

Abstract
Background and PurposeRetinoids, through their activation of retinoic acid receptors (RARs) and retinoid X receptors, regulate diverse cellular processes, and pharmacological intervention in their actions has been successful in the treatment of skin disorders and cancers. Despite the many beneficial effects, administration of retinoids causes irritating side effects with unknown mechanisms. Here, we demonstrate that LE135 [4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)benzoic acid], a selective antagonist of RARβ, is a potent activator of the capsaicin (TRPV1) and wasabi (TRPA1) receptors, two critical pain-initiating cation channels.
Experimental ApproachWe performed to investigate the excitatory effects of LE135 on TRPV1 and TRPA1 channels expressed in HEK293T cells and in dorsal root ganglia neurons with calcium imaging and patch-clamp recordings. We also used site-directed mutagenesis of the channels to determine the structural basis of LE135-induced activation of TRPV1 and TRPA1 channels and behavioural testing to examine if pharmacological inhibition and genetic deletion of the channels affected LE135-evoked pain-related behaviours.
Key ResultsLE135 activated both the capsaicin receptor (TRPV1) and the allyl isothiocyanate receptor (TRPA1) heterologously expressed in HEK293T cells and endogenously expressed by sensory nociceptors. Mutations disrupting the capsaicin-binding site attenuated LE135 activation of TRPV1 channels and a single mutation (K170R) eliminated TRPA1 activity evoked by LE135. Intraplantar injection of LE135 evoked pain-related behaviours. Both TRPV1 and TRPA1 channels were involved in LE135-elicited pain-related responses, as shown by pharmacological and genetic ablation studies.
Conclusions and ImplicationsThis blocker of retinoid acid signalling also exerted non-genomic effects through activating the pain-initiating TRPV1 and TRPA1 channels.
Keywords: LE135, TRPV1, TRPA1, nociception, thermal hyperalgesia, mechanical allodynia
Introduction
Primary sensory neurons in dorsal root, trigeminal and nodose ganglia initiate pain in response to noxious thermal, mechanical and chemical stimuli (Patapoutian et al., 2009). Transient receptor potential (TRP) channels are calcium-permeable non-selective cation channels with highly divergent properties that are widely expressed in the primary sensory neurons (Dubin and Patapoutian, 2010). TRP channels play key roles in the pathogenesis of both inflammatory and neuropathic pain (Jaggi and Singh, 2011).
Among nearly 30 mammalian TRP channels, TRPV1 and TRPA1 (channel nomenclature follows Alexander et al., 2013a) are both polymodal detectors integrating painful stimuli and playing central roles in pain sensation under physiological and pathological conditions including inflammation and neuropathy. TRPV1 channels are activated by a variety of physical and chemical stimuli including capsaicin, noxious heat (>43°C) and low pH (5.2) (Tominaga et al., 1998). Expression of these channels is up-regulated in inflamed human skin and vulva, which correlates with inflammatory hyperalgesia (Gopinath et al., 2005). Selective TRPV1 blockers, such as AMG9810 [(2E)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide], reduce both hyperalgesia and allodynia in rodent models of pathological nociception and inflammatory pain (Gavva et al., 2005). TRPA1 channels are expressed in a subset of TRPV1-positive nociceptors integrating actions of many exogenous and endogenous noxious stimuli, such as the natural pungent compound allyl isothiocyanate (AITC) and oxidative oxygen radicals (Bautista et al., 2013). There is compelling evidence for the involvement of TRPA1 channels in pain sensation in humans as a gain-of-function mutation in TRPA1 causes familial episodic pain syndrome (Kremeyer et al., 2010). Expression of these channels is increased in dorsal root ganglia (DRG) by Freund's complete adjuvant (CFA)-induced inflammation or nerve injury (Frederick et al., 2007; da Costa et al., 2010). Mice with genetic deletion (knockout) of TRPA1 channels show reduced thermal and mechanical pain responses to inflammatory mediator bradykinin (Bautista et al., 2006). Pharmacological inhibition of TRPA1 function reduces pain-related responses in both inflammatory and neuropathic pain models (Petrus et al., 2007; Eid et al., 2008).
Retinoids are structurally related derivatives of vitamin A and are required for normal vision as well as cell proliferation and differentiation (Kim, 2011; Mamede et al., 2011). Clinically, retinoids are effective in treating acute promyelocytic leukaemia and many skin disorders (Lowe and Plosker, 2000; Thacher et al., 2000; Geria et al., 2011; Mamede et al., 2011). All-trans retinoic acid (ATRA or tretinoin) is the first Food and Drug Administration (FDA)-approved topical retinoid with documented efficacy to treat acne vulgaris, the most common skin condition in the United States (Hsu et al., 2011). Retinoids were also approved by the FDA to be used as an anti-ageing treatment in 1996. The pleiotropic effect of retinoids is mediated by the retinoid nuclear receptors comprising the retinoic acid receptors (RARs) (α, β and γ isotypes) and the retinoid X receptors (RXRs) (α, β and γ isotypes), in the form of RAR/RXR heterodimers (Thacher et al., 2000 receptor nomenclature follows Alexander et al., 2013b).
Despite the many beneficial effects, topical application of retinoids often causes severe local irritation associated with itching and stinging (Le et al., 1997; Leyden, 1998; Lowe and Plosker, 2000; Akhavan and Bershad, 2003; Thielitz and Gollnick, 2008; Geria et al., 2011; Hsu et al., 2011; Yoon et al., 2011). Retinoids also cause severe headache and bone pain when used systemically to treat cancers (White et al., 1992; Vergne et al., 2000; Mawson, 2009). Previous studies also show that oral or intrathecal application of ATRA induces nociceptive behavioural effects in rodents (Romero-Sandoval et al., 2004; Alique et al., 2006). Our recent studies demonstrated that retinoids selectively activated the capsaicin receptor TRPV1 and produce sensory hypersensitivity (Yin et al., 2013). Also, some pan antagonists of RAR attenuated retinoid-induced irritation (Alique et al., 2006). Against this background of pro-nociceptive actions of retinoid receptor agonists, we were surprised to find that a selective RAR receptor antagonist, LE135 [4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b][1,4]diazepin-13-yl)benzoic acid], activated both TRPA1 and TRPV1 channels and produced pain-related behaviours.
Methods
Animals
All animal care and experimental procedures were in accordance with the animal care and use protocol approved by The University of Texas Health Science Center at Houston Animal Welfare Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 118 animals were used in the experiments described here. Trpv1+/+ and congenic Trpv1−/− mice on the C57BL/6J background were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and were bred at the University of Texas Health Science Center at Houston. Trpa1+/+ and congenic Trpa1−/− mice on the C57BL/6J background were described previously (Cruz-Orengo et al., 2008) and bred at the University of Texas Health Science Center at Houston. Mice were housed in a temperature- and humidity-controlled environment on a 12:12 h dark–light cycle with free access to food and water.
Molecular biology, HEK293T cell culture and transfection
HEK293T cells (ATCC, Manassas, VA, USA) were grown as a monolayer using passage numbers less than 30 and maintained in DMEM (Life Technologies, Grand Island, NY, USA), supplemented with 10% FBS (Life Technologies), 100 units·mL−1 penicillin and 100 μg·mL−1 streptomycin in a humidified incubator at 37°C with 5% CO2. The cells were transiently transfected with complementary DNA for mouse TRPV1 (mTRPV1), TRPV1 mutants, or human TRPA1 (hTRPA1) and TRPA1 mutants using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) with a ratio of 0.8:2. After transfection, cells were maintained in DMEM at 37°C for 24 h before use. All TRPV1 and TRPA1 mutants were made using the QuickChange II XL mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA, USA), according to the manufacturer's directions. All mutations were confirmed by DNA sequencing.
Isolation and short-term culture of mouse DRG neurons
Mouse spinal columns were removed and placed in ice-cold HBSS; neurons were acutely dissociated and maintained as described (Hu et al., 2009). In brief, laminectomies were performed and bilateral DRG were dissected out. After removal of connective tissues, DRG were transferred to a 1 mL Ca2+/Mg2+-free HBSS containing 2 μL saturated NaHCO3, 0.35 mg l-cysteine and 20 U papain (Worthington, Lakewood, NJ, USA), and incubated at 37°C for 10 min. The suspension of DRG was centrifuged, the supernatant was removed, 1 mL Ca2+/Mg2+-free HBSS containing 4 mg collagenase type II and 1.25 mg dispase type II (Worthington) was added and incubated at 37°C for 10 min. After digestion, neurons were pelleted, suspended in neurobasal medium containing 2% B-27 supplement, 1% l-glutamine, 100 U·mL−1 penicillin plus 100 μg·mL−1 streptomycin, and 50 ng·mL−1 nerve growth factor, plated on a 12 mm coverslip coated with poly-l-lysine (10 μg·mL−1) and cultured under a humidified atmosphere of 5% CO2/95% air at 37°C for 18–24 h before use.
Ratiometric measurement of intracellular free Ca2+
Cultured DRG neurons were loaded with 4 μM Fura-2 AM (Life Technologies) in culture medium at 37°C for 60 min. Cells were then washed three times and incubated in HBSS at room temperature for 30 min before use. Fluorescence at 340 and 380 nm excitation wavelengths was recorded on an inverted Nikon Ti-E microscope equipped with 340, 360 and 380 nm excitation filter wheels using NIS-Elements imaging software (Nikon Instruments Inc., Melville, NY, USA). Fura-2 ratios (F340/F380) were used to reflect changes in intracellular Ca2+ upon stimulation. Values were obtained from 100–250 cells in time-lapse images from each coverslip.
Patch-clamp recording
Whole-cell patch-clamp recordings were performed using an EPC 10 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) at room temperature (22–24°C) on the stage of an inverted phase-contrast microscope equipped with a filter set for green fluorescence protein visualization. Pipettes pulled from borosilicate glass (BF 150-86-10; Sutter Instrument Company, Novato, CA, USA) with a Sutter P-97 pipette puller had resistances of 2–4 MΩ when filled with pipette solution containing 140 mM CsCl, 2 mM EGTA, and 10 mM HEPES with pH 7.3 and 315 mOsm·L−1. To avoid Ca2+-dependent desensitization of TRPV1 channels, a Ca2+-free extracellular solution was used for whole-cell recording which contained (in mM): 140 NaCl, 5 KCl, 0.5 EGTA, 1 MgCl2, 10 glucose and 10 HEPES (pH was adjusted to 7.4 with NaOH, and the osmolarity was adjusted to ≈340 mOsm·L−1 with sucrose). The whole-cell membrane currents were recorded using voltage ramp from −100 to +100 mV for 500 ms at holding potential of 0 mV. Data were acquired using PatchMaster software (HEKA Elektronik). Currents were filtered at 2 kHz and digitized at 10 kHz. Data were analysed and plotted using Clampfit 10 (Molecular Devices, Sunnyvale, CA, USA). Values are given as means ± SEM; n represents the number of measurements.
Pain behavioural assays
Each mouse was placed individually in clear Plexiglas chambers (8 × 8 × 12 cm) and acclimated for at least 1 h to the testing environment prior to all experiments. To measure pain-related behaviours, the left hindpaws of mice were injected intraplantarly with 20 μL vehicle (0.9% saline + 5% DMSO + 2.5‰ Tween 80; Sigma, St Louis, MO, USA) with or without chemicals. Time spent on nocifensive behaviour (flicking and licking injected paw) was recorded for 5 min. Mechanical or thermal hyperalgesia assays were performed as described (Caterina et al., 2000; Petrus et al., 2007). Paw withdraw latencies in response to radiant heat were measured using the Hargreaves apparatus (Plantar Analgesia meter; IITC, Woodland Hills, CA, USA). For assessment of thermal nociception, left hindpaw withdraw latencies were measured before (0 min) and 15, 30, 60, 90 and 120 min after injections. The thermal intensity was adjusted to obtain paw withdrawal latencies of about 10–15 s under basal conditions . An automatic 20 s cut-off was used to prevent tissue damage. For assessment of mechanical allodynia, starting with the 0.4 g filament, von Frey filaments ranging from 0.04 to 4 g bending force were applied to the plantar skin of the left hindpaw using the up–down method to determine threshold sensitivity. Von Frey threshold was measured at 15, 30, 60, 90 and 120 min post-injection. AMG9810 (10 mg·kg−1, i.p.) was given 30 min before paw injections of retinoids to test its analgesic effect. To reduce the effects of baseline variability among animals, withdrawal responses were expressed as differences from baseline across groups (Bedi et al., 2010). All experiments were performed without knowledge of genotype and treatment.
Data analysis
All data are presented as means ± SEM for n independent observations. Student's t-test was used to analyse statistical significance between control and experimental groups. anova and repeated measures tests were used to test hypotheses about effects in multiple groups occurring over time. P < 0.05 was considered significant differences between means.
Materials
AITC was from ACROS (Geel, Belgium); LE135 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA); papain and collagenase (type 2) were from Worthington. All other compounds were from Sigma.
Results
LE135 activates TRPV1 channels expressed in heterologous cells and DRG neurons
Previous studies showed that ATRA induces sensitization of nociceptive responses in rodents (Romero-Sandoval et al., 2004; Alique et al., 2006), suggesting that retinoids might alter sensory neuron excitability. Our recent studies show that ATRA produces an excitatory response when administered to mouse DRG cultures by activating the capsaicin receptor TRPV1 (Yin et al., 2013). Interestingly, it was also reported that inhibition of RARs by pan RAR inhibitors could reduce ATRA-evoked nociceptive response (Alique et al., 2006), suggesting that RAR inhibitors could potentially inhibit ATRA-induced activation of TRPV1 in DRG neurons. We tested this possibility by using a selective RAR inhibitor, LE135, which binds to, but does not activate, RARs, thus displaying antagonist activities at RARs. We used calcium imaging as readout of neuronal excitability. Surprisingly, bath application of LE135 alone evoked a robust intracellular calcium response in nearly 25% of DRG neurons tested, all of which also responded to capsaicin (Figure 1A–C), demonstrating that LE135 directly activated the TRPV1-positive nociceptors. Interestingly, the proportion of LE135-responsive neurons was also substantially reduced but not abolished by genetic ablation of TRPV1 function (Figure 1A–C). These results indicate that TRPV1 channels were involved in LE135-induced excitatory effect, but that those channels were not the sole target for LE135 in nociceptors.
Figure 1.
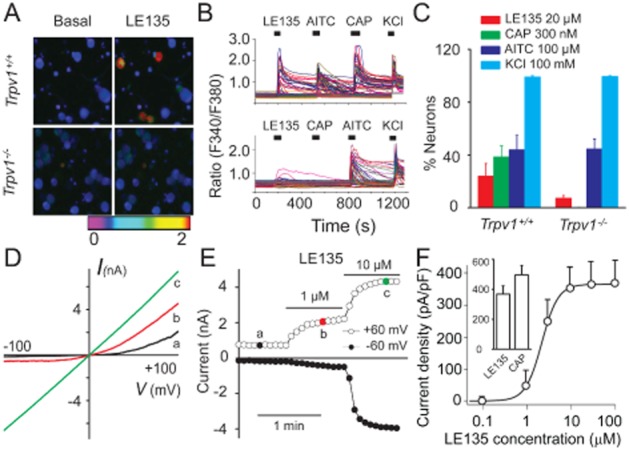
LE135 activates both recombinant and native TRPV1 channels. Representative images (A) and traces (B) illustrate that LE135-elicited intracellular Ca2+ responses are decreased in Trpv1−/− DRG neurons compared with that from Trpv1+/+ mice. AITC evoked similar intracellular Ca2+ responses in Trpv1+/+ and Trpv1−/− DRG neurons. Each trace corresponds to the change of fluorescence ratio in a single neuron in response to 20 μM LE135, 300 nM capsaicin (CAP), 100 μM AITC and 100 mM KCl at indicated times. (C) Percentage of DRG neurons responding to LE135, capsaicin, AITC and KCl in neurons isolated from Trpv1+/+ or Trpv1−/− mice (n ≥ 350 per genotype). Representative current–voltage relationships (D) and the time course (E) of LE135 (1 and 10 μM)-activated outward (at +60 mV) and inward (at −60 mV) currents in a TRPV1-expressing HEK293T cell. (F) Concentration–response curve of LE135-activated inward currents at −60 mV in WT TRPV1 is fitted with the logistic equation: Y = Ymin + (Ymax − Ymin)/(1 + 10∧[(log EC50 − X)*Hill slope)], where Y is the response at a given concentration, Ymax and Ymin are the maximum and minimum response, X is the logarithmic value of the concentration and Hill slope is the slope factor of the curve. EC50 is the concentration that gives a response halfway between Ymax and Ymin. The graph inset illustrates the maximal responses (current densities) evoked by saturating concentrations of LE135 (100 μM) and capsaicin (10 μM).
We then asked if LE135 could directly activate TRPV1 channels using whole-cell patch-clamp recordings on TRPV1-expressing HEK293T cells. Indeed, LE135 produced a concentration-dependent activation of TRPV1 channels with an EC50 of around 2.5 μM (Figure 1D–F). The current–voltage relationship showed outward rectification at low concentrations but became linear when high concentrations of LE135 (>10 μM) were applied (Figure 1D), as reported for the response of TRPV1 channels to capsaicin (Caterina et al., 1997; Tominaga et al., 1998). The LE135-activated current was partially reversible after washout (not shown). The maximal response evoked by 100 μM LE135 was about 74% of the response evoked by 10 μM capsaicin (Figure 1F). These results demonstrated that LE135 activated both recombinant and native TRPV1 channels.
Structural requirements for LE135 activation of TRPV1 channels
The TRPV1 channel is a molecular sensor that integrates many different noxious stimuli, such as those provided by capsaicin, protons or noxious temperatures, through distinct protein domains (Latorre et al., 2007). Next, we asked if LE135 shared a common mechanism with other stimulators, to activate TRPV1 channels. We made TRPV1 mutants in which activation by capsaicin (R115A, Y512A, S513Y, Y512A/S513Y, M548L and T551I), proton (E601Q and E649Q) or temperature (N629K, N653T/N654T) was selectively and severely impaired (Figure 2C) (Jordt et al., 2000; Jordt and Julius, 2002; Jara-Oseguera et al., 2008; Grandl et al., 2010). We also made a point mutation (S503A) in which a key TRPV1 phosphorylation site is disrupted (Figure 2C) (Jara-Oseguera et al., 2008). Mutants were then individually transfected into HEK293T cells, and concentration–response curves of LE135-induced whole-cell currents were made for each mutant with EC50 value comparison between each mutant and the wild-type (WT) TRPV1 channel. Interestingly, only those mutants affecting the response to capsaicin exhibited a substantially attenuated response to LE135, whereas mutants affecting proton and temperature activation of the channel did not alter LE135 response (Figure 2A,B,D). These results suggested that those amino acid residues required for capsaicin activation were likely to be involved in the interactions of LE135 with TRPV1 channels.
Figure 2.
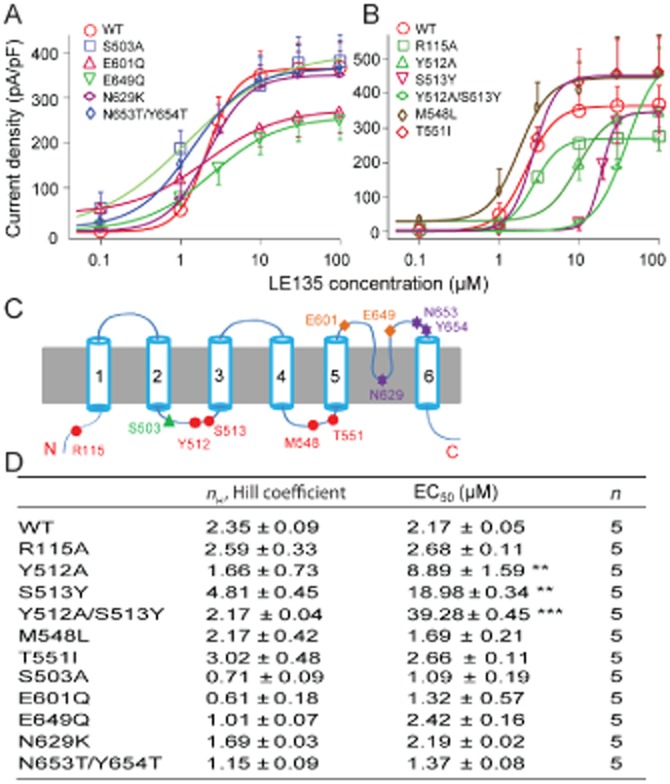
Structural basis of LE135 activation of TRPV1 channels. (A) Concentration–response curves of LE135-activated inward currents at −60 mV in WT and TRPV1 channel mutants with disrupted protein phosphorylation, proton or heat activation domains. (B) Concentration–response curves of LE135-activated inward currents at −60 mV in WT and TRPV1 mutants carrying single or double point mutations in the vanilloid-binding pocket. (C) Schematic diagram illustrates structural elements required for activation/modulation of TRPV1 channels by capsaicin (red filled circle), protein phosphorylation (green triangle), protons (orange diamond) and heat (purple star). (D) EC50 values and Hill coefficients of LE135-activated response in WT and TRPV1 mutants derived from concentration–response curves in (A) and (B). **P < 0.01 and ***P < 0.001 versus WT. Concentration–response curves are fitted with the logistic equation as described in Figure 1.
LE135 evokes TRPV1-dependent acute nocifensive responses and thermal hyperalgesia
TRPV1 is an excitatory pain-initiating channel expressed by small-diameter sensory neurons, activation of which produces nocifensive responses in rodents (Caterina et al., 1997). Knowing that LE135 is a potent TRPV1 channel activator, we tested if intraplantar injection of LE135 evokes TRPV1-mediated nociception in mice. As expected, LE135 induced a robust pain response as reflected by the time spent on flicking, licking and biting injected hindpaws. Interestingly, the selective TRPV1 antagonist, AMG9810, abolished LE135-evoked acute pain response, and the LE135-induced nociception was completely absent in Trpv1−/− mice (Figure 3).
Figure 3.
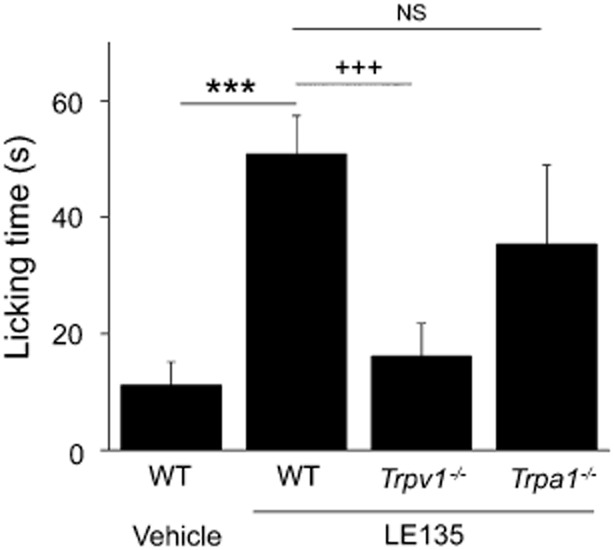
TRPV1 channels mediate LE135-induced nocifensive responses. Bar chart illustrates that in contrast to vehicle, intraplantar injection of LE135 (100 nmol/20 μL) produced robust flinching and licking responses that were significantly reduced in Trpv1−/− mice. LE135-evoked nocifensive responses were not significantly affected in Trpa1−/− mice compared with WT mice. ***, +++P < 0.001 versus vehicle and Trpv1−/− respectively; NS, not significant versus Trpa1−/−. n = 6–10 animals per condition.
Besides TRPV1 channels, the TRPA1 channels have also emerged as critical pain-initiating channels in sensory nociceptors. TRPA1 channels are exclusively located in TRPV1-positive small-diameter DRG neurons (Story et al., 2003; Jordt et al., 2004). Recent studies show that TRPA1 and TRPV1 channels may form heteromers and interact with each other functionally in the pain pathway (Akopian, 2011). Therefore, we examined if TRPA1 channels were also involved in LE135-induced nociception. However, we found similar nocifensive responses in WT and the Trpa1−/− mice after intraplantar injection of LE135. Therefore, TRPV1 but not TRPA1 channels are likely to be the primary mediator of LE135-induced nocifensive behaviour in our model.
It is also well known that TRPV1 channels are essential to inflammatory thermal hyperalgesia in both acute and chronic inflammatory pain models (Caterina et al., 2000; Davis et al., 2000; Kanai et al., 2007; Yu et al., 2008). We tested LE135 in the model of thermal hyperalgesia, using the plantar test (Hargreaves' method) and found that LE135 significantly reduced the paw withdrawal latency in response to radiant heat in WT mice (Figure 4), which was completely abolished by administration of AMG9810. Furthermore, such LE135-induced thermal pain behaviour was absent in the Trpv1−/− mice (Figure 4). Therefore, it appears that TRPV1 channels were sufficient and necessary for LE135-induced acute nocifensive responses and thermal hyperalgesia.
Figure 4.
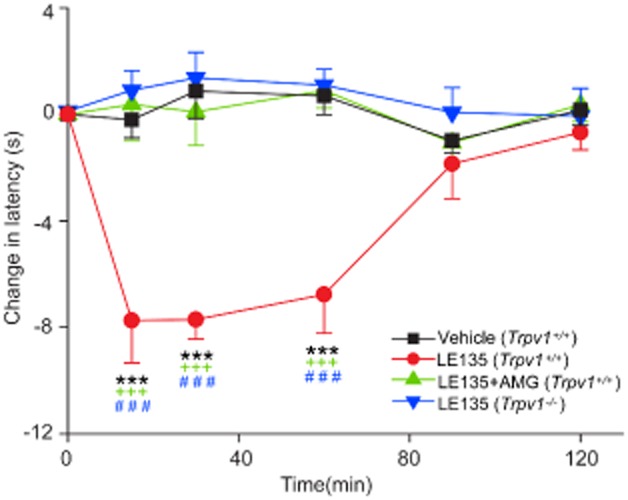
Pharmacological or genetic blockade of TRPV1 function abolishes LE135-induced thermal hyperalgesia. Time course of thermal hypersensitivity in animals treated with LE135. Intraplantar injection of LE135 (30 nmol/10 μL, red trace) induced thermal hyperalgesia in Trpv1+/+ mice as reflected by a decrease in paw withdrawal latency. Administration of AMG9810 (10 mg kg−1, i.p.,) removed the effect of LE135. LE135-elicited thermal hypersensitivity was also abolished in Trpv1−/− mice. *** P < 0.001 versus vehicle; +++ P < 0.001 versus AMG9810; and ### P < 0.001 versus Trpv1−/−. Please note that no effect was observed upon injection of vehicle alone. n = 5–10 animals per condition.
LE135-induced mechanical allodynia requires activation of both TRPV1 and TRPA1 channels
TRPV1 channels play an important role in inflammatory mechanical allodynia (Gavva et al., 2005; Yu et al., 2008; Hillery et al., 2011; Kitagawa et al., 2012). On testing LE135 in a model of mechanical allodynia, using the von Frey method (Caterina et al., 2000; Petrus et al., 2007), we found that intraplantar injection of LE135 substantially reduced the mechanical threshold of the injected hindpaws. Unexpectedly, the effect of LE135 was not significantly attenuated in the Trpv1−/− mice as we initially expected (Figure 5). Furthermore, AMG9810 had little, if any, effect on LE135-induced decrease of mechanical threshold (Figure 5). We therefore tested the involvement of TRPA1 channels because these channels are critically involved in both somatic and visceral mechanical hypersensitivity (Petrus et al., 2007; Lennertz et al., 2012; Sisignano et al., 2012), using mice with genetic deletion of TRPA1 channels (Trpa1−/− mice). Surprisingly, LE135-evoked mechanical allodynia was the same in the WT and Trpa1−/− mice. However, after pre-treatment with AMG9810 for 30 min, LE135-induced mechanical hypersensitivity in Trpa1−/− mice was totally abolished (Figure 5). The results suggested that activation of both TRPV1 and TRPA1 channels contributed to LE135-mediated mechanical hypersensitivity and genetic deletion of TRPA1 or TRPV1 function alone was not sufficient to abolish LE135-induced mechanical allodynia because of the functional redundancy in the TRPV1+/TRPA1+ nociceptors.
Figure 5.
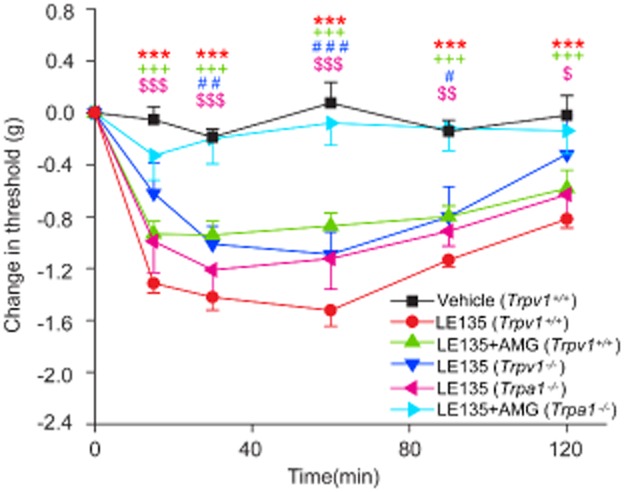
Both TRPV1 and TRPA1 channels contribute to LE135-induced mechanical allodynia. Time course of mechanical allodynia in animals treated with LE135. Intraplantar injection of LE135 (30 nmol/10 μL) produced mechanical allodynia as reflected by a decrease in paw withdrawal threshold in Trpv1+/+ mice, which was not significantly reduced by i.p. injection of AMG9810 (10 mg·kg−1), or genetic deletion of TRPA1 or TRPV1 channels. However, LE135-elicited mechanical hypersensitivity was completely abolished in the Trpa1−/− mice pre-treated with AMG9810 (10 mg·kg−1). ***P < 0.001 versus LE135; +++P < 0.001 versus AMG9810; #P < 0.05, ##P < 0.01, ###P < 0.001 versus Trpv1−/−; and $$P < 0.01, $$$P < 0.001 versus Trpa1−/−.
LE135 directly activates TRPA1 channels
In light of the fact that TRPA1 channels were required for LE135-induced mechanical allodynia in vivo, we next investigated if LE135 could directly activate TRPA1 channels expressed in heterologous cells and cultured DRG neurons. Whole-cell patch-clamp recordings showed that LE135 activated TRPA1 current in a concentration-dependent manner in TRPA1-expressing HEK293T cells with an EC50 of about 20 μM (Figure 6A–C). The maximal response evoked by 100 μM LE135 was about 41% of the response evoked by 100 μM AITC (Figure 6C). The I-V curves of LE135-activated currents show a strong outward rectification, a characteristic of TRPA1 currents (Figure 6A) (Jordt et al., 2004). Consistent with being an activator of both TRPA1 and TRPV1 channels, LE135-evoked intracellular calcium response was abolished in Trpa1−/− DRG neurons in the presence of AMG9810 or Trpv1−/− DRG neurons in the presence of HC030031 [2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N-(4-isopropylphenyl)acetamide], a selective TRPA1 antagonist (Figure 6D–F) (Eid et al., 2008). Furthermore, a combination of AMG9810 and HC030031 also completely abolished LE135-induced intracellular calcium response in WT DRG neurons (not shown). These results show that the excitatory effects of LE135 in DRG neurons were mediated by both TRPV1 and TRPA1, two critical pain-initiating channels.
Figure 6.
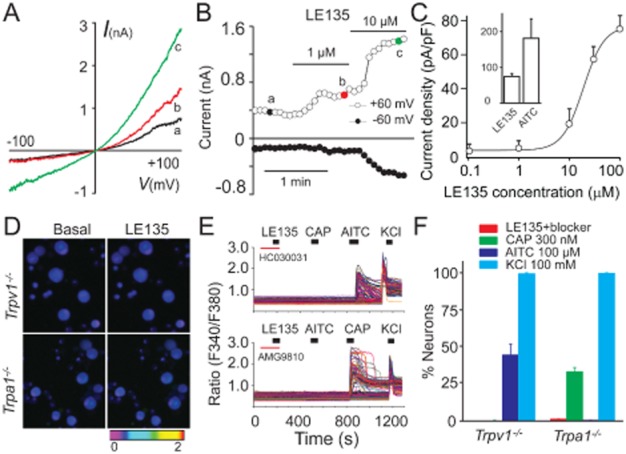
LE135 directly activates TRPA1 channels. LE135 (1 and 10 μM) activated an outward current at +60 mV and an inward current at −60 mV in a TRPA1-expressing HEK293T cell. Current traces in (A) show representative current–voltage relationships of LE135-activated currents. The time course of the current is shown in (B). (C) Concentration–response curve of LE135-activated inward currents taken at −60 mV in TRPA1-expressing HEK293T cells is fitted with the logistic equation as described in Figure 1. The graph inset illustrates the maximal responses (current densities) evoked by saturating concentrations of LE135 (100 μM) and AITC (100 μM). Representative pictures (D) and traces (E) illustrate that LE135-elicited intracellular Ca2+ responses are abolished in Trpv1−/− DRG neurons pre-treated with TRPA1 antagonist HC030031 (upper panel) or Trpa1−/− DRG neurons pre-treated with TRPV1 antagonist AMG9810 (lower panel). Each trace corresponds to the change of fluorescence ratio in a single neuron. (F) Percentage of DRG neurons responding to LE135, capsaicin (CAP), AITC and KCl in neurons isolated from Trpv1−/− or Trpa1−/− mice (n ≥ 350 per genotype).
A lysine residue is required for LE135 activation of TRPA1 channels
Previous studies have shown that covalent modification of three key cysteine residues (C621, C641 and C665) located at the N-terminus of TRPA1 channels mediates their activation by electrophilic compounds, such as AITC and 4-hydroxynonenal (4-HNE) (Hinman et al., 2006; Macpherson et al., 2007; Trevisani et al., 2007). To test if LE135 shares the same activation mechanism, we constructed concentration–response curves for LE135-activated currents in HEK293T cells transfected with either WT or the TRPA1-3C mutant. Surprisingly, there was only a slight increase of the EC50 value in the TRPA1-3C mutant compared with that of WT TRPA1 (Figure 7A,B), suggesting that these cysteine residues were not essential to LE135 activation of TRPA1 channels.
Figure 7.
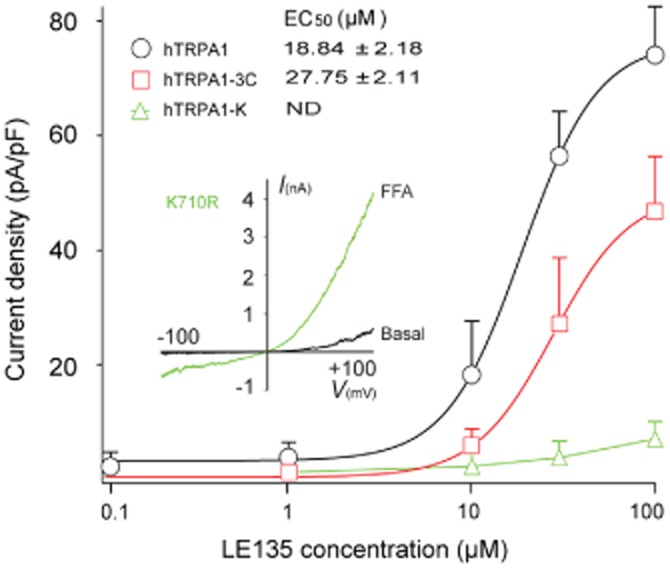
A single lysine residue is required for LE135 activation of TRPA1 Channels. Concentration–response curves of LE135-activated currents in WT and TRPA1 mutants carrying a single point mutation K710R or triple cysteine mutations (C621S, C641S, and C665S). EC50 values of LE135-activated response in WT and TRPA1 mutants derived from concentration–response curves in (A) are indicated. Concentration–response curves are fitted with the logistic equation, as described in Figure 1. ND, not determined. The graph inset illustrates that the hTRPA1-K mutant channel was activated by a TRPA1 agonist, flufenamic acid (FFA; 300 μM).
Besides cysteines, a critical lysine residue, K710, has also been shown to be modified by AITC, and that a K710R mutation could abolish the residual response of the TRPA1-3C mutant to AITC (Hinman et al., 2006). On testing LE135 in HEK293T cells transfected with the K710R mutant, we found that the LE135-activated currents were almost abolished (Figure 7A,B). On the contrary, application of another TRPA1 agonist flufenamic acid (Hu et al., 2009) led to a robust activation of the K710R mutant, suggesting that the K710R mutant channel was functionally expressed in HEK293 cells. These observations suggested that activation of TRPA1 channels by LE135 critically involved the K710 residue in these channels.
Discussion
Our study provided evidence that LE135, a selective RARβ antagonist, induced acute nociception and inflammatory hyperalgesia through activation of two pain-initiating TRP channels in sensory nociceptors. LE135 interacts with cytosolic amino acid residues (the vanilloid-binding site) required for capsaicin to activate TRPV1 channels, and a cytosolic lysine residue to activate TRPA1 channels. Genetic deletion and pharmacological inhibition studies show that LE135 provoked nociceptive responses and elicited thermal hyperalgesia mainly through TRPV1 channels, but required both TRPA1 and TRPV1 channels for producing mechanical allodynia. These results suggest that LE135 is a potent activator of pain-initiating TRPV1 and TRPA1 channels on the membrane of sensory nociceptors, although this compound had been shown to be selective for the RARβ receptor over other retinoid nuclear receptors.
Both TRPA1 and TRPV1 channels are required for LE135-induced inflammatory mechanical allodynia
The role of TRPV1 channels in mechanical hypersensitivity is still controversial because TRPV1-deficient mice appear to have a mechanical threshold response similar to WT littermates with or without tissue injuries evoked by applying mustard oil to the hindpaws (Caterina et al., 2000). This observation can be explained by either a lack of involvement of TRPV1 channels in noxious mechanical hypersensitivity or that another mustard oil-sensitive protein in the pain pathway compensates for the absence of TRPV1 channels. In this case, the TRPA1 channels, which are activated by mustard oil (Bandell et al., 2004; Jordt et al., 2004), are an obvious alternative. The compensation hypothesis is further supported by later findings that acute pharmacological blockade of TRPV1 channels attenuated inflammatory mechanical allodynia (Gavva et al., 2005). Our results showed that mechanical allodynia evoked by LE135 was not affected by deletion of either TRPV1 or TRPA1 channels, individually. However, LE135-induced mechanical allodynia was completely abolished when the functions of both TRPA1 and TRPV1 channels were blocked using a combination of genetic and pharmacological manipulations. These observations support a model in which both TRPV1 and TRPA1 channels sense LE135 in vivo and are compensating for each other to maintain the mechanical allodynia caused by intraplantar injection of LE135. The other possibility is that sensitization of TRPV1 depends on the presence of TRPA1 channels and vice versa. Indeed, it has been proposed that the functions of TRPA1 and TRPV1 channels are closely associated, and that these two nociceptive channels might form functional heteromers (Akopian, 2011). Therefore, LE135 becomes a useful tool to dissect the in vivo function of pain-initiating TRP channels.
TRPV1 channels are essential to LE135-induced acute nocifensive response and thermal hyperalgesia
It is well established that TRPV1 channels are required for inflammatory thermal hyperalgesia in both acute and chronic inflammatory pain models generated by intraplantar injection of carrageenan or CFA (Caterina et al., 2000; Davis et al., 2000; Yu et al., 2008). Our results show that LE135, like other TRPV1 activators, produces robust thermal pain behaviours that were abolished by either genetic or pharmacological blockade of TRPV1 channels. Therefore, TRPV1 channels are necessary and sufficient for LE135-induced thermal pain response, although we cannot exclude the possibility that TRPA1 activation by LE135 in the same DRG neurons might also modulate the neurogenic inflammation. Interestingly, mustard oil, another TRPA1 channel activator, induces inflammatory thermal hyperalgesia that is also mediated by TRPV1 channels (Bandell et al., 2004; Jordt et al., 2004), suggesting that TRPV1 channels are key downstream, thermal pain mediators integrating signalling events evoked by many different noxious chemicals, including TRPA1 channel activators. Therefore, it is possible that LE135 activation of TRPA1 channels could also be an upstream event for thermal hyperalgesia.
Although TRPA1 channels are required for LE135-evoked mechanical allodynia, genetic deletion of TRPA1 channels did not affect the acute nociceptive response produced by paw injection of LE135. Furthermore, nocifensive responses to LE135 were abolished in Trpv1−/− mice, suggesting that TRPV1 channels were sufficient and necessary for LE135-induced acute pain. The discrepancy in the role of TRPA1 channels in acute nociceptive response and mechanical allodynia might also result from the eight-fold lower potency of LE135 to activate TRPA1 channels compared with its potency at TRPV1 channels (EC50 values: 2.5 μM for TRPV1 and 20 μM for TRPA1).
Molecular mechanisms underlying LE135 activation of TRPA1 and TRPV1 channels
Like other TRP channels, both TRPV1 and TRPA1 are sensors for a variety of thermal, physical and chemical cues in the environment (Clapham, 2003; Damann et al., 2008). Different classes of activators act through distinct protein domains to gate these channels allosterically (Latorre et al., 2007). Our study shows that LE135 shares the same activation mechanisms as capsaicin and other structurally related vanilloids as disruption of the ‘capsaicin-binding pocket’ substantially attenuated the EC50 values of LE135 activation of TRPV1 channels. On the contrary, mutations affecting proton and temperature activation of TRPV1 channels did not affect the responses to LE135.
Besides the traditional ‘lock and key’ mechanism of ligand–receptor interaction, TRPA1 channels are also activated by covalent modification of cysteines and lysines at the cytosolic N-terminal region. Electrophilic compounds, such as AITC, 4-HNE and acrolein form disulfide bonds, covalently modifying cysteines, especially C621, C641 and C665, of human TRPA1. Our results show that the K710 residue was critically involved in LE135 activation of TRPA1 channels because responses to LE135 were abolished in HEK293T cells transfected with the K710R mutant. On the contrary, LE135-activated TRPA1 current was only slightly attenuated in HEK293T cells transfected with the hTRPA1-3C mutant, which contrasts with AITC-activated TRPA1 response (Hinman et al., 2006; Macpherson et al., 2007). Our results also suggest that different TRPA1 agonists could use distinct cysteine or lysine residues to activate TRPA1 channels even though they might all share the covalent modification mechanism.
Our previous studies show that many retinoids, including AMG580 and 4-hydroxy(phenyl)retinamide [4-HPR], selectively activate TRPV1 but not TRPA1 channels (Yin et al., 2013). Although structurally related to AMG580, LE135 activates both TRPV1 and TRPA1 channels and produces pain-related behaviours. Therefore, the diversity in the molecular structures of retinoid receptor ligands are likely to be responsible for LE135-mediated excitatory action on TRPA1 channels. There are many examples in pahamrcology of compounds with disparate effects on different receptors. Our results indicated that although LE135 displayed antagonist activities at nuclear RARs, it also activated membrane-bound pain-initiating ion channels in the peripheral nociceptors. One important property of TRPV1 and TRPA1 channels is that they undergo a Ca2+-dependent desensitization, which shuts down their functions after initial exposure of their respective agonists (Efendiev et al., 2013; Ibarra and Blair, 2013). We therefore speculate that RAR antagonists might inhibit retinoid-induced irritation by desensitizing the pain-initiating TRPV1 and TRPA1 channels.
In summary, our study revealed molecular and cellular mechanisms underlying activation of two pain-producing TRP channels by LE135, that mediated the inflammatory pain and nociception induced by this compound. Our findings disclosed an important pharmacological property of LE135 that could apply to other structurally related retinoid receptor ligands. Such off-target effects through activation of TRPA1 and TRPV1 channels have not been described earlier and should be considered in future studies in which LE135 and related retinoid drugs are used as selective antagonists of RARβ nuclear receptors.
Acknowledgments
We are grateful to Gina Story (Washington University in St. Louis) for providing the Trpa1−/− mice. We thank Dr Richard Clark for the critical reading of this article and our colleagues for helpful comments and discussions. This work was supported partly by grants from the National Institutes of Health RO1RGM101218A, Mission Connect/The Institute for Rehabilitation and Research (TIRR) Foundation (013-108), The University of Texas Health Science Center to H. H., and the National Natural Sciences Foundation of China to S. Y. (81373379) and J. L. (31000420).
Glossary
- 4-HNE
4-hydroxynonenal
- 4-HPR
4-hydroxy(phenyl)retinamide
- AITC
allyl isothiocyanate
- AM580
4-[(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)carboxamido]benzoic acid
- AMG9810
(2E)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide
- ATRA
all-trans retinoic acid
- CFA
Freund's complete adjuvant
- DRG
dorsal root ganglia
- HC030031
2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N-(4-isopropylphenyl)acetamide
- LE135
4-(7,8,9,10-tetrahydro-5,7,7,10,10-pentamethyl-5H-benzo[e]naphtho[2,3-b] [1,4]diazepin-13-yl)benzoic acid
- RARs
retinoic acid receptors
- RXRs
retinoid X receptors
- TRP
transient receptor potential
- WT
wild type
Conflict of interest
There is no conflict of interest to declare.
References
- Akhavan A, Bershad S. Topical acne drugs: review of clinical properties, systemic exposure, and safety. Am J Clin Dermatol. 2003;4:473–492. doi: 10.2165/00128071-200304070-00004. [DOI] [PubMed] [Google Scholar]
- Akopian AN. Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol. 2011;12:89–94. doi: 10.2174/138920111793937952. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013b;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alique M, Lucio FJ, Herrero JF. Vitamin A active metabolite, all-trans retinoic acid, induces spinal cord sensitization. II. Effects after intrathecal administration. Br J Pharmacol. 2006;149:65–72. doi: 10.1038/sj.bjp.0706826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: a gatekeeper for inflammation. Annu Rev Physiol. 2013;75:181–200. doi: 10.1146/annurev-physiol-030212-183811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, et al. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci. 2010;30:14870–14882. doi: 10.1523/JNEUROSCI.2428-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- da Costa DS, Meotti FC, Andrade EL, Leal PC, Motta EM, Calixto JB. The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain. 2010;148:431–437. doi: 10.1016/j.pain.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Cruz-Orengo L, Dhaka A, Heuermann RJ, Young TJ, Montana MC, Cavanaugh EJ, et al. Cutaneous nociception evoked by 15-delta PGJ2 via activation of ion channel TRPA1. Mol Pain. 2008;4:30. doi: 10.1186/1744-8069-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damann N, Voets T, Nilius B. TRPs in our senses. Curr Biol. 2008;18:R880–R889. doi: 10.1016/j.cub.2008.07.063. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120:3760–3772. doi: 10.1172/JCI42843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Bavencoffe A, Hu H, Zhu MX, Dessauer CW. Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J Biol Chem. 2013;288:3929–3937. doi: 10.1074/jbc.M112.428144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, et al. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain. 2008;4:48. doi: 10.1186/1744-8069-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick J, Buck ME, Matson DJ, Cortright DN. Increased TRPA1, TRPM8, and TRPV2 expression in dorsal root ganglia by nerve injury. Biochem Biophys Res Commun. 2007;358:1058–1064. doi: 10.1016/j.bbrc.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Geria AN, Lawson CN, Halder RM. Topical retinoids for pigmented skin. J Drugs Dermatol. 2011;10:483–489. [PubMed] [Google Scholar]
- Gopinath P, Wan E, Holdcroft A, Facer P, Davis JB, Smith GD, et al. Increased capsaicin receptor TRPV1 in skin nerve fibres and related vanilloid receptors TRPV3 and TRPV4 in keratinocytes in human breast pain. BMC Womens Health. 2005;5:2. doi: 10.1186/1472-6874-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandl J, Kim SE, Uzzell V, Bursulaya B, Petrus M, Bandell M, et al. Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain. Nat Neurosci. 2010;13:708–714. doi: 10.1038/nn.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillery CA, Kerstein PC, Vilceanu D, Barabas ME, Retherford D, Brandow AM, et al. Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood. 2011;118:3376–3383. doi: 10.1182/blood-2010-12-327429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman A, Chuang HH, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P, Litman GI, Brodell RT. Overview of the treatment of acne vulgaris with topical retinoids. Postgrad Med. 2011;123:153–161. doi: 10.3810/pgm.2011.05.2294. [DOI] [PubMed] [Google Scholar]
- Hu H, Bandell M, Petrus MJ, Zhu MX, Patapoutian A. Zinc activates damage-sensing TRPA1 ion channels. Nat Chem Biol. 2009;5:183–190. doi: 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra Y, Blair NT. Benzoquinone reveals a cysteine-dependent desensitization mechanism of TRPA1. Mol Pharmacol. 2013;83:1120–1132. doi: 10.1124/mol.112.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggi AS, Singh N. Therapeutic targets for the management of peripheral nerve injury-induced neuropathic pain. CNS Neurol Disord Drug Targets. 2011;10:589–609. doi: 10.2174/187152711796235041. [DOI] [PubMed] [Google Scholar]
- Jara-Oseguera A, Simon SA, Rosenbaum T. TRPV1: on the road to pain relief. Curr Mol Pharmacol. 2008;1:255–269. doi: 10.2174/1874467210801030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Julius D. Molecular basis for species-specific sensitivity to ‘hot’ chili peppers. Cell. 2002;108:421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Tominaga M, Julius D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci U S A. 2000;97:8134–8139. doi: 10.1073/pnas.100129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hara T, Imai A, Sakakibara A. Differential involvement of TRPV1 receptors at the central and peripheral nerves in CFA-induced mechanical and thermal hyperalgesia. J Pharm Pharmacol. 2007;59:733–738. doi: 10.1211/jpp.59.5.0015. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH. Retinoic acid, immunity, and inflammation. Vitam Horm. 2011;86:83–101. doi: 10.1016/B978-0-12-386960-9.00004-6. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Miyai A, Usui K, Hamada Y, Deai K, Wada M, et al. Pharmacological characterization of (3S)-3-(hydroxymethyl)-4-(5-methylpyridin-2-yl)-N-[6-(2,2,2-trifluoroethoxy)pyrid in-3-yl]-3,4-dihydro-2H-benzo[b][1,4]oxazine-8-carboxamide (JTS-653), a novel transient receptor potential vanilloid 1 antagonist. J Pharmacol Exp Ther. 2012;342:520–528. doi: 10.1124/jpet.112.194027. [DOI] [PubMed] [Google Scholar]
- Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R, Brauchi S, Orta G, Zaelzer C, Vargas G. ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium. 2007;42:427–438. doi: 10.1016/j.ceca.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Le TK, De Mon P, Schalkwijk J, van der Valk PG. Effect of a topical corticosteroid, a retinoid and a vitamin D3 derivative on sodium dodecyl sulphate induced skin irritation. Contact Dermatitis. 1997;37:19–26. doi: 10.1111/j.1600-0536.1997.tb00369.x. [DOI] [PubMed] [Google Scholar]
- Lennertz RC, Kossyreva EA, Smith AK, Stucky CL. TRPA1 mediates mechanical sensitization in nociceptors during inflammation. PLoS ONE. 2012;7:e43597. doi: 10.1371/journal.pone.0043597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ. Topical treatment of acne vulgaris: retinoids and cutaneous irritation. J Am Acad Dermatol. 1998;38:S1–S4. doi: 10.1016/s0190-9622(98)70138-0. [DOI] [PubMed] [Google Scholar]
- Lowe MN, Plosker GL. Bexarotene. Am J Clin Dermatol. 2000;1:245–250. doi: 10.2165/00128071-200001040-00006. discussion 251–242. [DOI] [PubMed] [Google Scholar]
- Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede AC, Tavares SD, Abrantes AM, Trindade J, Maia JM, Botelho MF. The role of vitamins in cancer: a review. Nutr Cancer. 2011;63:479–494. doi: 10.1080/01635581.2011.539315. [DOI] [PubMed] [Google Scholar]
- Mawson AR. Bone pain, growth failure, and skin rash after an upper respiratory illness in a boy with autism: possible association with altered retinoid metabolism. Clin Pediatr (Phila) 2009;48:21–25. doi: 10.1177/0009922808320697. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ. Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov. 2009;8:55–68. doi: 10.1038/nrd2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Alique M, Moreno-Manzano V, Molina C, Lucio FJ, Herrero JF. The oral administration of retinoic acid enhances nociceptive withdrawal reflexes in rats with soft-tissue inflammation. Inflamm Res. 2004;53:297–303. doi: 10.1007/s00011-004-1261-5. [DOI] [PubMed] [Google Scholar]
- Sisignano M, Park CK, Angioni C, Zhang DD, von Hehn C, Cobos EJ, et al. 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J Neurosci. 2012;32:6364–6372. doi: 10.1523/JNEUROSCI.5793-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Thacher SM, Vasudevan J, Chandraratna RA. Therapeutic applications for ligands of retinoid receptors. Curr Pharm Des. 2000;6:25–58. doi: 10.2174/1381612003401415. [DOI] [PubMed] [Google Scholar]
- Thielitz A, Gollnick H. Topical retinoids in acne vulgaris: update on efficacy and safety. Am J Clin Dermatol. 2008;9:369–381. doi: 10.2165/0128071-200809060-00003. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne P, Bertin P, Bonnet C, Scotto C, Treves R. Drug-induced rheumatic disorders: incidence, prevention and management. Drug Saf. 2000;23:279–293. doi: 10.2165/00002018-200023040-00002. [DOI] [PubMed] [Google Scholar]
- White KL, Wiley JS, Frost T, McKendrick JJ, Hermann RP, Seldon M, et al. All-trans retinoic acid in the treatment of acute promyelocytic leukaemia. Aust N Z J Med. 1992;22:449–454. [PubMed] [Google Scholar]
- Yin S, Luo J, Qian A, Du J, Yang Q, Zhou S, et al. Retinoids activate the irritant receptor TRPV1 and produce sensory hypersensitivity. J Clin Invest. 2013;123:3941–3951. doi: 10.1172/JCI66413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Kim YK, Chung JH. High-concentration all-trans retinoic acid induces dermal inflammation and reduces the accumulation of type I procollagen in human skin in vivo. Br J Dermatol. 2011;165:669–672. doi: 10.1111/j.1365-2133.2011.10435.x. [DOI] [PubMed] [Google Scholar]
- Yu L, Yang F, Luo H, Liu FY, Han JS, Xing GG, et al. The role of TRPV1 in different subtypes of dorsal root ganglion neurons in rat chronic inflammatory nociception induced by complete Freund's adjuvant. Mol Pain. 2008;4:61. doi: 10.1186/1744-8069-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]