

Abstract
Background and PurposeProstamides are lipid mediators formed by COX-2-catalysed oxidation of the endocannabinoid anandamide and eliciting effects often opposed to those caused by anandamide. Prostamides may be formed when hydrolysis of anandamide by fatty acid amide hydrolase (FAAH) is physiologically, pathologically or pharmacologically decreased. Thus, therapeutic benefits of FAAH inhibitors might be attenuated by concomitant production of prostamide F2α. This loss of benefit might be minimized by compounds designed to selectively antagonize prostamide receptors and also inhibiting FAAH.
Experimental ApproachInhibition of FAAH by a series of selective antagonists of prostamide receptors, including AGN 204396, AGN 211335 and AGN 211336, was assessed using rat, mouse and human FAAH in vitro, together with affinity for human recombinant CB1 and CB2 receptors. Effects in vivo were measured in a model of formalin-induced inflammatory pain in mice.
Key ResultsThe prostamide F2α receptor antagonists were active against mouse and rat FAAH in the low μM range and behaved as non-competitive and plasma membrane-permeant inhibitors. AGN 211335, the most potent inhibitor of rat FAAH (IC50 = 1.2 μM), raised exogenous anandamide levels in intact cells and also bound to cannabinoid CB1 receptors. Both AGN 211335 and AGN 211336 (0.25–1 mg·kg−1, i.p.) inhibited the formalin-induced nociceptive response in mice.
Conclusions and ImplicationsSynthetic compounds with indirect agonist activity at cannabinoid receptors and antagonist activity at prostamide receptors can be developed. Such compounds could be used as alternatives to selective FAAH inhibitors to prevent the possibility of prostamide F2α-induced inflammation and pain.
Linked ArticlesThis article is part of a themed section on Cannabinoids 2013. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-6
Keywords: endocannabinoid, anandamide, FAAH, PGs, prostamides, pain
Introduction
Fatty acid amide hydrolase (FAAH; McKinney and Cravatt, 2005) is a serine hydrolase belonging to the amidase family of enzymes. It was established as the main hydrolytic enzyme for the endocannabinoid N-arachidonoyl-ethanolamine (anandamide, AEA) and the sleep-inducing factor oleoylamide (oleamide; Cravatt et al., 1996) after initial studies identifying this enzymic activity in porcine brain (Ueda et al., 1995) and mouse neuroblastoma cells (Maurelli et al., 1995). Following its cloning, it became clear that FAAH also recognizes other bioactive fatty acid amides, including oleamide and AEA congeners (the fatty acid primary amides and ethanolamides respectively), the N-acyltaurines and the N-acylglycines, with higher affinity towards monounsaturated and polyunsaturated members of these lipid families. Since the discovery of these lipid mediators, it has become clear that (i) AEA, via either cannabinoid receptor (CB1 or CB2), or transient receptor potential vanilloid type-1 (TRPV1) channels; (ii) N-acylethanolamines such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), via direct actions at peroxisome proliferator-activated receptor-α (PPAR-α) and TRPV1 channels and indirect actions at CB1 and CB2 receptors; and (iii) N-acylglycines, particularly N-arachidonoylglycine (NAGly), acting at yet unidentified molecular targets, exert anti-nociceptive and anti-hyperalgesic actions in various animal models of acute and chronic pain (Maione et al., 2013; receptor nomenclature follows Alexander et al., 2013). Therefore, selective FAAH inhibitors have been developed as potential novel analgesics (Blankman and Cravatt, 2013). However, one such compound, PF-04457845, was recently tested in a clinical trial against osteoarthritic pain and, although capable of elevating the blood levels of FAAH substrates in the treated subjects, it did not produce any significant amelioration of pain scores.
One possible mechanism that might limit the efficacy of FAAH inhibitors lies in the now well-established concept that arachidonic acid-containing fatty acid amides such as AEA and NAGly can also be metabolized through oxygenation catalysed by cyclooxygenase-2 (COX-2) (Prusakiewicz et al., 2002; Kozak et al., 2003). With AEA as a substrate, COX-2 leads to the formation of PGH2-ethanolamide, which can then be reduced to a whole series of PG-ethanolamides, also known as ‘prostamides’ (Burk and Woodward, 2007; Figure 1). Among these compounds, which are inactive at cannabinoid receptors or TRPV1 channels, and are unable to be hydrolysed to the corresponding PGs (Matias et al., 2004), prostamide F2α is biosynthesized by PGF synthases from PGH2-ethanolamide (Koda et al., 2004; Moriuchi et al., 2008) and was recently shown to be formed in vivo in the spinal cord of mice with knee inflammation (Gatta et al., 2012). In contrast to AEA and other cannabinoid receptor agonists, prostamide F2α contributed to pain transmission in these animals (Gatta et al., 2012). Thus, in theory, FAAH inhibition, by elevating the endogenous levels of AEA in tissues under conditions where COX-2 is up-regulated, such as during inflammation, might lead to the formation of the hyperalgesic prostamide F2α. In turn, this would counteract the anti-hyperalgesic effects of AEA or other FAAH substrates.
Figure 1.
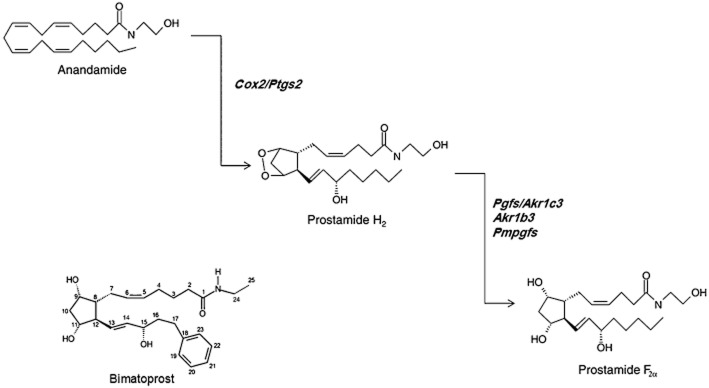
Biosynthetic scheme for prostamide F2α and chemical structure of its synthetic analogue, bimatoprost. Abbreviations of the genes encoding for biosynthetic enzymes: Cox2/Ptgs, COX-2/PG synthase; Pgfs/Akr1c3, PG F synthase/α-keto-reductase 1c3; Pmpgfs; prostamide and PG F synthase; Akr1b3, α-keto-reductase 1b3.
There is pharmacological evidence, at molecular and in vitro and in vivo levels, suggesting that prostamide F2α, as well as its synthetic analog bimatoprost, do not act at the same GPCRs for PGF2α, that is, the FP receptors (Woodward et al., 2008). However, a heterodimeric association between the wild-type FP receptor and a particular alternative splicing variant (Alt4) of such receptors did transduce some of the pharmacological actions of prostamide F2α and bimatoprost in vitro (Liang et al., 2008). Accordingly, synthetic compounds that specifically antagonize the pharmacological actions of bimatoprost and prostamide F2α, without affecting those of PGF2α, have been developed (Woodward et al., 2007; 2008; Liang et al., 2008; Jones et al., 2009). In view of the pro-algesic actions of endogenous prostamide F2α in mice with knee inflammation, these antagonist compounds, and particularly AGN 211336, might be useful for the treatment of pain (Gatta et al., 2012). As some of these selective prostamide receptor antagonists have a chemical structure resembling that of some FAAH inhibitors, and with either one or two potential leaving groups for serine hydrolase activity (Figure 2), it is possible that these compounds could also act as inhibitors of FAAH. Indeed, a FAAH inhibitor that at the same time was capable of antagonizing prostamide F2α effects should, in principle, be more efficacious against pain than selective FAAH inhibitors or prostamide receptor antagonists, acting individually.
Figure 2.

Chemical structures of the eight compounds investigated in this study.
With this background in mind, all the available prostamide receptor antagonists were screened for FAAH inhibitory activity in three different animal species, as well as for their possible affinity for human recombinant CB1 and CB2 receptors. We report here that AGN 211335 and AGN 211336 inhibit FAAH from various species and that the former compound also weakly binds to human CB1 receptors. As predicted by this activity, both compounds, injected i.p. in mice, inhibited formalin-induced pain.
Methods
Animals
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 56 animals (8 cats and 48 mice) were used in the experiments described here. For the studies using tissues from cats, these were performed at Covance (Madison, WI, USA). Class A laboratory bred cats were housed communally in USDA and AAALAC approved facilities, with standards exceeding those for enrichment and group housing. Water was freely available and food was standard cat nutritional diet; they were kept on a 12 h light-dark cycle.
For the nociceptive studies in mice, animal care was in compliance with Ethical Guidelines of the IASP and European Community (E.C. L358/1 18/12/86) on the use and protection of animals in experimental research and the experimental procedures were in accordance with the Italian and European regulations governing the care and treatment of laboratory animals.
Assays of the compounds against prostanoid receptors
For prostanoid antagonist assays, standard agonists BW 245C for DP1, PGE2 for EP1–4, 17-phenyl-PGF2α for FP, carbaprostacyclin for IP receptors and U-46619 for TP receptors were purchased from Cayman (Ann Arbor, MI, USA). Stable cell lines (HEK-293 cells for EP1, EP2, EP4, FP, and TP receptors and COS-7 cells for EP3 and TP receptors) over-expressing human recombinant prostanoid DP1, EP1–4, FP, IP and TP receptors, were previously established by others at Allergan (Matias et al., 2004). To measure the response of Gs and Gi-coupled prostanoid receptors as a Ca2+ signal, chimeric G protein cDNAs were used as previously described (Matias et al., 2004). Ca2+ signalling studies were performed using a FLIPR system (Molecular Devices, Sunnyvale, CA, USA) in the 96-well format as published previously (Matias et al., 2004). Briefly, cells were seeded at a density of 5 × 104 cells per well in Biocoat poly-D-lysine-coated blackwall, clear-bottom 96-well plates (BD Biosciences, Franklin Lakes, NJ, USA) and allowed to attach overnight in an incubator at 37°C. The cells were then washed twice with HBSS-HEPES buffer (Hanks' balanced salt solution without bicarbonate and phenol red, 20 mM HEPES, pH 7.4). After 60 min of dye loading with Fluo-4AM (Invitrogen, Carlsbad, CA, USA) at a final concentration of 2 μM, the plates were washed four times with HBSS-HEPES buffer. Putative antagonists were added to each well to give final concentrations of 10 μM. After 4.5 min, a 7-point serial dilution of the standard agonist for the corresponding receptor was injected to provide final concentrations of 10 pM to 10 μM in 10-fold serial dilution increments for cells expressing human recombinant DP1, EP1, EP2, EP3, EP4, FP and IP receptors. The dose range for the standard agonist for human recombinant TP receptors was from 1 pM to 1 μM. HBSS-HEPES buffer was used as the negative control. Cells were excited with an argon laser at 488 nm, and emission was measured through a 510–570 nm emission filter. Standard agonists purchased from Cayman Chemical were as follows: DP = BW 245C, EP1-EP4 = PGE2, FP = 17-phenyl-PGF2α, IP = carbaprostacyclin and TP = U-46619. The peak fluorescence change in each well containing drug was exported and expressed relative to vehicle controls with the standard agonist at 10−6 M (the positive control). To obtain concentration–response curves, compounds were tested in triplicate in each plate over the desired concentration range in at least three separate assays to give n = 3. Values of Kb in nM were calculated from the equation Kb = [antagonist concentration]/(IC50/EC50-1).
Assays of the compounds against prostamide receptors
For prostamide F2α receptor antagonist assays, the cat isolated iris was used as previous studies have shown this to be a particularly abundant source of prostamide F2α receptors (Matias et al., 2004). Adult cats were of either sex were killed by i.v. overdose of sodium pentobarbital (390 mg mL−1; Anthony, Arcadia, CA, USA) The eyes were enucleated immediately after death and placed on ice The eyes were placed cornea side up on an indented wax plate. An incision was made at the corneal-scleral junction. The iris was removed following two radial incisions such that each eye provided two iridial preparations. The iris sphincter was mounted vertically under 50 to 100 mg tension in a jacketed 10 mL organ bath. Smooth muscle tension of the isolated iris sphincter was measured isometrically with force displacement transducers (Grass FT-03; Grass Technologies, Warwick, RI, USA) and recorded on a Grass polygraph (Model 7; Grass Technologies). The organ baths contained Krebs solution maintained at 37°C by a heat exchanger and circulating pump. The Krebs solution was gassed with 95% O2, 5% CO2 to give a pH of 7.4 and had the following composition: 118.0 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.9 mM CaCl2, 1.18 mM MgSO4, 25.0 mM NaHCO3, 11.7 mM glucose and 1 μM indomethacin. A 60 min stabilization period was provided before commencing each experiment. Agonist activity at prostamide F2α receptors was manifested as contractile responses and measured as such. The experiment was designed so that a direct, four-way comparison for antagonist versus prostamide, vehicle versus prostamide, antagonist versus corresponding PG and vehicle versus corresponding PG was provided in tissue preparations obtained from a single animal. One cumulative dose–response curve to agonist was obtained in each tissue. Vehicle (ethanol) and antagonist (AGN 204396) were given 30 min before the agonist dose–response curves were constructed. The response to a standard concentration of PGF2α (100 nM) was determined at the beginning and end of each dose–response curve, with appropriate washout, and responses were calculated as % of this reference contraction.
In order to calculate the pA2 values, the mean concentration–response curve was plotted as log concentration response −1 (CR-1) versus log antagonist concentration using GraphPad Prism 4 software (GraphPad Software, Inc., La Jolla, CA, USA). An adaptation of the prostamide F2α concentration–response curve in the presence of vehicle was required for analysis. Thus, each point on the prostamide F2α versus vehicle concentration–response curve represents the mean of each of the six separate antagonist experiments. As the slope of the CR-1 versus log [antagonist] plot did not significantly differ from unity, the slope was constrained to 1. The effects of antagonists were statistically analysed by comparing the mid-points (EC50) of the agonist concentration–response curves in the presence or absence of antagonists using GraphPad Prism 4.
FAAH assays with cell-free membranes or human recombinant FAAH
FAAH activity was measured in membranes from either rat brain or mouse neuroblastoma N18TG2 cells or in a commercially available source of human recombinant FAAH (Cayman). AEA hydrolysis was measured by incubating the enzymic source (2 μg per sample for human recombinant FAAH and 70 μg per sample for the 10 000× g membrane fraction of tissues or cells) in Tris–HCl 50 mM, at pH 9.5 at 37°C for 30 min, with synthetic N-arachidonoyl-[14C]-ethanolamine ([14C]-AEA, 55 mCi·mmol−1, ARC, St. Louis, MO, USA), diluted as required with un-labelled AEA (Tocris Bioscience, Avonmouth, Bristol, UK). After incubation, the amount of [14C]-ethanolamine produced was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with two volumes of CHCl3/MeOH 1:1 (by vol.). Data are expressed as means ± SD of two separate experiments of the concentration exerting 50% inhibition of [14C]-AEA hydrolysis (IC50) calculated by fitting sigmoidal concentration–response curves by GraphPad.
FAAH assay in intact RBL-2H3 cells
RBL-2H3 cells (American Type Culture Collection, Manassas, VA, USA) were cultured according to the supplier's protocol. Cells were seeded at ∼90% confluency in 100 mm dishes and incubated for 30 min at 37°C with [14C]-AEA (2 μM; 60,000 cpm per sample) in EMEM medium in presence or absence of increasing concentration of AGN 211335 and AGN 211336 (0.1-1-10 μM). URB-597 (1–10 μM) was used as positive control. In some experiments, cells were pretreated with compounds for 20 min at 37°C before the incubation with [14C]-AEA. The reaction was stopped by extraction with two volumes of CHCl3/MeOH (1:1, v/v). FAAH activity was analysed by measuring the radioactivity associated both with the aqueous phase (which contains [14C]-ethanolamine produced) and the organic phase (which contains non-hydrolysed [14C]-AEA) of cell extracts.
Effect of compounds on FAAH substrates in intact RBL-2H3 cells
RBL-2H3 cells were cultured according to the supplier's protocol. Cells were seeded at ∼90% confluency in 100 mm dishes and incubated with AGN 211335 (2.5 μM) both 20 min before and during incubation with ionomycin (4 μM; Sigma-Aldrich, St. Louis, MO, USA). After stimulation, cells plus medium were extracted with CHCl3/CH3OH (2:1, v/v). AEA and the endogenous AEA congeners, PEA and OEA, were extracted with two volumes of CHCl3/CH3OH (2:1, v/v), pre-purified on silica and quantified by isotope dilution-liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry (LC-APCI-MS), as described by Gatta et al., (2012).
Effect of AGN 211335 and AGN 211336 on 2-arachidonoylglycerol (2-AG) hydrolysis
The two most potent and selective FAAH inhibitors were also tested for their selectivity against the enzymic hydrolysis of another endocannabinoid, 2-arachidonoylglycerol (2-AG), by cellular fractions of COS-7 cells, which contain high levels of monoacylglycerol lipase (Bisogno et al., 2009). Briefly, the 10 000× g cytosolic and membrane fractions from COS-7 cells, were incubated in Tris–HCl 50 mM, at pH 7.0 at 37°C for 20 min, with synthetic 2-arachidonoyl-[3H]-glycerol (40 Ci·mmol−1, HARTMANNANALYTIC GmbH, Germany) diluted with 2-AG (Cayman Chemicals) to a final concentration of 20 μM. Protein concentrations and incubation time were established in pilot experiments to be within the range of values when activity varies linearly with protein content and time, respectively, whereas the concentration of substrate used was near the apparent Km of the 2-AG hydrolysing activity in COS-7 cells. After incubation with 2-arachidonoyl-[3H]-glycerol, the amount of [3H]-glycerol produced was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with 2 volumes of CHCl3/MeOH (1:1, v/v).
CB1 and CB2 receptor binding assays
Membranes from HEK-293 cells stably transfected with the human recombinant CB1 receptor (Bmax = 2.5 pmol·mg−1 protein using [3H]-CP-55 940) or human recombinant CB2 receptor (Bmax = 4.7 pmol·mg−1·protein using [3H]-CP-55 940) were incubated with [3H]-CP-55 940 (0.14 nM, Kd = 0.12 nM and 0.084 nM, Kd = 0.19 nM, respectively, for CB1 and CB2 receptors) as the high affinity ligand and displaced with 10 μM WIN 55212-2 as the heterologous competitor for non-specific binding (Ki values 9.2 and 2.1 nM, respectively, for CB1 and CB2 receptors). All compounds were tested following the procedure described by the manufacturer (Perkin Elmer, Monza, MB, Italy). Displacement curves were generated by incubating drugs with [3H]-CP-55 940 for 90 min at 30°C. Ki values were calculated by applying the Cheng-Prusoff equation to the IC50 values (obtained by GraphPad) for the displacement of the bound radioligand by increasing concentrations of the test compound. Data are expressed as means ± SD of Ki values obtained from two separate experiments.
Effect of AGN 211335 and AGN 211336 on human recombinant TRPV1 receptors
HEK-293 cells stably over-expressing the human recombinant TRPV1 were selected by G-418 (Geneticin, 600 μg·mL−1; Life Technologies, Monza, MB, Italy), grown on 100-mm-diameter Petri dishes as monolayers in minimum essential medium supplemented with non-essential amino acids, 10% FBS, and 2 mM glutamine, and maintained under 5% CO2 at 37°C. On the day of the experiment, the cells were loaded for 1 h at 25°C with the cytoplasmic calcium indicator Fluo-4AM (Invitrogen) at 4 μM in DMSO containing 0.02% Pluronic F-127 (Invitrogen). After loading, cells were washed twice in Tyrode's buffer (145 mM NaCl, 2.5 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 10 mM d-glucose and 10 mM HEPES, pH 7.4), resuspended in the same buffer and transferred to a quartz cuvette of the spectrofluorimeter (Perkin-Elmer LS50B; PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) under continuous stirring (about 100 000 cells per assay). [Ca2+]i was determined before and after the addition of various concentrations of test compounds by measuring cell fluorescence (excitation λ = 488 nm; emission λ = 516 nm). All determinations were at least in triplicate.
Effect of AGN 211335 and AGN 211336 in the mouse model of inflammatory pain induced by formalin
All efforts were made to minimize animal suffering and to reduce the number of animals used, according to IASP guidelines. Male C57/BL6 mice received formalin (1.25% in saline, 30 μL) in the dorsal surface of one side of the hind-paw. Each mouse was randomly assigned to one of the experimental groups (n = 6) and placed in a Plexiglass cage and allowed to move freely for 15–20 min. A mirror was placed at a 45° angle under the cage to allow full view of the hind-paws. Lifting, favouring, licking, shaking and flinching of the injected paw were recorded as nociceptive responses (Abbott et al., 1995). The total time of the nociceptive response was measured every 5 min and expressed in min (mean ± SEM). The data represent the sum of the time spent engaging in nociceptive behaviour over the 5 min period. Recording of nociceptive behaviour commenced immediately after formalin injection and was continued for 60 min. Mice received vehicle (10% DMSO in saline) or different doses of AGN 21135 or AGN 21136 (0.25, 0.5 and 1 mg·kg−1, i.p.) 10 min before formalin injection. Significant differences between groups were evaluated by using anova followed by the Newman–Keuls's post hoc test.
Data analysis
Results are shown as means ± SEM, unless otherwise stated. Differences between group means were assessed by one-way ANOVA, with either Bonferroni's or Newman–Keuls's post hoc test. P < 0.05 was taken to show significance.
Materials
AGN 211334, AGN 211335 and AGN 211336 were prepared in Selcia Laboratories (Ongar, Essex United Kingdom) following the methods described previously (Woodward et al., 2011b). AGN 204396 and AGN 204397 were also prepared following the methods described previously (Krauss and Woodward, 2006). AGN 205492, AGN 205493 and AGN 205494 were prepared from the common intermediate AGN 197727 (Burk and Woodward, 2007) following standard chemical procedures (Figure 3). Bimatoprost was provided by Allergan Inc. (Irvine, CA, USA), whereas PGF2α was purchased from Cayman Chemicals (Ann Arbor, MI, USA).
Figure 3.
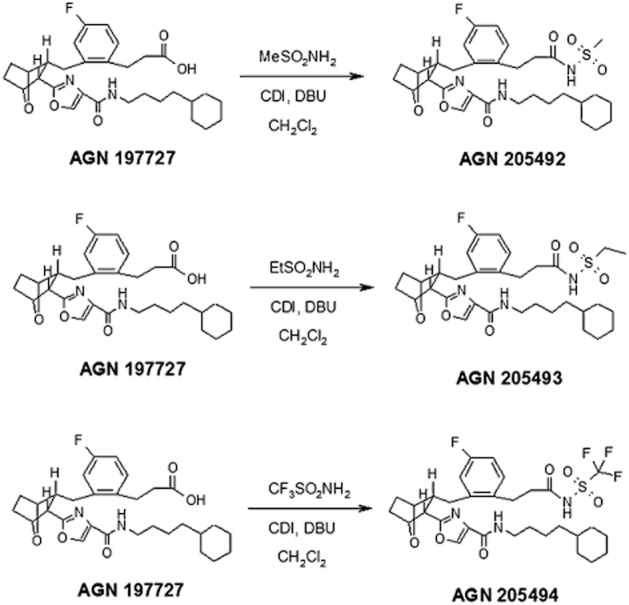
Synthetic methods for the preparation of AGN 205492, AGN 205493 and AGN 205494. MeSO2NH2, methylsulfonamide; EtSO2NH2, ethylsulfonamide; CDI, 1,1′-carbonyldiimidazole; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Results
Activity of compounds at prostamide receptors and their selectivity versus prostanoid receptors
All compounds tested acted as antagonists for prostamide F2α-induced contractions of the isolated iris (Table 1). All compounds exhibited high inhibitory efficacy against TP thromboxane receptors. AGN 205492, AGN 205493 and AGN 205494 exerted antagonistic activity also at FP, DP, EP1, EP3, EP4 and IP receptors. AGN 205494 also antagonized EP2 receptors Only those compounds that did not possess an acyl sulfonamide moiety in their chemical structure (Figure 2), that is, AGN 204396, AGN 204397, AGN 211334, AGN 211335 and AGN 211336, were selective towards the other prostanoid receptors tested here (Table 1), with the exception of the TP receptor.
Table 1.
Effect of the eight prostamide receptor antagonists examined in this study on various prostanoid receptors and the prostamide F2α receptor
| Compound | Prostamide F2α receptors (Kb, μM) | FP (Kb, μM) | DP (Kb, μM) | EP1 (Kb, μM) | EP2 (Kb, μM) | EP3 (Kb, μM) | EP4 (Kb, μM) | IP (Kb, μM) | TP (Kb, μM) |
|---|---|---|---|---|---|---|---|---|---|
| AGN 205492 | 0.17 | 0.20 | 0.55 | 1.2 | NA | 4.5 | 0.32 | 4 | <0.001 |
| AGN 205493 | 0.05 | 0.15 | 0.09 | 0.25 | NA | 3.9 | 0.46 | 4.2 | <0.001 |
| AGN 205494 | 0.5 | 0.03 | 0.009 | 0.03 | 3 | 0.66 | 0.03 | 0.48 | <0.001 |
| AGN 204396 | 2.6 | NA | NA | NA | NA | NA | NA | NA | 0.001 |
| AGN 204397 | 3.1 | NA | NA | NA | NA | NA | NA | NA | 0.001 |
| AGN 211334 | 0.24 | NA | NA | NA | NA | NA | NA | NA | 0.02 |
| AGN 211335 | 0.35 | NA | NA | NA | NA | NA | NA | NA | 0.10 |
| AGN 211336 | 0.3 | NA | NA | NA | NA | NA | NA | NA | 0.11 |
See Methods for the description of assay conditions.
Effect of compounds on FAAH in cell-free preparations and human recombinant FAAH
The effects of the prostamide receptor antagonists on FAAH activity in membranes from either rat brain or mouse neuroblastoma N18TG2 cells or human recombinant FAAH are shown in Table 2. All compounds except AGN 204396 and AGN 204397, exhibited IC50 < 10 μM against rat FAAH. However, when tested with a 20 min pre-incubation of rat brain membranes, the potency of these two compounds was increased, thus suggesting that they acted as non-competitive irreversible inhibitors. Accordingly, also the activity of the two most potent compounds, that is, AGN 211335 and AGN 211336, increased significantly following a 20 min pre-incubation of rat brain membranes, with the former compound being slightly more potent (Table 2). Furthermore, when tested against increasing concentrations (1, 5, 10, 50 and 150 μM) of the [14C]-AEA substrate using rat brain membranes, and the effect was assessed through a Lineweaver–Burk analysis, AGN 211335 (10 μM) reduced the Bmax (from 3.5 ± 0.2 to 0.2 ± 0.05 nmol·min−1·mg−1) without significantly affecting the apparent Km (from 24 ± 5 to 35 ± 4 μM, data are means ± SD of two experiments in duplicate) of the enzyme (Figure 4), thus confirming a non-competitive mechanism of action. All compounds, except AGN 211334, were also tested on mouse FAAH, where they again exhibited IC50 values < 10 μM, with the exception of AGN 204396. However, both AGN 211335 and, particularly, AGN 211336 were significantly less potent and efficacious when tested on human recombinant FAAH from a commercial source (Table 2).
Table 2.
Effect of the eight prostamide receptor antagonists examined in this study on AEA hydrolysis by FAAH in various species and preparations
| Compound | Rat brain FAAH (IC50, μM) | Rat brain FAAH (% inhibition at max concentration tested in μM, shown in parentheses) | Rat brain FAAH following pre-incubation (IC50, μM) | Rat brain FAAH following pre-incubation (% inhibition at max concentration tested in μM, shown in parentheses) | Mouse N18TG2 cell FAAH (IC50, μM) | Human recombinant FAAH (% inhibition at 10 μM) |
|---|---|---|---|---|---|---|
| AGN 205492 | 8.3 ± 0.4 | 63.8 (10) | ND | ND | 9.7 ± 0.6 | ND |
| AGN 205493 | 4.8 ± 0.3 | 72.5 (10) | ND | ND | 8.7 ± 0.5 | ND |
| AGN 205494 | 3.7 ± 0.2 | 75.9 (10) | ND | ND | 5.9 ± 0.5 | ND |
| AGN 204396 | 22.2 ± 0.8 | 51.7 (25) | 3.9 ± 0.1 | 84.9 (10) | 13.4 ± 0.7 | ND |
| AGN 204397 | 12.8 ± 0.5 | 73.1 (25) | 6.6 ± 0.4 | 67.9 (10) | 8.5 ± 0.4 | ND |
| AGN 211334 | 7.5 ± 0.4 | 73.9 (10) | ND | ND | ND | ND |
| AGN 211335 | 3.0 ± 0.05 | 76.8 (5) | 1.2 ± 0.3 | 96.9 (5) | 3.4 ± 0.3 | 48.5 |
| AGN 211336 | 3.6 ± 1.1 | 71.9 (5) | 1.3 ± 0.3 | 97.8 (5) | 3.1 ± 0.2 | 24.8 |
| URB597 | – | – | 0.01 ± 0.003 | 98.1 (0.1) | – | 0.1 ± 0.02 |
Data are expressed as means ± SD of two separate experiments conducted in duplicate of the concentrations exerting 50% inhibition of [14C]-AEA hydrolysis (IC50) calculated by fitting sigmoidal concentration–response curves by GraphPad. For some experiments, the average effect observed with the maximal tested concentration (indicated between parentheses) was also shown (without SD for the sake of clarity). ND, not determined.
Figure 4.
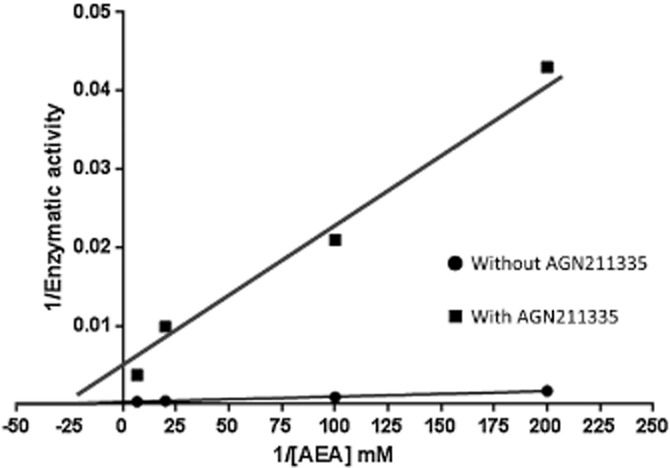
Lineweaver–Burk analysis of the effect of AGN 211335 (10 μM) on [14C]-anandamide hydrolysis by rat brain membranes. Five different concentrations of substrate were used (1, 5, 10, 50, 150 μM), but the data for lowest one (1 μM) is not shown in order to avoid compressing the data from the higher concentrations, close to the Y and X axes.
Effect of AGN 211335 and AGN 211336 on AEA inactivation and FAAH substrate levels in intact cells
The two most potent compounds against rat brain FAAH, that is, AGN 211335 and AGN 211336, were also tested in intact RBL-2H3 cells to indirectly assess their plasma membrane permeability. When these cells were incubated for 30 min at 37°C with [14C]-AEA, there was a strong decrease of AEA-associated radioactivity, and a concomitant increase of [14C]-ethanolamine, compared with [14C]-AEA incubated only with empty 100 mm dishes. However, when AGN 211335 or AGN 211336 was incubated with the cells for 20 min before and during [14C]-AEA incubation, there was a dose dependent increase of [14C]-AEA and a corresponding decrease of [14C]-ethanolamine associated with cell homogenates. Thus both AGN compounds behaved like the selective FAAH inhibitor, URB597 (Figure 5A). Interestingly, all three compounds appeared to elevate [14C]-AEA levels to a greater extent than that expected from their reduction of [14C]-ethanolamine levels, suggesting that they might affect also [14C]-AEA binding to cells and plastic.
Figure 5.
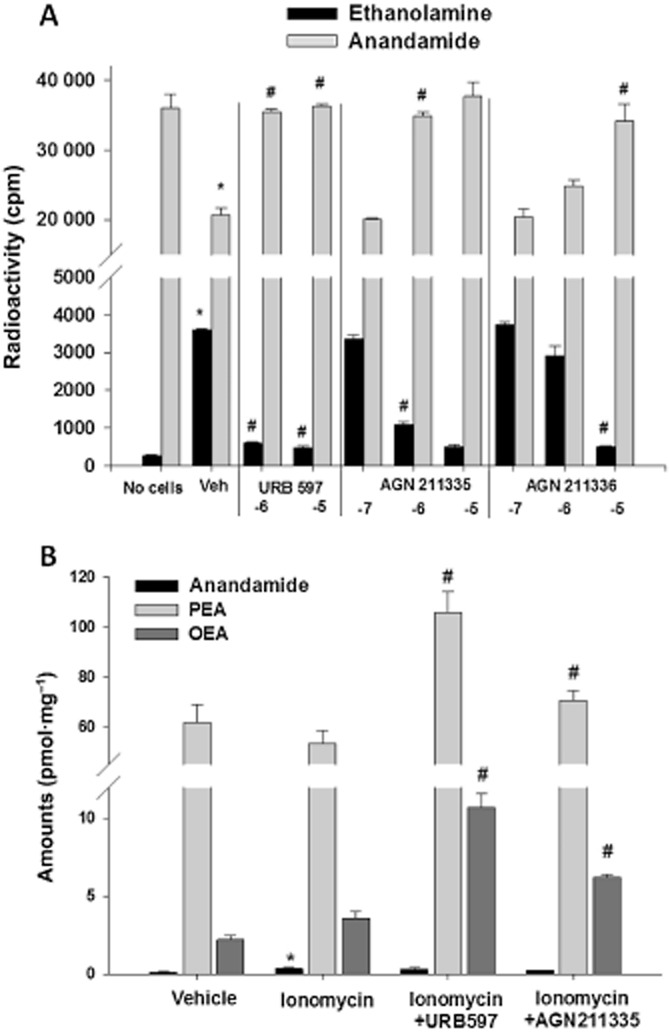
Effect of AGN 211335 and AGN 211336, and of the selective FAAH inhibitor URB597, on the hydrolysis of anandamide by intact RBL-2H3 cells. (A) The amounts (in cpm) of residual [14C]-anandamide or [14C]-ethanolamine formed from [14C]-anandamide hydrolysis by cells are shown. *P < 0.05 versus ‘no cells’; #P < 0.05 versus corresponding vehicle (DMSO, 0.1%) control, as assessed by anova followed by Bonferroni's test. −7, −6 and −5 denote the log of the concentrations used (M) for each compound. (B) The amounts, in pmol (mg of extracted lipids)-1 of anandamide, PEA and OEA in cells treated with vehicle (DMSO, 0.1%) or ionomycin (4 μM) with or without URB597 (1 μM) or AGN 211335 (2.5 μM). *P < 0.05 versus corresponding vehicle; #P < 0.05 versus corresponding ionomycin, as assessed by anova followed by Bonferroni's test. Incubations and cell processing were carried out as described in the Methods. Data are means ± SEM of n = 3 experiments.
When incubated with cells for 20 min before and during ionomycin stimulation to trigger endogenous AEA biosynthesis, AGN 211335 (2.5 μM), like the selective FAAH inhibitor URB597 (1 μM), did not further elevate AEA levels but instead raised the levels of two other FAAH substrates, PEA and OEA, which were not up-regulated by ionomycin alone (Figure 5B). These data suggest that AGN 211335 can inhibit inactivation of exogenous AEA and of endogenous PEA and OEA, catalysed by FAAH in intact cells.
Affinity of compounds for CB1 and CB2 receptors
When tested in displacement assays against the specific binding of [3H]-CP55 490 to human recombinant CB1 or CB2 receptors, AGN 211335 and AGN 211336 exhibited some affinity only for the former. However, only AGN 211335 possessed sub-μM affinity (Table 3).
Table 3.
Effect of AGN 211335 and AGN 211336 on the human recombinant CB1 and CB2 receptors
| Compound | Ki on CB1 (μM) | Max concentration tested on CB1 μM (% inhibition) | Ki on CB2 (μM) | Max concentration tested on CB2 μM (% inhibition) |
|---|---|---|---|---|
| AGN 211335 | 2.4 ± 0.1 | 10 (65) | >10 | 10 (42) |
| AGN 211336 | 0.8 ± 0.4 | 10 (75) | 1.9 ± 0.4 | 10 (57) |
Displacement curves were generated by incubating drugs with [3H]-CP-55 940 for 90 min at 30°C. Ki values were calculated by applying the Cheng-Prusoff equation to the IC50 values (obtained by GraphPad) for the displacement of the bound radioligand by increasing concentrations of the test compound. Data are expressed as means ± SD of Ki values obtained from two separate experiments conducted in duplicate.
Lack of activity of AGN 211335 and AGN 211336 in inhibiting 2-AG hydrolysis or stimulating human TRPV1 receptors
Neither AGN 211335 nor AGN 211336 exerted strong inhibitory activity against 2-AG hydrolysis in COS-7 cells (maximal inhibition at 10 μM was 13.5 ± 2.3 and 10.8 ± 1.9% respectively, whereas the monoacylglycerol lipase inhibitor OMDM169 at 1 μM produced an 82.2 ± 4.5% inhibition, means ± SD of n = 3 determinations). Furthermore, neither compound, up to 20 μM, produced any measurable effect on [Ca2+]i in HEK-293 cells transfected with the human TRPV1 channel, whereas capsaicin produced a strong TRPV1-mediated potentiation with EC50 = 30 nM (data not shown).
Activity of AGN 211335 and AGN 211336 in the formalin model of inflammatory pain
Nociceptive responses to subcutaneous formalin induced an early, short-lasting first phase (0–7 min) followed by a quiescent period and then a second, prolonged phase (15–60 min) of tonic hyperalgesia. Systemic administration of AGN 21135 and AGN 21136 (0.25, 0.5 and 1 mg·kg−1, i.p.) reduced, in a dose-dependent manner, both the first and, particularly, the second phase of the formalin-induced nociceptive behaviour, the effect becoming statistically significant with the two highest doses tested (Figure 6).
Figure 6.
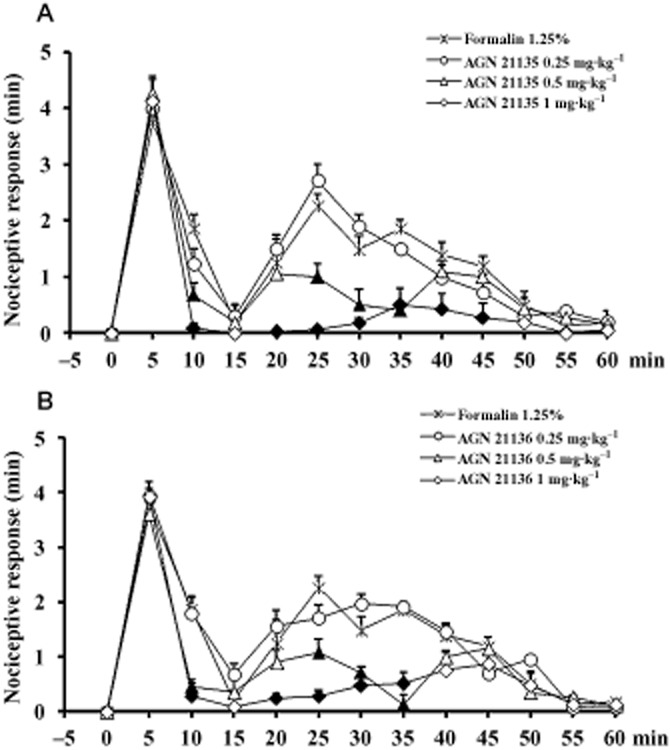
Effect of AGN21135 and AGN 21136 (0.25, 0.5 and 1 mg·kg−1, i.p.) in the formalin model of pain in mice. The total time of the nociceptive response was measured every 5 min and expressed in min. Data are means ± SEM from six mice and were analysed by one-way anova followed by Newman–Keuls' post hoc test. Filled symbols denote statistically significant differences from values with formalin only (P < 0.05).
Discussion
Prostamides are COX-2-derived metabolites of AEA with no activity at cannabinoid receptors or TRPV1 channels (two preferential targets for AEA at sub-micromolar concentrations) and which exhibit no meaningful activity at wild-type prostanoid FP receptors (Matias et al., 2004; Woodward et al., 2008). They are not hydrolysed by FAAH, the main hydrolytic enzyme for AEA (Matias et al., 2004). Prostamide F2α, to date the most studied of the prostamides, appears to interact with a dedicated subset of prostamide receptors (Matias et al., 2004; Woodward et al., 2008; 2011a). Specifically, the receptors for prostamide F2α and its synthetic derivative bimatoprost, have been identified as heterodimers between wild-type FP receptors and an alternative FP splicing variant (Liang et al., 2008). Among the pharmacological responses triggered by prostamide F2α receptors (Woodward et al., 2008), hyperexcitation of spinal cord nociceptive neurons contributes to sustained pain under conditions of inflammation (Gatta et al., 2012), a pathological setting that is (i) opposed by activation of cannabinoid CB1 or CB2 receptors (Guindon and Hohmann, 2009); (ii) accompanied by raised levels of prostamide F2α in the spinal cord or dorsal root ganglia (Duggan et al., 2011; Gatta et al., 2012); and (iii) often also accompanied by reduction of FAAH expression/activity and up-regulation of COX-2 (Jhaveri et al., 2007). Indeed, prostamides have also been identified in much higher amounts in several tissues from FAAH null mice (Weber et al., 2004), which are characterized by elevated basal levels of AEA. Therefore, it is possible that not only up-regulation of COX-2, but also reduction of FAAH activity can lead to up-regulation of prostamide F2α levels and activation of prostamide F2α receptors. Hence, FAAH inhibition with selective FAAH blockers as has been proposed as a potential therapeutic approach for several pathological conditions, including pain (Petrosino and Di Marzo, 2010; Blankman and Cravatt, 2013), might be accompanied by prostamide F2α receptor activation and subsequent worsening of pain and inflammation. One way to circumvent this potential problem, which could limit the efficacy of FAAH inhibitors as analgesics, would be to develop compounds that at the same time inhibit FAAH and antagonize prostamide F2α receptors. A similar approach has proved successful, for example, with ‘dual’ FAAH/TRPV1 blockers (Maione et al., 2007; 2013; like prostamide F2α receptors, TRPV1 is be involved in pain transduction (Gatta et al., 2012)). However, the cellular levels of prostamide F2α are also elevated under inflammatory conditions not necessarily accompanied by reduced FAAH activity (Duggan et al., 2011), or under physiological conditions in cells that constitutively express high levels of COX-2, such as pre-adipocytes, where they inhibit differentiation into adipocytes (Silvestri et al., 2013). Therefore, antagonists of prostamide F2α receptors might be therapeutically useful, regardless of whether or not they inhibit FAAH, although their potential simultaneous ‘indirect’ activation of cannabinoid receptors should still be seen as an added value towards increased efficacy, for example, against inflammatory pain. The present study provides new evidence that compounds developed as antagonists of prostamide F2α and bimatoprost receptors can also inhibit FAAH to a varying extent.
Of the compounds tested here against rat brain FAAH, AGN 211335 and AGN 211336, which were among the most potent at antagonizing the prostamide F2α receptor in the feline iris and selective in terms of a panel of prostanoid receptors (Woodward et al., 2007; Liang et al., 2008; present data), proved to be the most potent at inhibiting the enzyme, with IC50 values at around 1 μM when assessed with a pre-incubation protocol. These pharmacodynamic properties might be due to the fact that these two compounds not only lack an acyl sulfonamide moiety, but, unlike all other compounds tested here except AGN 211334, also exhibit an extra ether function in their chemical structure. Furthermore, in the series of compounds that differ only in the nature of one of the two alkylamide substituents, that is, AGN 211334, AGN 211335 and AGN 211336, the inhibitory activity against FAAH was highest with the propyl and lowest with the ethyl group. Comparing AGN 211335 and AGN 211336, the former compound appeared to be slightly more efficacious at inhibiting AEA hydrolysis also in intact RBL-2H3 cells, which suggests that these compounds are also equally efficacious at crossing the plasma membrane. Importantly, neither compound exerted significant inhibitory activity against the hydrolysing enzymes for the other major endocannabinoid, 2-AG, in COS-7 cell cytosolic fractions. However, all compounds were still active as thromboxane TP receptor antagonists, which is a desired effect for anti-inflammatory compounds but may cause undesired cardiovascular effects.
AGN 211335 was further evaluated and exhibited non-competitive inhibition of rat brain FAAH and also enhancement of ionomycin-induced OEA and PEA levels in RBL-2H3 cells. AEA levels were not significantly enhanced, possibly because, unlike those of OEA and PEA, they were already maximally stimulated by ionomycin. AGN 211335 was also the most efficacious against the human enzyme, with an IC50 approaching 10 μM, whereas AGN 211336 exhibited almost no measurable activity at human recombinant FAAH. It is worthwhile noting that the commercial human recombinant FAAH used in this study was also significantly less sensitive to the potent inhibitor URB597 (IC50 0.1 μM vs. 0.01 μM in rat brain membranes, with a 20 min pre-incubation, data not shown). Based on previously published data with other ‘dual’ or ‘multiple’ target FAAH inhibitors, such as the dual TRPV1/FAAH blocker N-arachidonoyl-serotonin (Maione et al., 2007; 2013), it was therefore possible to hypothesize that the activity of AGN 211335 and AGN 211336 at FAAH is sufficient to produce analgesic actions in vivo. This possibility was addressed experimentally here in mice, by using the formalin model of acute pain. As predicted, both compounds exerted similar analgesic actions, both in the first phase of acute pain, and particularly in the second phase of pain sensitization and inflammatory pain. These findings should encourage new studies of AGN 211335 and AGN 211336 also in other models of inflammatory and chronic pain.
Of the two most potent ‘dual’ FAAH/prostamide receptor blockers identified in this study, AGN 211335 was also the one with highest (although with a Ki still in the high nanomolar range) affinity at CB1 receptors, as assessed in a specific binding assay, whereas both compounds were completely inactive in a functional assay of human recombinant TRPV1 channel activity. While it still remains to be determined whether its affinity for CB1 receptors represents agonist or antagonist/inverse agonist activity, the ability of AGN 211335 to bind to this receptor might improve further its efficacy against inflammatory pain, as both CB1 receptor agonists and inverse agonists are known to counteract this condition (Comelli et al., 2010; Wiley et al., 2011), but also produce unwanted central effects. In this sense, we propose that future studies aiming at obtaining new ‘dual’ FAAH/prostamide receptor blockers with improved activity at both targets should start from the chemical modification of AGN 211336, rather than AGN 211335 because ‘indirect’ (i.e. via elevation of endocannabinoid levels only in those tissues where their degradation is pathologically enhanced), rather than direct, activation of CB1 receptors is still seen as a safer approach to treat pain and inflammation (Petrosino and Di Marzo, 2010; Blankman and Cravatt, 2013). On the other hand, it is unlikely that by antagonizing prostamide F2α receptors, AGN 211335 and AGN 211336 redirect prostamide F2α activity towards TRPV1 channels, thus diminishing their analgesic potential because prostamide F2α only showed very little activity at these channels, which was significantly lower also than that of AEA (Matias et al., 2004).
It is worthwhile noting that COX-2 also catalyses the oxygenation of the other most studied endocannabinoid, 2-AG. The resulting metabolites, known as PG glycerol esters, have also been suggested to act on non-cannabinoid, non-prostanoid receptors, and shown to produce pro-inflammatory actions (Nirodi et al., 2004; Hu et al., 2008; see Woodward et al., 2008). Therefore, it would be interesting in the future to also develop compounds with dual antagonist activity on yet to be identified PG glycerol ester receptors and 2-AG hydrolytic enzymes such as monoacylglycerol lipase. Importantly, selective inhibition of COX-2-mediated oxygenation of endocannabinoids was recently reported to produce anxiolytic effects through the enhancement of the brain levels of AEA and 2-AG and indirect activation of CB1 receptors (Hermanson et al., 2013). However, the potential role in these effects of the concomitant reduction of the brain concentrations of prostamides and PG glycerol esters, and of the subsequent reduction of the activity of their receptors, has not yet been investigated, nor has the role of these mediators in anxiety. In this context, and also in consideration of the affective component of pain perception, it might be interesting to study in the future the effect on anxiogenic behaviours of the FAAH/prostamide receptor blockers identified in the present study.
In conclusion, we have provided here the first examples of ‘dual’ FAAH inhibitors/prostamide receptor blockers. Further studies are now needed to evaluate the potential of these compounds to treat chronic inflammatory conditions and to improve their pharmacokinetic and pharmacodynamic properties.
Acknowledgments
The authors are grateful to Roberta Verde and Teresa Aveta for technical assistance with LC-MS analyses in intact RBL-2H3 cells, and Dr. Luciano De Petrocellis for performing the TRPV1 functional assays.
Glossary
- 2-AG
2-arachidonoylglycerol
- AEA
anandamide
- COX-2
cyclooxygenase-2 (prostaglandin-endoperoxide synthase 2)
- FAAH
fatty acid amide hydrolase
- NAGly
N-arachidonoylglycine
- OEA
oleylethanolamide; PEA
palmitoylethanolamide
Conflict of interest
J. W. and D. W. are employees of Allergan, USA. J. M. is an employee of Selcia, UK. A. L. and V. D. receive funding from Allergan, USA.
References
- Abbott FV, Franklin KB, Westbrook RF. The formalin test: scoring properties of the first and second phases of the pain response in rats. Pain. 1995;60:91–102. doi: 10.1016/0304-3959(94)00095-V. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, et al. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta. 2009;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RM, Woodward DF. A historical perspective and recent advances in prostamide research and therapeutics. Curr Opin Drug Discov Devel. 2007;10:413–421. [PubMed] [Google Scholar]
- Comelli F, Bettoni I, Colombo A, Fumagalli P, Giagnoni G, Costa B. Rimonabant, a cannabinoid CB1 receptor antagonist, attenuates mechanical allodynia and counteracts oxidative stress and nerve growth factor deficit in diabetic mice. Eur J Pharmacol. 2010;637:62–69. doi: 10.1016/j.ejphar.2010.03.061. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Duggan KC, Hermanson DJ, Musee J, Prusakiewicz JJ, Scheib JL, Carter BD, et al. R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol. 2011;7:803–809. doi: 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta L, Piscitelli F, Giordano C, Boccella S, Lichtman A, Maione S, et al. Discovery of prostamide F2alpha and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. Plos ONE. 2012;7:e31111. doi: 10.1371/journal.pone.0031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, et al. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16:1291–1298. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol. 2007;152:624–632. doi: 10.1038/sj.bjp.0707433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Giembycz MA, Woodward DF. Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J Pharmacol. 2009;158:104–145. doi: 10.1111/j.1476-5381.2009.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koda N, Tsutsui Y, Niwa H, Ito S, Woodward DF, Watanabe K. Synthesis of prostaglandin F ethanolamide by prostaglandin F synthase and identification of Bimatoprost as a potent inhibitor of the enzyme: new enzyme assay method using LC/ESI/MS. Arch Biochem Biophys. 2004;424:128–136. doi: 10.1016/j.abb.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JJ, Rowlinson SW, Prudhomme DR, Marnett LJ. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochemistry. 2003;42:9041–9049. doi: 10.1021/bi034471k. [DOI] [PubMed] [Google Scholar]
- Krauss AH, Woodward DF. Prostamide Receptor Antagonists. 10893054. Irvine, CA: Allergan Sales, Inc; 2006. [Google Scholar]
- Liang Y, Woodward DF, Guzman VM, Li C, Scott DF, Wang JW, et al. Identification and pharmacological characterization of the prostaglandin FP receptor and FP receptor variant complexes. Br J Pharmacol. 2008;154:1079–1093. doi: 10.1038/bjp.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Costa B, Di Marzo V. Endocannabinoids: a unique opportunity to develop multitarget analgesics. Pain. 2013 doi: 10.1016/j.pain.2013.03.023. doi: pii:S0304-3959(13)00118-8.10.1016/j.pain.2013.03.023. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Matias I, Chen J, De Petrocellis L, Bisogno T, Ligresti A, Fezza F, et al. Prostaglandin ethanolamides (prostamides): in vitro pharmacology and metabolism. J Pharmacol Exp Ther. 2004;309:745–757. doi: 10.1124/jpet.103.061705. [DOI] [PubMed] [Google Scholar]
- Maurelli S, Bisogno T, De Petrocellis L, Di Luccia A, Marino G, Di Marzo V. Two novel classes of neuroactive fatty acid amides are substrates for mouse neuroblastoma ‘anandamide amidohydrolase’. FEBS Lett. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- Moriuchi H, Koda N, Okuda-Ashitaka E, Daiyasu H, Ogasawara K, Toh H, et al. Molecular characterization of a novel type of prostamide/prostaglandin F synthase, belonging to the thioredoxin-like superfamily. J Biol Chem. 2008;283:792–801. doi: 10.1074/jbc.M705638200. [DOI] [PubMed] [Google Scholar]
- Nirodi CS, Crews BC, Kozak KR, Morrow JD, Marnett LJ. The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proc Natl Acad Sci U S A. 2004;101:1840–1845. doi: 10.1073/pnas.0303950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Di Marzo V. FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs. 2010;11:51–62. [PubMed] [Google Scholar]
- Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem Biophys Res Commun. 2002;296:612–617. doi: 10.1016/s0006-291x(02)00915-4. [DOI] [PubMed] [Google Scholar]
- Silvestri C, Martella A, Poloso NJ, Piscitelli F, Capasso R, Izzo A, et al. Anandamide-derived prostamide F2alpha negatively regulates adipogenesis. J Biol Chem. 2013;288:23307–23321. doi: 10.1074/jbc.M113.489906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J Biol Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- Weber A, Ni J, Ling KH, Acheampong A, Tang-Liu DD, Burk R, et al. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J Lipid Res. 2004;45:757–763. doi: 10.1194/jlr.M300475-JLR200. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Breivogel CS, Mahadevan A, Pertwee RG, Cascio MG, Bolognini D, et al. Structural and pharmacological analysis of O-2050, a putative neutral cannabinoid CB(1) receptor antagonist. Eur J Pharmacol. 2011;651:96–105. doi: 10.1016/j.ejphar.2010.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Krauss AH, Wang JW, Protzman CE, Nieves AL, Liang Y, et al. Identification of an antagonist that selectively blocks the activity of prostamides (prostaglandin-ethanolamides) in the feline iris. Br J Pharmacol. 2007;150:342–352. doi: 10.1038/sj.bjp.0706989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Carling RW, Cornell CL, Fliri HG, Martos JL, Pettit SN, et al. The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Pharmacol Ther. 2008;120:71–80. doi: 10.1016/j.pharmthera.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011a;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Wang JW, Cornell CL, Fliri HG, Martos JL, Pettit SN. Prostamide Receptor Antagonists. 11869697. Irvine CA: Allergan Sales, Inc; 2011b. [Google Scholar]