnyvale, CA).
Determination of NF-κB activity
Nuclear factor kappa-B (NF-κB) (p65) activation was detected in liver nuclear extracts by using an ELISA-based transcription factor assay kit as described previously.26 Nuclear protein, 10 μg, was obtained from liver nuclear extracts and added to a 96-well plate to which oligonucleotide containing the NF-κB consensus binding sequence had been immobilized (Active Motif North America, Carlsbad, CA). Antibody directed against the p65 subunit was added followed by a secondary horseradish peroxidase conjugate antibody. Developing solution utilizing a colorimetric readout was used. The plate was read by a spectrophotometer at 450 nm with a reference wavelength of 655 nm. A wild-type and mutated oligonucleotides were used for NF-κB binding to monitor the specificity of the assay (data not shown).
Statistical analysis
Statistical analysis of plasma cytokines, adiponectin, 15d-PGJ2 levels, and were performed using the Mann Whitney Rank Sum test and are expressed in the text as median with interquartile range. Statistical analyses of lung myeloperoxidase activity were compared by analysis of variance with Holm-Sidak method. The t-test was used to compare plasma glucose levels, densitometric analysis of lung IκBα and PPARγ, and lung NF-κB p65 activity and are expressed in the text as mean ± SEM. N = 3–4 animals were used for each group. A value of p ≤ 0.05 was considered significant.
Results
Pioglitazone reduces lung injury and lung neutrophil infiltration after induction of polymicrobial sepsis
To determine the pioglitazone effects in polymicrobial sepsis, mice were subjected to CLP and sacrificed at various time points. As early as 6h after CLP, vehicle-treated mice exhibited marked lung injury characterized by extravasation of red cells, alveolar edema and accumulation of inflammatory cells (Figure 1C). This was associated with a significant increase in lung neutrophil infiltration quantified by myeloperoxidase (MPO) assay. Specifically, MPO activity was increased at 6 and 18h after CLP in vehicle-treated mice (182 ± 27 and 177 ± 22 U/100mg tissue, respectively) when compared to sham mice (113 ± 19 and 13 ± 5 U/100mg tissue, respectively, p<0.05) (Figure 2A). Pioglitazone-treatment revealed a marked reduction of inflammatory cells in the lung with the return to normal lung architecture (Figure 1D). This was associated with a significant reduction in lung neutrophil infiltration compared with vehicle treatment at 6 h after CLP (111 ± 9 U/100mg tissue, p<0.05) (Figure 2A). To investigate the mechanism through which neutrophil infiltration is increased after CLP, expression of the adhesion molecule, ICAM-1, was investigated by Western blot analysis. Lung expression of ICAM-1 was increased at 18h after CLP in vehicle-treated mice compared to control mice (Figure 2B). Treatment with pioglitazone reduced ICAM-1 expression in the lung although this was not statistically significant (p=0.1).
Figure 1.
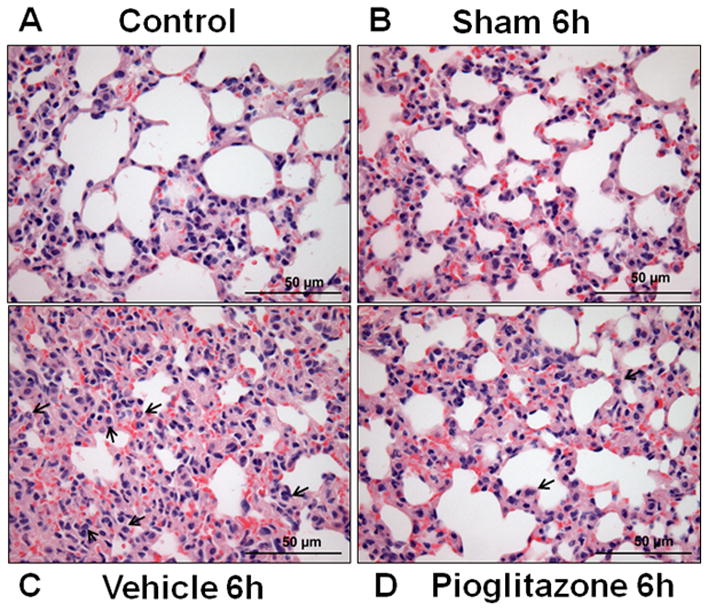
Pioglitazone improves lung injury after CLP. (A) Lung from control mice revealing normal architecture. (B) Lung from 6h sham mice. (C) Lung from vehicle-treated mice showing interstitial hemorrhage, neutrophil infiltration and obliteration of normal architecture. Black arrows denote neutrophil infiltration (D) Lung from pioglitazone-treated mice reveals reduction of hemorrhage and reduction of neutrophil infiltration and amelioration of lung injury. Black arrows denote neutrophil infiltration. 40x magnification.
Figure 2.
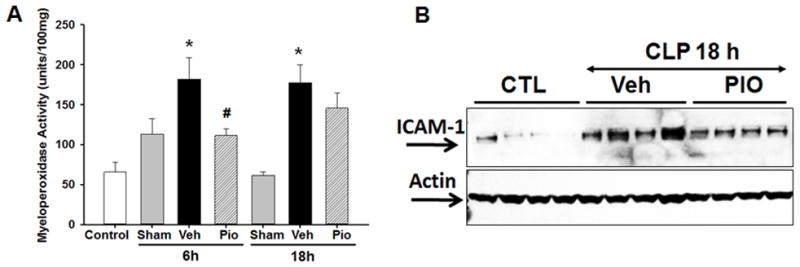
Effect of pioglitazone on lung injury after CLP. (A) Myeloperoxidase activity was determined at 6 and 18h after CLP. *p<0.05 vs. sham and #p<0.05 vs. vehicle. n=3–4 mice/group and samples were run in duplicate. (B) Representative Western blot analysis for lung expression of ICAM-1 at 18h after CLP. n=4 mice/group.
Pioglitazone reduces pro-inflammatory cytokines and increases adiponectin levels
To further investigate the effect of pioglitazone on the systemic inflammatory response we measured plasma cytokine levels. The plasma cytokines, TNFα and IL-6, were increased in vehicle-treated mice at 6h after CLP [74 pg/ml (IQR 10.3 – 165.1) and 36,588 pg/ml (IQR 32,891 – 37,095) respectively] compared to sham mice [10 pg/ml and 144 pg/ml respectively, p<0.05] (Figure 3). Pioglitazone treatment reduced TNFα and IL-6 plasma levels at 6h after CLP [16 pg/ml (6 – 38) and 17,866 pg/ml (5,001 – 34,792) respectively, p<0.05]. Adiponectin, an adipose tissue-derived cytokine, has a peroxisome proliferator response element (PPRE) in its promoter region.27 Consequently changes in PPARγ activity may be reflected as changes in adiponectin.10 Adiponectin levels were lower in septic vehicle-treated mice at both 3 and 6 hours after CLP (Figure 4). However, treatment with pioglitazone increased plasma adiponectin levels (Figure 4).
Figure 3.
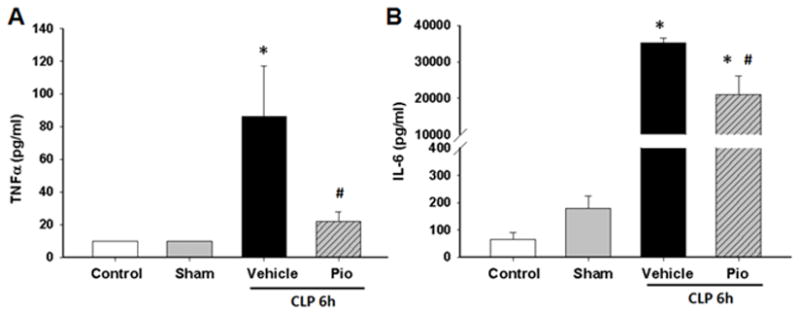
Pioglitazone reduces plasma cytokines after CLP. Plasma cytokine levels (A) TNFα and (B) IL-6 were measured at 6h after CLP. *p<0.05 vs. control and #p<0.05 vs. vehicle. n=3–4 mice/group and samples were run in duplicate.
Figure 4.
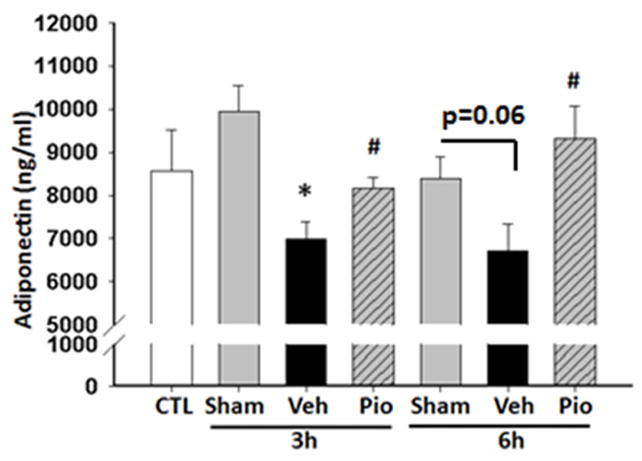
Pioglitazone increases plasma adiponectin levels after CLP. Plasma adiponectin were measured at 3 and 6h after CLP. *p<0.05 vs. control and #p<0.05 vs. vehicle. n=3–4 mice/group and samples were run in duplicate.
Pioglitazone improves glucose control
Pioglitazone improves glucose control and thus is used clinically as a treatment for diabetes. Therefore we investigated the glucose effects of pioglitazone following CLP. Vehicle-treated mice had a significant decrease in blood glucose levels at 6h after CLP compared to sham mice (60 ± 4 mg/dl vs. 160 ± 12 mg/dl respectively, p<0.01). However, pioglitazone treatment significantly increased glucose levels at 6h after CLP compared with mice treated with vehicle alone (101 ± 13 mg/dl, p<0.05).
Pioglitazone increases 15d-PGJ2 plasma levels
The cyclopentenone prostaglandin, 15d-PGJ2, is an endogenous ligand for PPARγ, so we investigated whether pioglitazone alters plasma 15d-PGJ2 levels in polymicrobial sepsis. Plasma levels of 15d-PGJ2 were markedly diminished in vehicle treated mice at 6h after CLP when compared to sham [2,420 pg/ml (IQR 1,975 – 3,348) vs. 4,011 pg/ml (IQR 3,546 – 5,695), p<0.05] (Figure 5). However, mice treated with pioglitazone had significantly higher plasma levels of 15d-PGJ2 compared to vehicle treated mice [4,332 pg/ml (IQR 3,271 – 5,464), p<0.05]. These results demonstrate that treatment with synthetic, exogenous ligand of PPARγ also affect the endogenous ligand.
Figure 5.
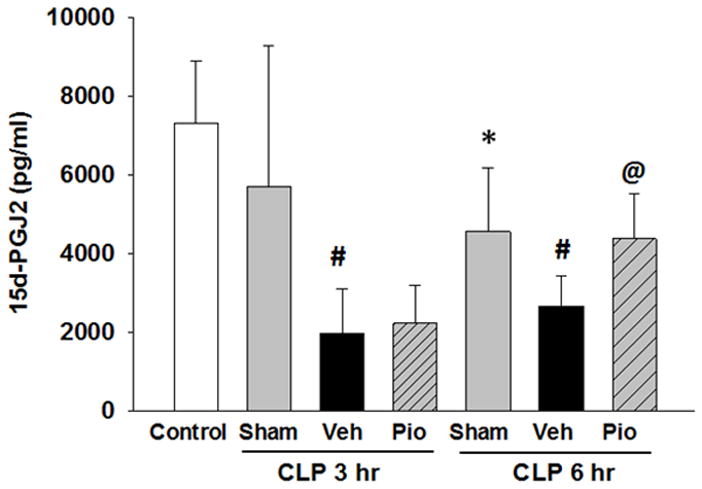
Plasma levels of 15d-PGJ2 are altered after CLP and restored by pioglitazone. Plasma levels of 15d-PGJ2 were evaluated by ELISA at 3 and 6h after CLP. *p<0.05 vs. control, #p<0.05 vs. sham, @p<0.05 vs. vehicle. n=3–4 mice/group and samples were run in duplicate.
Pioglitazone increases lung PPARγ protein expression
Since pioglitazone treatment altered inflammatory cytokine production and improved glucose control we sought to determine whether these effect were associated with changes in PPARγ expression in the lung. At Western blot analysis, expression of PPARγ was decreased in the nuclear compartment in lungs of vehicle-treated mice at 18h after CLP (Figure 6). Treatment with pioglitazone increased lung PPARγ protein expression after CLP compared to vehicle treated mice.
Figure 6.
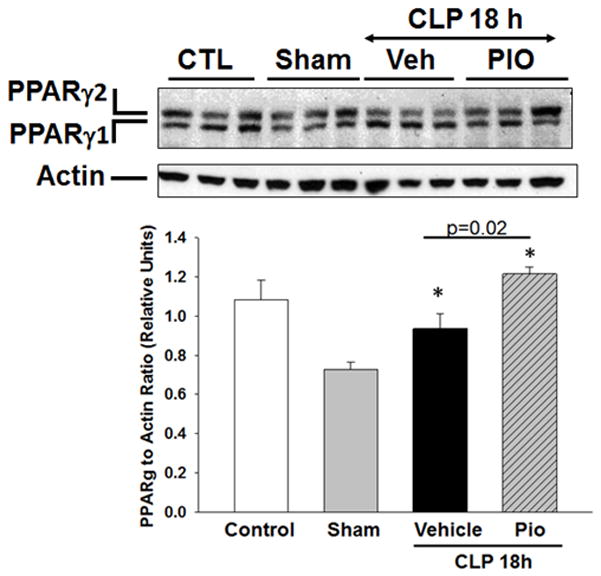
Effect of pioglitazone treatment on nuclear PPARγ expression in the lung. Representative Western blot analysis for lung expression of the two isoforms of PPARγ (PPARγ1 and PPARγ2) and relative densitometric analysis. *p<0.05 vs. sham. n=3 mice/group.
Pioglitazone inhibits NF-κB activity in the lung after CLP
To investigate the mechanism of action of pioglitazone, we evaluated the activation of the NF-κB pathway. Vehicle treated mice exhibited marked IκBα protein degradation in the lung as measured by Western blot analysis at 18h after CLP (Figure 7). The reduction in IκBα protein expression was associated with a significant increase in NF-κB p65 DNA binding activity in the lung at 18h after CLP compared with controls (0.39 relative units ± 0.03 vs. 0.21 relative units ± 0.03, p<0.05). However, treatment with pioglitazone increased lung IκBα protein expression and reduced NF-κB DNA binding at 18h after CLP vs. vehicle treatment (0.16 relative units ± 0.1, p<0.05).
Figure 7.
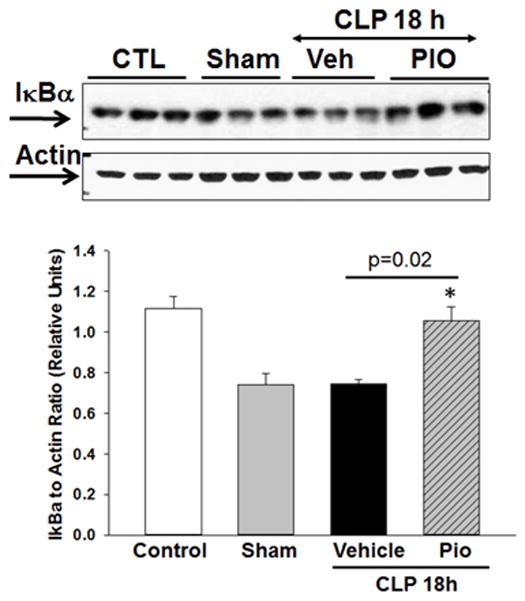
Effect of pioglitazone treatment on IκBα expression in the lung. Representative Western blot analysis for lung expression of cytosol IκBα and relative densitometric analysis. *p<0.05 vs. sham. n=3 mice/group.
Discussion
There are very few therapies which improve outcomes in patients with sepsis. Therefore it is imperative that other therapeutic options be explored. Activators of PPARγ may be novel therapeutic agents in sepsis.28 Preclinical work form our laboratory has demonstrated that PPARγ expression is downregulated on the endothelium of thoracic aortas and in the lung in polymicrobial sepsis and this downregulation is associated with poor survival.11, 12 Furthermore, in animal models of sepsis treatment with the PPARγ ligand, 15d-PGJ2, decreases neutrophil infiltration and adhesion molecule expression in the lung and small intestine.11
Pioglitazone and rosiglitazone are FDA-approved medications that are used clinically in diabetes. These medications also activate PPARγ and may prove as novel therapies in clinical sepsis. However, concerns have been raised regarding the long-term use of rosiglitazone in adults and its association with heart failure and death.29 Although there have been important concerns regarding the use of rosiglitazone in adults and its association with heart failure and death, a large meta-analysis demonstrated that pioglitazone is associated with a significantly lower risk of death, myocardial infarction or stroke in patients with type 2 DM compared with placebo.30 In order to have a translatable model that is clinically relevant and given the concerns regarding rosiglitazone we have used pioglitazone as an activator of PPARγ.
PPARs are ligand-dependent transcription factors and nuclear receptors that influence cellular responses by altering gene expression. Although PPARs were initially described as important in triglyceride and cholesterol homeostasis these receptors are also important in regulating the inflammatory response.31 PPARγ can trans-activate and trans-repress target genes through ligand-dependent and independent mechanisms.32–35 The insulin-sensitizing drugs, thiazolidinediones (TZDs), and the cyclopentenone prostaglandin, 15d-PGJ2, are specific PPARγ agonists.36–38 The endogenous ligand, 15d-PGJ2, can repress the expression of inflammatory genes in activated macrophages including TNFα and COX-2.39 Data from our laboratory and others demonstrate that 15d-PGJ2 has anti-inflammatory effects on NF-κB activation. This inhibition occurs through PPARγ-dependent and independent mechanisms.40–43 One mechanism by which 15d-PGJ2 has effects is through binding of the electrophilic carbon in the cyclopentenone ring to cellular proteins, modifying signaling pathways.44 This mechanism may account for the direct repression of NF-κB by 15d-PGJ2.45 Clinically, 15d-PGJ2 production may predict PPARγ activation in vivo. 15d-PGJ2 can be measured in urine, synovial fluid and plasma.46, 47 Reduced levels of 15d-PGJ2 from articular chondrocytes correlate with the degree of inflammation in patients with rheumatoid and osteo-arthritis and were lower than levels from patients undergoing joint surgery for traumatic fracture. Our data demonstrate that plasma levels of 15d-PGJ2 are decreased in an experimental sepsis and correlate with an increase in inflammatory mediators. However, treatment with pioglitazone restores 15d-PGJ2 and decreases inflammation.
The exogenous activators of PPARγ, pioglitazone and rosiglitazone, exert anti-inflammatory effects in experimental models of inflammation. Activation of PPARγ by rosiglitazone inhibited LPS release of high mobility group box 1 (HMGB1), a late inflammatory mediator in sepsis, from RAW 264.7 macrophages.48 In a mouse model of endotoxin, rosiglitazone improved survival and cardiac dysfunction.49 And in experimental polymicrobial sepsis, rosiglitazone decreased leukocyte rolling and adhesion in the brain microvasculature after CLP compared to vehicle treated mice.50 Tsujimura et al. demonstrated that pretreatment with pioglitazone for seven days prior to CLP improved sepsis survival compared to pretreatment with vehicle only.51 In the current study, pioglitazone decreased pro-inflammatory plasma cytokines and resulted in a reduction in neutrophil infiltration and lung injury following induction of sepsis. Mice treated with pioglitazone had higher plasma 15d-PGJ2 levels and increased PPARγ protein expression in the lung. Mechanistically, in the lung, these effects were associated with NF-κB inhibition. Our current findings support these data and demonstrate that pioglitazone, even when given after the induction of sepsis, inhibits anti-inflammatory mediators and may exert anti-inflammatory effects by modulating NF-κB activity.
Based on the presence of a direct binding site for PPARγ in its promoter region, adiponectin is a target for PPARγ.52 Adiponectin is an adipocyte-derived protein which is secreted into human plasma.53–56 The adiponectin protein exists as multimer complexes in plasma: a low-molecular weight trimer, a middle-molecular weight hexamer, and a high-molecular weight protein (HMWA).57 In humans, the HMWA has been proposed as the most potent form mediating the metabolic actions of adiponectin.58, 59 In addition to its metabolic actions adiponectin has anti-inflammatory effects and plays a role in the innate and adaptive immune response. In an animal model of polymicrobial sepsis, plasma adiponectin levels were lower after sepsis and inversely correlated with plasma TNFα and endotoxin levels.60 Adiponectin knockout mice had higher inflammatory cytokine production and higher mortality after polymicrobial sepsis compared to wild-type mice.61, 62 Adiponectin reduces LPS-induced lung injury and when adiponectin is knocked out, mice treated with LPS have increased systemic and lung TNFα levels and worse lung injury.63 Data from the current study support these findings that plasma adiponectin are decreased in experimental sepsis and this decrease is associated with increased systemic and lung inflammation. In our study, treatment with pioglitazone, however, was able to restore plasma adiponectin levels.
Adiponectin is also altered in patients with sepsis. In separate human studies, healthy volunteers demonstrated no significant change in plasma adiponectin levels after endotoxin; however, the adiponectin receptors, AdipoR1 and AdipoR2, were decreased in monocytes after endotoxemia injection.64, 65 Data from our laboratory utilizing a cohort of critically ill children with sepsis demonstrated contrasting findings to these studies but are consistent with published data in other inflammatory conditions. We found that on the first day of hospitalization plasma high molecular weight adiponectin levels are increased in children with septic shock compared to control subjects.10 A similar increase in adiponectin was demonstrated in patients with inflammation from arthritis and inflammatory bowel disease.66, 67 These findings suggest that adiponectin may play a role in sepsis. Adiponectin has a PPRE in their promoter regions therefore changes in PPARγ activity may be reflected in changes in plasma adiponectin.27 Multiple studies have demonstrated that treatment with thiazolidinediones increase plasma adiponectin levels and increase adiponectin mRNA expression in adipocytes and adipose tissue.68–70
Further studies are needed to determine the exact mechanisms which account for these protective effects and whether pioglitazone protection in sepsis is through alterations in glucose, adiponectin and PPARγ or through alternative actions. Although not investigated in this study, the anti-inflammatory effects of pioglitazone may be a result of an increase in adiponectin and not directly related to PPARγ effects. Adiponectin inhibits LPS-induced TNFα production by macrophages.71 Adiponectin can also directly bind to LPS extracellularly.60, 72 In a dose dependent manner, recombinant adiponectin suppressed limulus amoebocyte lysate activity through direct binding of LPS.60 15d-PGJ2 is derived from arachidonic acid via the cyclo-oxygenase (COX) pathway. Therefore increases in COX expression may result in 15d-PGJ2 expression. Shibata et al. demonstrated that adiponectin exerts anti-inflammatory protection in myocardial ischemia-reperfusion injury through COX-2 dependent action.73, 74 Inhibiting COX-2 in cardiac myocytes following myocardial ischemia reperfusion injury resulted in a reduction in adiponectin-stimulated prostaglandin E2 production. Additionally, inhibiting COX-2 reversed the suppressive effect of adiponectin on TNFα plasma levels.73 This suggests that adiponectin protects the myocardium in ischemia-reperfusion injury through COX-2 suppression of TNFα. It is possible, in our current study, that pioglitazone could increase plasma 15d-PGJ2 through increasing adiponectin and COX-2 expression and subsequently increasing 15d-PGJ2.
The goal of the current work was to understand whether pioglitazone protected lung injury as a result of the systemic inflammatory response in sepsis. A limitation of our study is the lack of survival data. To be clinically relevant, these studies need to be combined with antibiotic therapy. However, for the current study, to investigate a direct effect of pioglitazone therapy on sepsis-induced acute lung injury, pioglitazone was given as the sole therapy without any other interfering medication. Longer term therapeutic interventional studies will be needed to investigate whether pioglitazone has a survival benefit.
In conclusion, our study demonstrates that polymicrobial sepsis leads to hypoglycemia, elevated plasma levels of pro-inflammatory cytokines and lung injury. Mechanistically, this corresponds with IκBα protein degradation and an increase in NF-κB activity in the lung. Treatment with pioglitazone, when given after the induction of sepsis, improves glucose levels and reduces pro-inflammatory plasma cytokine levels. Furthermore, pioglitazone reduces lung injury most likely through inhibition of NF-κB activity. These findings suggest that pioglitazone may be a beneficial and novel therapy in clinical sepsis.
Acknowledgments
Funding
This work was supported, in part, by the National Institutes of Health [K08 GM093135 (JK) and R01 GM067202 (BZ)].
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167:695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 3.Weycker D, Akhras KS, Edelsberg J, Angus DC, Oster G. Long-term mortality and medical care charges in patients with severe sepsis. Crit Care Med. 2003;31:2316–23. doi: 10.1097/01.CCM.0000085178.80226.0B. [DOI] [PubMed] [Google Scholar]
- 4.Elixhauser A, Machlin SR, Zodet MW, et al. Health care for children and youth in the United States: 2001 annual report on access, utilization, quality, and expenditures. Ambul Pediatr. 2002;2:419–37. doi: 10.1367/1539-4409(2002)002<0419:hcfcay>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 5.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 6.van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–67. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 7.Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–77. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 8.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 9.Opal SM, Laterre PF, Francois B, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–62. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan JM, Denenberg A, Monaco M, Nowell M, Wong H, Zingarelli B. Changes in peroxisome proliferator-activated receptor-gamma activity in children with septic shock. Intensive Care Med. 2010;36:123–30. doi: 10.1007/s00134-009-1654-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24:59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 12.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. 2003;171:6827–37. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 13.Lv X, Song JG, Li HH, et al. Decreased hepatic peroxisome proliferator-activated receptor-gamma contributes to increased sensitivity to endotoxin in obstructive jaundice. World J Gastroenterol. 2011;17:5267–73. doi: 10.3748/wjg.v17.i48.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou M, Wu R, Dong W, Simms HH, Wang P. Hepatic peroxisome proliferator-activated receptor-gamma (PPAR-gamma) is downregulated in sepsis. SHOCK. 2004;21:39. [Google Scholar]
- 15.Forst T, Karagiannis E, Lubben G, et al. Pleiotrophic and anti-inflammatory effects of pioglitazone precede the metabolic activity in type 2 diabetic patients with coronary artery disease. Atherosclerosis. 2008;197:311–7. doi: 10.1016/j.atherosclerosis.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Martens FM, Visseren FL, de Koning EJ, Rabelink TJ. Short-term pioglitazone treatment improves vascular function irrespective of metabolic changes in patients with type 2 diabetes. J Cardiovasc Pharmacol. 2005;46:773–8. doi: 10.1097/01.fjc.0000187176.13403.05. [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Wei J, Guan Y, Jin N, Mao J, Wang X. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone reduces clinical inflammatory responses in type 2 diabetes with coronary artery disease after coronary angioplasty. Metabolism. 2005;54:590–7. doi: 10.1016/j.metabol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Fedson DS. Treating Influenza with Statins and Other Immunomodulatory Agents. Antiviral Res. 2013 doi: 10.1016/j.antiviral.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Aldridge JR, Jr, Moseley CE, Boltz DA, et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:5306–11. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moseley CE, Webster RG, Aldridge JR. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respi Viruses. 2010;4:307–11. doi: 10.1111/j.1750-2659.2010.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haraguchi G, Kosuge H, Maejima Y, et al. Pioglitazone reduces systematic inflammation and improves mortality in apolipoprotein E knockout mice with sepsis. Intensive Care Med. 2008;34:1304–12. doi: 10.1007/s00134-008-1024-9. [DOI] [PubMed] [Google Scholar]
- 22.National Research Council Committee for the Update of the Guide for the C and Use of Laboratory A. 2011. [Google Scholar]
- 23.Zingarelli B, Piraino G, Hake PW, et al. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol. 2010;177:1834–47. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Zhu Z, Zhu B, Ma Z. Pioglitazone attenuates allergic inflammation and induces production of regulatory T lymphocytes. Am J Rhinol Allergy. 2010;24:454–8. doi: 10.2500/ajra.2010.24.3522. [DOI] [PubMed] [Google Scholar]
- 25.Birnbaum Y, Long B, Qian J, Perez-Polo JR, Ye Y. Pioglitazone limits myocardial infarct size, activates Akt, and upregulates cPLA2 and COX-2 in a PPAR-gamma-independent manner. Basic Res Cardiol. 2011;106:431–46. doi: 10.1007/s00395-011-0162-3. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan JM, Nowell M, Lahni P, O’Connor MP, Hake PW, Zingarelli B. Short-Term High Fat Feeding Increases Organ Injury and Mortality After Polymicrobial Sepsis. Obesity (Silver Spring) 2012 doi: 10.1038/oby.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang B, Brown KK, Chen L, et al. Serum adiponectin as a biomarker for in vivo PPARgamma activation and PPARgamma agonist-induced efficacy on insulin sensitization/lipid lowering in rats. BMC Pharmacol. 2004;4:23. doi: 10.1186/1471-2210-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan JM, Zingarelli B. Novel Therapeutic Agents in Pediatric Sepsis: Peroxisome Proliferator Receptor gamma (PPAR gamma) Agonists. Open Inflamm J. 2011;4:120–4. doi: 10.2174/1875041901104010120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 30.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–8. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 31.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 32.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M. p300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1997;272:33435–43. doi: 10.1074/jbc.272.52.33435. [DOI] [PubMed] [Google Scholar]
- 33.Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- 34.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghisletti S, Huang W, Ogawa S, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–9. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 37.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–4. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer CN, Hsu MH, Griffin HJ, Johnson EF. Novel sequence determinants in peroxisome proliferator signaling. J Biol Chem. 1995;270:16114–21. doi: 10.1074/jbc.270.27.16114. [DOI] [PubMed] [Google Scholar]
- 39.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 40.Straus DS, Pascual G, Li M, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–9. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3- kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173:5196–208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- 42.Yang XY, Wang LH, Chen T, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan J, Cook JA, O’Connor M, Zingarelli B. Peroxisome proliferator-activated receptor gamma is required for the inhibitory effect of ciglitazone but not 15-deoxy-Delta 12,14-prostaglandin J2 on the NFkappaB pathway in human endothelial cells. Shock. 2007;28:722–6. doi: 10.1097/SHK.0b013e318055683a. [DOI] [PubMed] [Google Scholar]
- 44.Atsmon J, Sweetman BJ, Baertschi SW, Harris TM, Roberts LJ., 2nd Formation of thiol conjugates of 9-deoxy-delta 9, delta 12(E)-prostaglandin D2 and delta 12(E)-prostaglandin D2. Biochemistry. 1990;29:3760–5. doi: 10.1021/bi00467a023. [DOI] [PubMed] [Google Scholar]
- 45.Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARgamma. J Biol Chem. 2000;275:28028–32. doi: 10.1074/jbc.M001387200. [DOI] [PubMed] [Google Scholar]
- 46.Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma. J Clin Invest. 2003;112:945–55. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shan ZZ, Masuko-Hongo K, Dai SM, Nakamura H, Kato T, Nishioka K. A potential role of 15-deoxy-delta(12,14)-prostaglandin J2 for induction of human articular chondrocyte apoptosis in arthritis. J Biol Chem. 2004;279:37939–50. doi: 10.1074/jbc.M402424200. [DOI] [PubMed] [Google Scholar]
- 48.Hwang JS, Kang ES, Ham SA, et al. Activation of peroxisome proliferator-activated receptor gamma by rosiglitazone inhibits lipopolysaccharide-induced release of high mobility group box 1. Mediators Inflamm. 2012;2012:352807. doi: 10.1155/2012/352807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drosatos K, Khan RS, Trent CM, et al. PPARgamma Activation Prevents Sepsis-Related Cardiac Dysfunction and Mortality in Mice. Circ Heart Fail. 2013 doi: 10.1161/CIRCHEARTFAILURE.112.000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Araujo CV, Estato V, Tibirica E, Bozza PT, Castro-Faria-Neto HC, Silva AR. PPAR gamma activation protects the brain against microvascular dysfunction in sepsis. Microvasc Res. 2012;84:218–21. doi: 10.1016/j.mvr.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Tsujimura Y, Matsutani T, Matsuda A, et al. Effects of pioglitazone on survival and omental adipocyte function in mice with sepsis induced by cecal ligation and puncture. J Surg Res. 2011;171:e215–21. doi: 10.1016/j.jss.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 52.Iwaki M, Matsuda M, Maeda N, et al. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52:1655–63. doi: 10.2337/diabetes.52.7.1655. [DOI] [PubMed] [Google Scholar]
- 53.Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem. 1996;120:803–12. doi: 10.1093/oxfordjournals.jbchem.a021483. [DOI] [PubMed] [Google Scholar]
- 54.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 55.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–9. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 56.Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1) Biochem Biophys Res Commun. 1996;221:286–9. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- 57.Pajvani UB, Du X, Combs TP, et al. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem. 2003;278:9073–85. doi: 10.1074/jbc.M207198200. [DOI] [PubMed] [Google Scholar]
- 58.Pajvani UB, Hawkins M, Combs TP, et al. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem. 2004;279:12152–62. doi: 10.1074/jbc.M311113200. [DOI] [PubMed] [Google Scholar]
- 59.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–92. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsuchihashi H, Yamamoto H, Maeda K, et al. Circulating concentrations of adiponectin, an endogenous lipopolysaccharide neutralizing protein, decrease in rats with polymicrobial sepsis. J Surg Res. 2006;134:348–53. doi: 10.1016/j.jss.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 61.Teoh H, Quan A, Bang KW, et al. Adiponectin deficiency promotes endothelial activation and profoundly exacerbates sepsis-related mortality. Am J Physiol Endocrinol Metab. 2008;295:E658–64. doi: 10.1152/ajpendo.90384.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uji Y, Yamamoto H, Tsuchihashi H, et al. Adiponectin deficiency is associated with severe polymicrobial sepsis, high inflammatory cytokine levels, and high mortality. Surgery. 2009;145:550–7. doi: 10.1016/j.surg.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 63.Konter JM, Parker JL, Baez E, et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol. 2012;188:854–63. doi: 10.4049/jimmunol.1100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keller P, Moller K, Krabbe KS, Pedersen BK. Circulating adiponectin levels during human endotoxaemia. Clin Exp Immunol. 2003;134:107–10. doi: 10.1046/j.1365-2249.2003.02264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson PD, Mehta NN, Wolfe ML, et al. Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab. 2007;92:2272–9. doi: 10.1210/jc.2006-2545. [DOI] [PubMed] [Google Scholar]
- 66.Otero M, Lago R, Gomez R, et al. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:1198–201. doi: 10.1136/ard.2005.046540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:100–5. doi: 10.1097/01.MIB.0000200345.38837.46. [DOI] [PubMed] [Google Scholar]
- 68.Maeda N, Takahashi M, Funahashi T, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–9. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 69.Combs TP, Wagner JA, Berger J, et al. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology. 2002;143:998–1007. doi: 10.1210/endo.143.3.8662. [DOI] [PubMed] [Google Scholar]
- 70.Yu JG, Javorschi S, Hevener AL, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51:2968–74. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 71.Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723–32. [PubMed] [Google Scholar]
- 72.Peake PW, Shen Y, Campbell LV, Charlesworth JA. Human adiponectin binds to bacterial lipopolysaccharide. Biochem Biophys Res Commun. 2006;341:108–15. doi: 10.1016/j.bbrc.2005.12.162. [DOI] [PubMed] [Google Scholar]
- 73.Shibata R, Sato K, Pimentel DR, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ikeda Y, Ohashi K, Shibata R, et al. Cyclooxygenase-2 induction by adiponectin is regulated by a sphingosine kinase-1 dependent mechanism in cardiac myocytes. FEBS Lett. 2008;582:1147–50. doi: 10.1016/j.febslet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]