

Abstract

2-[(Diphenylmethyl)sulfinyl]acetamide (modafinil, (±)-1) is a unique dopamine uptake inhibitor that binds the dopamine transporter (DAT) differently than cocaine and may have potential for the treatment of psychostimulant abuse. To further investigate structural requirements for this divergent binding mode, novel thio- and sulfinylacetamide and ethanamine analogues of (±)-1 were synthesized wherein (1) the diphenyl rings were substituted with methyl, trifluoromethyl, and halogen substituents and (2) substituents were added to the terminal amide/amine nitrogen. Halogen substitution of the diphenyl rings of (±)-1 gave several amide analogues with improved binding affinity for DAT and robust selectivity over the serotonin transporter (SERT), whereas affinity improved at SERT over DAT for the p-halo-substituted amine analogues. Molecular docking studies, using a subset of analogues with DAT and SERT homology models, and functional data obtained with DAT (A480T) and SERT (T497A) mutants defined a role for TM10 in the substrate/inhibitor S1 binding sites of DAT and SERT.
Inhibition of dopamine (DA) reuptake is proposed to be the mechanism underlying the reinforcing effects of abused psychostimulant drugs such as cocaine and methamphetamine. Modafinil (2-[(diphenylmethyl)sulfinyl]acetamide, (±)-1; Figure 1) is used clinically for the treatment of sleep disorders and inhibits DA reuptake, with no evidence of abuse liability in humans.1,2 Recent attention has focused on a distinctive binding mode at the dopamine transporter (DAT) to explain this curious pharmacological profile of (±)-1 and particularly its R-enantiomer (armodafinil, R-(−)-1).1,3 These studies independently demonstrated that (±)-1 binds the DAT in a unique fashion compared to cocaine, which may be related to its distinct behavioral profile. However, there are other reports suggesting additional mechanisms underlying the pharmacological actions of (±)-1 and in particular its effectiveness in attenuating psychostimulant drug seeking in animal models.4−9 Nevertheless, direct interaction with these other targets has not been demonstrated.2 One potential contribution to the preclinical pharmacology of (±)-1 is that it is a nonaminergic compound with limited water solubility, which can complicate investigation due to the large concentration of drug needed for in vitro and in vivo studies. The high doses of (±)-1 used in preclinical studies may indeed have direct or downstream interactions with numerous targets, including histaminergic, GABAergic, orexinergic, glutamatergic, adrenergic, and serotonergic neurons.2,5,8 However, whether or not these targets are related to therapeutic or behavioral outcomes remains unknown.
Figure 1.

(±)-1, its enantiomers, and the DAT-selective amino analogue 2.
In a previous study, we began to explore the structure–activity relationships (SARs) of (±)-1 analogues at the DAT, serotonin transporter (SERT), and norepinephrine transporter (NET) and identified one analogue, compound 2 (Figure 1), in which the terminal amide was replaced with a 3-phenylpropyl-substituted amine group, with enhanced DAT affinity.10 The DAT affinity for 2 was improved by 10-fold, compared to that for (±)-1, as was its water solubility. In addition, 2 demonstrated low micromolar binding affinities for SERT and NET, which prompted a systematic and comparative exploration of the SAR of the (±)-1 scaffold at all three monoamine transporters (MATs) with a series of novel analogues. Specifically, we wanted to further investigate the role of the terminal amide or substituted amine functions on DAT vs SERT and NET binding and also determine how additional diphenyl substitutions on the sulfinylethanamine or reduced thioethanamine template affected the binding affinities and modes. To this end, a series of thioacetamide and sulfinylacetamide analogues were prepared and compared to a set of thioethanamine and sulfinylethanamine analogues of (±)-1.
Chemistry
Scheme 1 outlines the synthesis of novel thioacetamide (compounds 4a–4z) and sulfinylacetamide (compounds 5a–5g) analogues of (±)-1. The thioacetamide analogues 4a–4z were generated via three different synthetic routes.
Scheme 1. Synthesis of Thioacetamide and Sulfinylacetamide Analogues of (±)-1.

Reagents and conditions: (a) 2-mercaptoacetamide or 2-mercapto-N-methylacetamide, TFA, room temperature (60 °C for substituted phenyl analogues), 20 h (procedure A); (b) (i) thioglycolic acid, TFA, room temperature (55–60 °C for substituted phenyl analogues), overnight; (ii) CH3I, K2CO3, acetone, reflux, overnight; (iii) NH4OH, NH4Cl, MeOH, 50 °C, 72 h (procedure B); (c) (i) thioglycolic acid, TFA, room temperature (55–60 °C for substituted phenyl analogues), overnight; (ii) CDI, THF, room temperature, 2 h; (iii) RNH2, THF, 0 °C to room temperature, overnight (procedure C); (d) H2O2 (30%), AcOH–MeOH (1:3), 40 °C, overnight.
The first route (procedure A) affords the thioacetamides in one step and was employed in the synthesis of compound 4a and the N-methylthioacetamides 4g–4i. As opposed to the previously reported two-step synthesis,11 thioacetamide 4a was synthesized in one step by coupling 2-mercaptoacetamide with diphenylmethanol in trifluoroacetic acid (TFA) at room temperature. Similarly, N-methylthioacetamides 4g–4i were synthesized via the coupling reaction between 2-mercapto-N-methylacetamide and diphenylmethanol (for 4g) or the corresponding bis(halophenyl)methanol (for 4h and 4i) in TFA. To improve product yields, the reactions for compounds 4h and 4i required heating to 60 °C.
The second synthetic route (procedure B), used for the synthesis of thioacetamides 4b–4f, required three steps, similar to previously described methods.10 Mono- or disubstituted diphenylmethanol was coupled with thioglycolic acid in TFA, followed by esterification with iodomethane in acetone under reflux conditions. The resulting methyl esters were converted into the primary amides 4b–4f through aminolysis with ammonium hydroxide in methanol.
The third route (procedure C)10 also involved three steps and was employed for the synthesis of N-substituted thioacetamides 4j–4z. First, diphenylmethanol or the appropriate bis(halophenyl)methanol was coupled with thioglycolic acid in TFA. The desired N-substituted thioacetamides 4j–4z were then synthesized by coupling the carboxylic acid to the corresponding primary amine via an in situ N,N′-carbonyldiimidazole coupling reaction. Oxidation of the appropriate thioacetamide (4b–4g and 4x) using hydrogen peroxide (H2O2; 30%) in an acetic acid–methanol solution mixture gave sulfinylacetamides 5a–5g.
Scheme 2 outlines the synthesis of the thioethanamine (6a–6i) and sulfinylethanamine (7a–7e) analogues of (±)-1. Thioethanamines 6a–6c were synthesized by coupling diphenylmethanol or the appropriate bis(halophenyl)methanol with cysteamine hydrochloride in glacial acetic acid in the presence of the Lewis acid catalyst boron trifluoride diethyl etherate (BF3·OEt2) (procedure D).12,13 The N-substituted thioethanamine 6d was synthesized by a reductive amination reaction between the hydrochloride salt of compound 6a and cyclopropanecarboxaldehyde using sodium cyanoborohydride as the reducing agent. Similarly, N-substituted thioethanamine 6e was synthesized by coupling n-butyl bromide to the free base of compound 6a in the presence of CsOH·H2O (Cs+ ions served as templating catalysts).14 N-substituted thioethanamines 6f–6h were synthesized by the reduction of thioacetamides 4u–4w using alane (LiAlH4–sulfuric acid mixture).10 Lastly, N-substituted thioethanamine 6i was prepared from thioacetamide 4x by reduction with borane in THF (BH3·THF). Sulfinylethanamines 7a–7e were synthesized from the appropriate thioethanamines (6a, 6e, or 6g–6i) by oxidation of the thioether function using either sodium periodate (NaIO4) in an ethanol–water solution (compounds 7a and 7b) or H2O2 (30%) in an acetic acid–methanol solution (compounds 7c–7e).
Scheme 2. Synthesis of Thioethanamine and Sulfinylethanamine Analogues of (±)-1.

Reagents and conditions: (a) cysteamine hydrochloride, BF3·OEt2, glacial AcOH, 80–90 °C, ∼20 min (40–50 min for substituted analogues (procedure D)); (b) (i) procedure D; (ii) cyclopropane carboxaldehyde, NaBH3CN, MeOH, 1,2-dichloroethane, room temperature, overnight; (c) (i) procedure D; (ii) BuBr, CsOH·H2O, 4 Å MS, DMF, room temperature, 20 h; (d) LiAlH4, H2SO4, THF; (e) BH3·THF, THF, reflux, overnight; (f) NaIO4, H2O, EtOH, 0 °C to room temperature, overnight; (g) H2O2 (30%), AcOH–MeOH (1:3), 40 °C, 24 h.
Results and Discussion
SARs at DAT, SERT, and NET
In a previous study, we showed that, in general, p-halogen substitution of the diphenylmethyl moiety of the (±)-1 structure gave analogues with improved binding affinities for the DAT over SERT and NET.10 Additionally, we confirmed that enantioselectivity at DAT for the R- and S-enantiomers was only ∼3-fold and that replacement of the sulfoxide (S=O) with a sulfide function may have minimal effects on DAT binding. Importantly, we discovered that reducing the terminal amide and appending a 3-phenylpropyl substituent resulted in compound 2 (Figure 1), which was identified as having higher binding affinities than the amide analogues at all three MATs. This result was particularly encouraging, as salts of amines present a solubility advantage over the parent amide (±)-1. In the current study, we further explored the effect of reducing the S=O, while adding increasingly bulky substituents at the amide nitrogen. Additionally, we expanded the library of amine analogues with and without the S=O motif. Our hypothesis was that SARs within this class of (±)-1 analogues would help unravel SAR differences between the MATs and also identify binding motifs related to the unique binding mode of this class of DAT inhibitors.
Binding affinities of all novel (±)-1 analogues were evaluated at the DAT, SERT, and NET in rat brain membranes using a slightly modified version of previously described methods15 and are detailed in the Experimental Methods. The results of the in vitro assays, grouped by functionality into amides and amines, are presented in Tables 1 and 2, respectively. All sulfinyl compounds were tested as racemic mixtures.
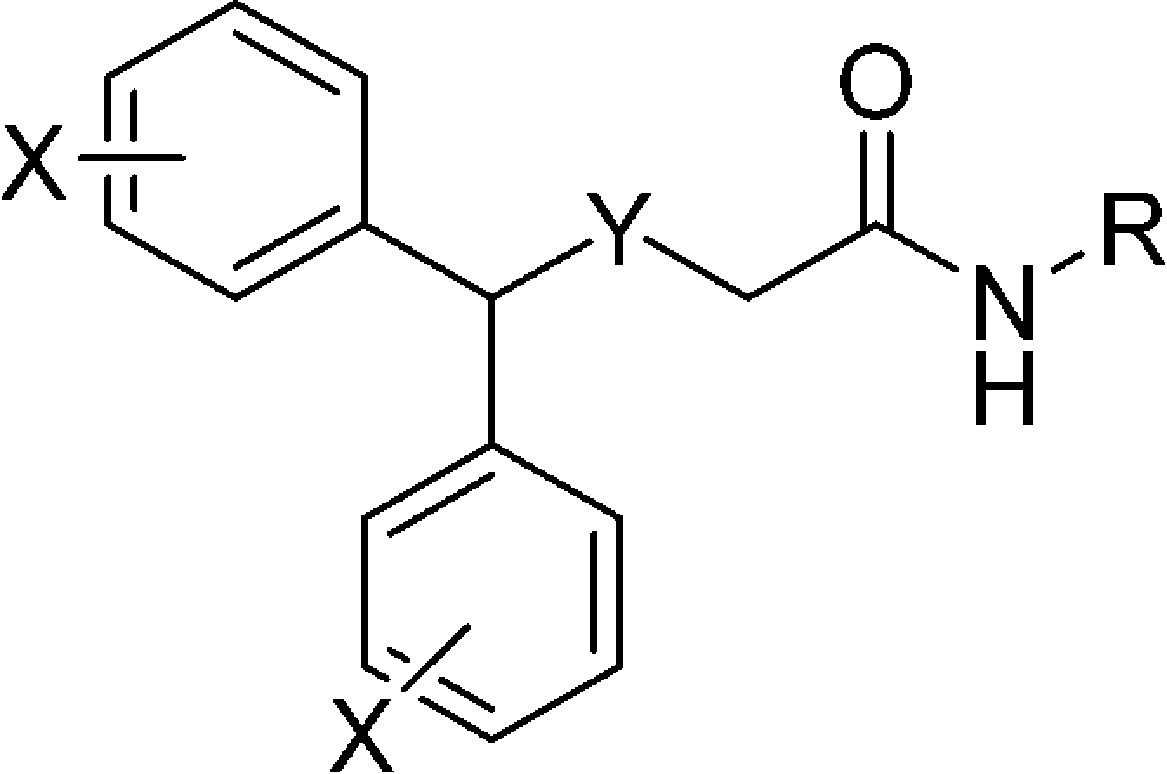
Table 1. MAT Binding Data for Thio- and Sulfinylacetamide Analoguesa.
|
Ki [SE interval] (nM) |
||||||
|---|---|---|---|---|---|---|
| compd | X | Y | R | DAT | SERT | NET |
| (±)-1 | H | S=O | H | 2600 [2430– 2780] | IAb | IAb |
| 4a | H | S | H | 12400 [10800–14300] | 14500 [11800–17700] | IA,b 285000 [117000–690000] |
| 4e | 3,3′-di-Cl | S | H | 275 [257–295] | IA,b 808000 [706000–924000] | 45400 [39600–52000] |
| 4g | H | S | methyl | 19300 [17900–20800] | IA,b 656000 [302000–1420000] | 27200 [25700–28900] |
| 4h | 4,4′-di-Cl | S | methyl | 4130 [3620–4710] | 10700 [7310–15700] | 9770 [9170–10400] |
| 4i | 4,4′-di-Br | S | methyl | 3010 [2770–3260] | 5720 [5320–6150] | 11000 [9540–12600] |
| 4j | H | S | allyl | 8370 [6680–10500] | IA,b 303000 [267000–344000] | IA,b 171000 [88300–332000] |
| 4k | H | S | n-propyl | 20700 [20300–21100] | IA,b 419000 [240000–729000] | 68000 [53200–86900] |
| 4l | 4,4′-di-F | S | n-propyl | 11700 [10300–13200] | 44200 [38700–50500] | 59700 [51200–69600] |
| 4m | 4,4′-di-Cl | S | n-propyl | 1240 [1120–1380] | 10100 [8900–11400] | 7540 [6830–8330] |
| 4n | 4,4′-di-Br | S | n-propyl | 590 [550–632] | 8900 [8150–9720] | 10600 [9980–11300] |
| 4o | H | S | cyclopropylmethyl | 13600 [11900–15600] | 20500 [17500–23900] | IAb |
| 4p | 4,4′-di-F | S | cyclopropylmethyl | 6700 [5730–7830] | 34000 [28800–40200] | 57000 [51500–63000] |
| 4q | 4,4′-di-Br | S | cyclopropylmethyl | 975 [852–1110] | 7030 [6040–8180] | IAb |
| 4r | H | S | n-butyl | 23600 [20500–27100] | IAb | IAb |
| 4s | 4,4′-di-F | S | n-butyl | 6400 [5820–7050] | 25500 [23300–28000] | 56100 [53900–58500] |
| 4t | 4,4′-di-Br | S | n-butyl | 722 [659–792] | 7090 [6990–8180] | 7580 [7210–7970] |
| 4u | H | S | 3-phenylpropyl | 2020 [1990–2050] | IAb | IAb |
| 4v | 4,4′-di-F | S | 3-phenylpropyl | 442 [385–509] | 3500 [2950–4160] | IAb |
| 4w | 4,4′-di-Cl | S | 3-phenylpropyl | 223 [191–260] | IAb | IAb |
| 4x | 4,4′-di-Br | S | 3-phenylpropyl | 238 [202–280] | 60700 [58400–63200] | 35500 [31700–39800] |
| 4y | H | S | 4-phenylbutyl | 1150 [1020–1290] | IAb | 7960 [7590–8350] |
| 4z | 4,4′-di-Br | S | 4-phenylbutyl | 405 [348–471] | IAb | IAb |
| 5a | 4,4′-di-CH3 | S=O | H | 12700 [12400–13100] | IAb | IAb |
| 5b | 4,4′-di-CF3 | S=O | H | 35400 [34100–36700] | NTc | NTc |
| 5c | 3,3′-di-F | S=O | H | 5930 [4990–7060] | IAb | IAb |
| 5d | 3,3′-di-Cl | S=O | H | 881 [763–1020] | IAb | IAb |
| 5e | H, 3-Br | S=O | H | 550 [542–557] | IAb | IAb |
| 5f | H | S=O | methyl | 13100 [12600–13700] | IAb | IAb |
| 5g | 4,4′-di-Br | S=O | 3-phenylpropyl | 1280 [1160–1400] | 892 [787–1010] | IAb |
| 8d | 4,4′-di-Cl | S | H | 2200 [2060–2390] | 38800 [36400–41300] | 51400 [46000–57500] |
Each Ki value represents data from at least three independent experiments, each performed in triplicate. Ki values were analyzed by PRISM. Binding assays are described in detail in the Experimental Methods.
IA = inactive, defined as <50% inhibition at 100 μM; however, in some cases a Ki value could be derived and is included.
NT = not tested.
Previously reported by Cao et al.10
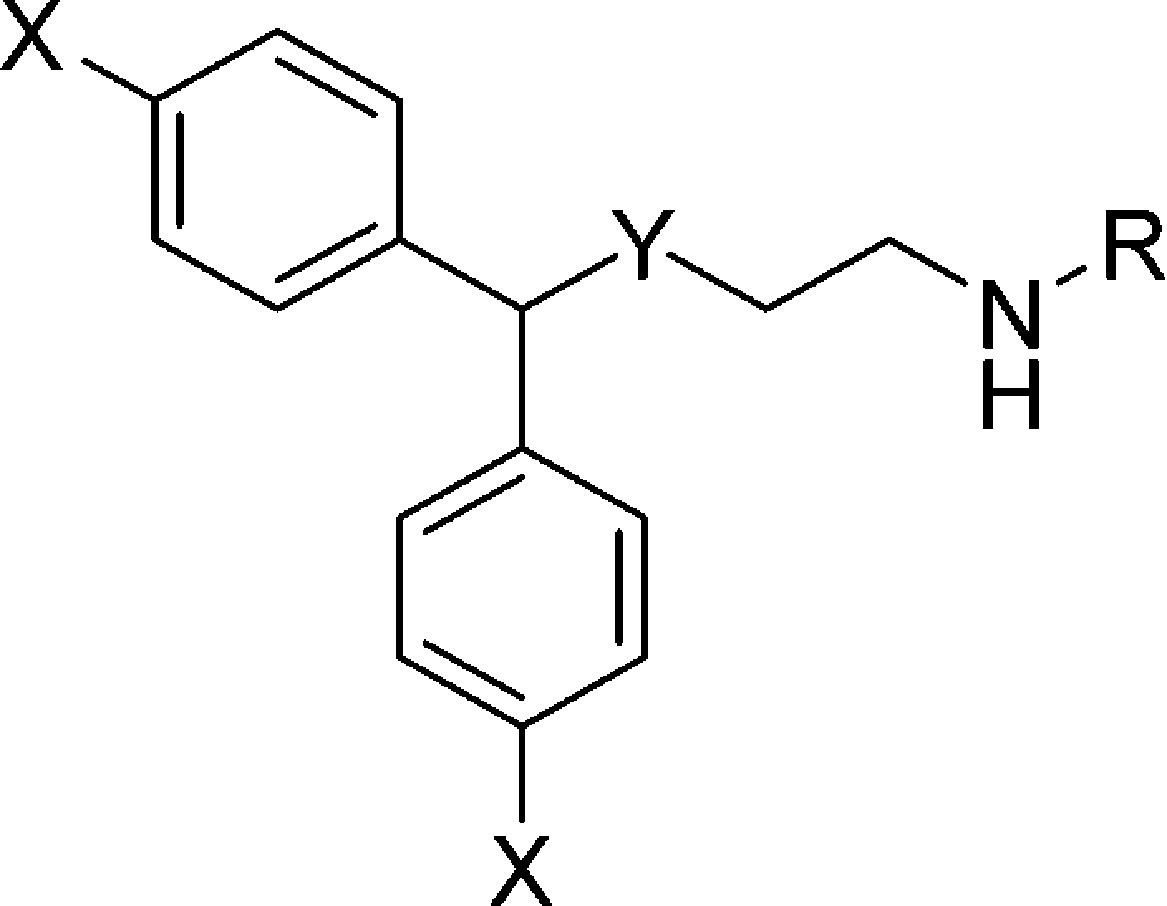
Table 2. MAT Binding Data for Thio- and Sulfinylethanamine Analoguesa.
|
Ki [SE interval] (nM) |
||||||
|---|---|---|---|---|---|---|
| compd | X | Y | R | DAT | SERT | NET |
| 6a | H | S | H | 142 [131–155] | 221 [191–257] | 980 [938–1020] |
| 6b | Cl | S | H | 296 [272–323] | 29.8 [28.2–31.5] | 6920 [6340–7550] |
| 6c | Br | S | H | 483 [434–536] | 26.1 [23.9–28.5] | 8540 [7980–9130] |
| 6d | H | S | cyclopropylmethyl | 435 [406–466] | 10000 [9570–10400] | 17300 [15400–19400] |
| 6e | H | S | n-butyl | 310 [275–350] | 5700 [5040–6440] | 11500 [10700–12300] |
| 6f | H | S | 3-phenylpropyl | 295 [268–325] | 927 [786–1090] | 5500 [5140–5880] |
| 6g | F | S | 3-phenylpropyl | 114 [97.4–132] | 354 [312–402] | 3850 [3830–3870] |
| 6i | Br | S | 3-phenylpropyl | 613 [564–667] | 163 [156–170] | 3160 [2950–3390] |
| 7a | H | S=O | H | 1110 [1020–1200] | 3380 [2970–3820] | 24500 [22800–26200] |
| 7b | H | S=O | n-butyl | 1570 [1490–1660] | 63600 [56100–72200] | 138000 [103000–184000] |
| 7c | F | S=O | 3-phenylpropyl | 183 [140–239] | 1280 [1100–1480] | 3270 [3130–3420] |
| 7d | Cl | S=O | 3-phenylpropyl | 645 [565–736] | 553 [513–595] | 5670 [5000–6420] |
| 7e | Br | S=O | 3-phenylpropyl | 1270 [1130–1420] | 557 [493–628] | 8650 [7450–10000] |
| 2b | H | S=O | 3-phenylpropyl | 192 [177–209] | 987 [870–1120] | 2320 [2060–2620] |
Each Ki value represents data from at least three independent experiments, each performed in triplicate. Ki values were analyzed by PRISM. Binding assays are described in detail in the Experimental Methods.
Previously reported by Cao et al.10
In Table 1, most of the thioacetamide and sulfinylacetamide analogues displayed micromolar affinities at the DAT, within ±10-fold of that of (±)-1 (Ki = 2600 nM). Reducing the S=O to the thioether 4a decreased DAT binding by ∼5-fold, while improving SERT affinity. When the diphenyl rings were unsubstituted, alkyl substitution of the terminal amide nitrogen decreased binding affinity at the DAT with or without the S=O motif (e.g., compounds 4a, 4g, 4j, 4k, 4o, 4r, and 5f). The exception to this trend was observed with compounds 4u and 4y, which displayed similar or nominally improved binding affinities (Ki = 2020 and 1150 in nM, respectively) in comparison to (±)-1. Within each series of N-substituted thioacetamides, binding affinity at the DAT generally increased with halogen substitution at the para-position of the diphenylmethyl moiety in the order H < F < Cl ≤ Br. This order is in agreement with previously reported data10 and applies to both the thioacetamides and sulfinylacetamides with or without substitution on the amide nitrogen. It has been proposed that if a ligand can establish a halogen bond interaction with a receptor in an optimal orientation, the Cl to Br to I substitution may result in an increase of affinity.16 Thus, the order we observed might be consistent with the halogen substituent forming a halogen bond with a polar residue of DAT. Additionally, substitution at other positions of the diphenyl rings followed this halogen substitution order, for example, compounds 5c–5e with halogen substituents in the meta-positions of the diphenyl rings. In general, the novel acetamides were selective for the DAT over the SERT and NET, except for compounds 4a and 5g, both of which displayed roughly equal affinities at the DAT and SERT (DAT:SERT affinity ratios of 1 and 1.4, respectively). Five amide analogues—4e, 4w, 4x, 4z, and 5e—were identified as the most DAT-selective compounds in the series (e.g., SERT:DAT affinity ratios of >2900 for 4e and 249 for 4x, with no displacement at the SERT for 4w, 4z, or 5e). The pronounced selectivity observed with compound 4e for DAT over SERT is remarkable, especially in comparison to its regioisomer, compound 8(10) (Table 1), which is only modestly selective for DAT over SERT (SERT:DAT affinity ratio = 18).
As shown in Table 2, removal of the amide carbonyl (C=O) function resulted in improved affinities at the DAT, SERT, and NET (compare compounds 6a and 7a to (±)-1), with several of the novel amino analogues having nanomolar binding affinities at the DAT in comparison to the micromolar affinity of compound (±)-1. With the thioethanamines, in contrast to the thioacetamides, DAT affinity generally increased with increasingly bulky substitution on the terminal amine nitrogen for analogues with unsubstituted diphenyl rings (see compounds 6d, 6e, and 6f). For analogues with halogen substituents on the diphenyl rings within a particular series, DAT binding affinities generally increased in a reverse order compared to that observed for the acetamides, viz., Br < Cl < F ≤ H with or without the S=O group. Overall, compounds 6g (Ki = 114 nM) and 6a (Ki = 142 nM) displayed the highest affinities at the DAT, with each displaying about 20-fold improved affinity compared to (±)-1. However, in terms of selectivity among the MATs, the most DAT-selective compounds in this series are 6d, 6e, and 7b (SERT:DAT = 23, 18, and 41, respectively; NET:DAT = 40, 37, and 88, respectively). Previously, we identified only one amino analogue of (±)-1 that was selective for the SERT over the DAT.10 In the series reported herein, we identified four additional compounds—6b, 6c, 6i, and 7e—that are SERT-selective, with compounds 6b and 6c displaying nanomolar affinities (Ki = 30 and 26 nM, respectively) at the SERT.
Molecular Docking and Mutagenesis Studies
To interpret SARs revealed by radioligand binding studies in the context of ligand–transporter interactions, we carried out molecular docking studies with both DAT and SERT homology models that are based on the crystal structure of the bacterial homologue, LeuT. These studies led to the identification of a key divergent position in transmembrane helix 10 (TM10), T497 in SERT and A480 in DAT, that appears to contribute to the DAT vs SERT selectivity. Previously A479 and A480 of DAT were found to be involved in the binding of benztropine (3α-(diphenylmethoxy)tropane) and its derivatives, the atypical DAT inhibitors, many of which do not exert cocaine-like subjective effects. In contrast, the mutation of these two residues did not significantly affect the binding of a cocaine analogue, WIN 35,428 (2β-carbomethoxy-3β-(4-fluorophenyl)tropane).17 In addition, it has been reported that the covalent modification on T497C of SERT by the cysteine-reactive MTSET (2-(trimethylammonium)ethyl methanethiosulfonate) disrupted activity.18
It is clear from the SARs described herein that reduction of the amide to a secondary or primary amine significantly improves binding affinities at all three MATs (e.g., 4a vs 6a). This effect appears to be most consistent at DAT, as all but a few analogues in Table 2 have submicromolar affinities. Interestingly, when the diphenyl ring system is substituted with either p-Cl or p-Br groups, the binding affinities at SERT are more improved than at DAT in all cases and most dramatically with compounds 6b and 6c, which bind with Ki values of ≤30 nM at SERT, suggesting a specific interaction at the para-position that may differ between these two transporters. To investigate this further, we carried out molecular docking studies with a group of representative compounds using the homology models of DAT and SERT based on the crystal structure of LeuT1,19 to compare the differences in their binding modes for these targets.
Previously, we proposed that the sulfoxide O interacted with the conserved Y156 in DAT.1 Interestingly, the residue immediately before Y156 is divergent among MATs: whereas in DAT this residue is phenylalanine (F155), the aligned position in SERT/NET is a tyrosine. Molecular docking studies revealed that while both F155 in DAT and Y175 in SERT directly interact with (±)-1, this molecule differentially affects how Y156 of DAT and Y176 of SERT are positioned when bound. Thus, the S=O is optimally positioned to interact with Y156 of DAT but not Y176 of SERT. If the S=O cannot properly interact with the conserved Tyr in SERT, the S=O contributes negatively to the binding affinity, and as a result (±)-1 has higher affinity for DAT than SERT. Conversely, absence of the sulfoxide oxygen should increase the affinity for SERT. Consistent with this prediction, in the presence of the carbonyl oxygen of the amide [(±)-1 vs compound 4a, Table 1], reducing the S=O decreased the binding affinity for DAT but increased the affinity for SERT. Nevertheless, when either of the phenyl rings was substituted with halogens (4e vs 5d) or the terminal amide was substituted (5f vs 4g), this trend was not obvious, underscoring the influence of these additional substituents on the binding mode in both DAT and SERT. Note the binding affinities at SERT are so low for these analogues it is impossible to determine a specific trend.
By reducing the amide carbonyl, the N becomes positively charged, resulting in an increase in affinity for all three MATs as described above [compare (±)-1 to 7a]. An interpretation is that the positive charge facilitates direct interaction between the N and the conserved negatively charged Asp involved in the Na1 binding site for all three transporters. Additionally, the combined effect of a global reduction of both the amide carbonyl and sulfoxide oxygens is even higher affinities at the DAT, SERT, and NET, suggesting that the impact of the charged N is dominant compared to removal of the sulfoxide O, especially for DAT and SERT (compare (±)-1 to 4a vs (±)-1 to 6a].
To test the hypothesis that these residues in TM10 are part of the primary substrate/inhibitor (S1)1,20 binding site and play different roles in DAT vs SERT binding for para-halogenated analogues in this series, we created two chimera mutants in DAT and SERT in which the residues are interchanged, resulting in DAT-A480T and SERT-T497A. The effect of the mutations on uptake inhibition potency for compounds with a Cl substituent in the para-position were measured on intact COS-7 cells transiently expressing WTs or the Ala- and Thr-substituted mutants (Tables 3 and 4). While this paper was being prepared, the crystal structure of Drosophila melanogaster DAT (dDAT) bound with the tricyclic antidepressant nortriptyline became available.21 The core of the dDAT structure “closely resembles that of LeuT”,21 which we used as the template to build the DAT and SERT homology models for this study, and shows the aligned Ala479 of TM10 is indeed in direct contact with the edge of one of the nortriptyline phenyl rings. Therefore, the dDAT structure supports our prediction that this TM10 position faces the S1 binding sites of SERT and DAT. Interestingly, the affinity of (±)-1 is increased in DAT-A480T (∼5-fold) and perhaps slightly in the SERT-T497A mutant, compared to those of their corresponding WTs. In addition, whereas the affinity of the p-Cl-substituted thioacetamide 4h (a secondary amide) is significantly decreased at SERT-T497A compared to SERT-WT (Table 4), suggesting a direct interaction between the p-Cl and the side chain of T497 (Figure 2), the affinity of 4h at DAT-A480T is essentially the same as that at DAT-WT, similar to 4g, which does not possess the p-Cl substituent (Table 3). The observed affinity is also consistent with an alternative explanation that the hydroxyl group of the Thr497 side chain forms an intrahelical H-bond with the protein backbone,22 while the α-methyl group is exposed to the binding site as a hydrophobic contact to accommodate the halogen substitution, especially for amide analogues (e.g., 4h; Table 4).
Table 3. [3H]DA Uptake Inhibition Potency for Selected Analogues Measured in Intact COS7 Cells Expressing the Human DAT Wild Type or the A480T Mutanta.
| compd | hDAT-WT Ki [SE interval] (nM) | n | hDAT-A480T Ki [SE interval] (nM) | n |
|---|---|---|---|---|
| DA (KM) | 1160 [980–1380] | 9 | 1930 [1510–2480] | 5 |
| (±)-1 | 13000 [10000–17000] | 6 | 3090 [2300–4200] | 3 |
| 4g | 5500 [4000–7600] | 4 | 3600 [2010–6300] | 3 |
| 4h | 3700 [2700–5100] | 5 | 2300 [1700–3100] | 3 |
| 6a | 390 [280–540] | 3 | 720 [620–830] | 4 |
| 6b | 1210 [960–1510] | 5 | 1370 [1240–1510] | 3 |
The inhibition potency for [3H]dopamine (DA) uptake was calculated from nonlinear regression analysis of uptake experiments performed on COS7 cells transiently transfected with cDNA of the human dopamine transporter (hDAT) wild type (WT) or the Ala480 to Thr mutant (A480T). The IC50 values used in the calculation of KM and Ki were calculated from the means of pIC50, and the indicated SE intervals were calculated from pIC50 ± SE. Nonspecific uptake was determined using 50 μM nomifensine.
Table 4. [3H]-5-HT Uptake Inhibition Potency for Selected Analogues Measured in Intact COS7 Cells Expressing the Human SERT Wild Type or the T497A Mutanta.
| compd | hSERT-WT Ki [SE interval] (nM) | n | hSERT-T497A Ki [SE interval] (nM) | n |
|---|---|---|---|---|
| 5-HT (KM) | 520 [360–760] | 7 | 1090 [840–1430] | 4 |
| (±)-1 | IAb | 3 | 570000 [497000–653000]c | 3 |
| 4g | IAb | 3 | IAb | 3 |
| 4h | 8300 [6000–11600] | 3 | 27000 [14000–53000] | 2 |
| 6a | 690 [590–810] | 4 | 630 [550–720] | 3 |
| 6b | 270 [230–330] | 3 | 170 [91–320] | 3 |
The inhibition potency for [3H]serotonin (5-HT) uptake was calculated from nonlinear regression analysis of uptake experiments performed on COS7 cells transiently transfected with cDNA of the human serotonin transporter (hSERT) wild type (WT) or the Thr497 to Ala mutant (T497A). The IC50 values used in the calculation of KM and Ki values were calculated from the means of pIC50, and the indicated SE intervals were calculated from pIC50 ± SE. Nonspecific uptake was determined using 5 μM paroxetine.
IA = inactive, defined as <50% inhibition at 100 μM.
According to our definition, (±)-1 would be IA. However, we were able to determine a Ki value, and although the affinity for the T497A mutant was very low, it was, in fact, higher than that at WT SERT, where no Ki could be determined.
Figure 2.
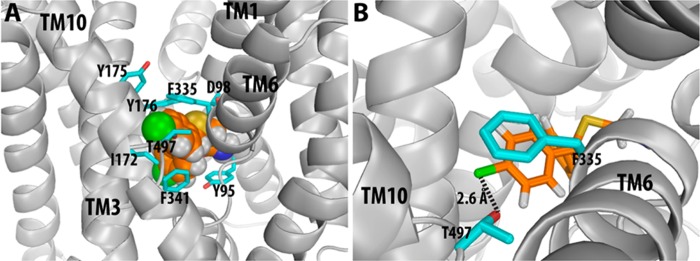
Docking of compound 4h in the S1 binding site of WT SERT. Panel A is an overall view of the binding pose of compound 4h in the binding site. Panel B is a zoom-in view showing the interaction with Thr497 from TM10. The dashed line indicates favorable halogen bonding between 4h and the side chain OH group of T497 in WT SERT, while a similar interaction between 4h and A497, in the mutant, is absent, resulting in a reduction in binding affinity.
These results support our hypothesis that halogen bond interactions at SERT T497 affect the binding affinities of these analogues. We also hypothesized that a change of affinity might result for the A480T DAT mutant; however, we found that the binding affinity was not affected by this mutation. Hence, these data suggest that the binding sites of DAT and SERT obviously have other divergences beyond this single residue position—the ways in which the rest of the binding sites of DAT and SERT change in response to the mutations are different—and simply switching the residues at this position is not enough to interconvert the specificity of the compounds. For example, in both DAT and SERT, the affinities of 6a and 6b (both primary amines) remain unchanged at the mutants, suggesting that the exact configuration near the terminal nitrogen, either amide or charged nitrogen, has a strong impact on the orientation of the diphenylmethyl moiety in both transporters. Taken together, this residue position of TM10 appears to be more important for binding of the amide derivatives of (±)-1 with a p-Cl substituent at the SERT, compared to binding at the DAT. If the amide function is reduced to an amine, the relative importance of interaction at these residues is diminished.
Consistent with this understanding, at the SERT, the difference in binding affinities for halogen-substituted analogues of (±)-1 is more pronounced in compounds lacking a charged N (e.g., >50-fold increase in SERT affinity for amide 4i vs 4g in Table 1 and only a 9-fold increase in SERT affinity for amine 6c vs 6a in Table 2). In both cases, improvements in DAT affinity were diminished compared to those in SERT affinity, with only a 6-fold improvement in DAT affinity between 4i and 4g and only a 3-fold improvement for DAT affinity between 6c and 6a. In contrast, moving the halogen substituent to the meta-position as in compounds 4e and 5d has little if any effect on SERT binding; hence, a decrease in or no change in binding affinity resulted compared to those of compound 4a and (±)-1, respectively. However, the halogen substituent in the meta-position appears to generate new interactions that favor binding to the DAT, further supporting the influence of other residue divergences in the binding sites of DAT and SERT on compound affinity. Thus, we propose that substitution at the meta-position may be more favorable for designing DAT-over-SERT-selective analogues of (±)-1 and may warrant further exploration.
Conclusion
A series of novel thio- and sulfinylacetamide and -ethanamine analogues of (±)-1, with or without substituents on the diphenyl rings, were synthesized to investigate the contributions of structural variations to selectivity across MATs. Previous SARs had suggested that the sulfinyl (S=O) function was not critical for binding to the DAT, but differential interactions with Y156 of DAT and Y176 of SERT may affect selectivity for SERT.1 In addition, we showed that reduction of the amide function to the amine not only improved water solubility, but also enhanced DAT affinity.10 In the current study, the earlier SARs were expanded. para- or meta-substitution of the phenyl rings of (±)-1 with Cl or Br gave several amide analogues with improved selectivity for DAT over SERT and NET, whereas selectivity was improved at SERT over DAT and NET for the amine analogues. Overall, we identified five highly DAT-selective amide analogues (e.g., 4e, 2900-fold over SERT) and two SERT-selective amine analogues with high affinity (Ki ≤ 30 nM). Computational modeling of DAT and SERT led to the identification of key amino acid residues in TM10 that form part of the S1 binding pocket in both DAT and SERT. By switching the T497 in SERT to Ala and the A480 in DAT to Thr and then testing a selected subgroup of analogues, the role of TM10 in DAT and SERT binding was further defined. Moreover, we propose that this TM10 position faces the S1 binding site and plays a role in the binding of this class of compounds to the DAT similar to the atypical DAT inhibitors exemplified by the benztropines, but not to cocaine.17 Interestingly, in the benztropine class of atypical DAT inhibitors similar observations were made in that (1) converting the tropane amine nitrogen to an amide significantly reduced the binding affinity (e.g., N-acetyl-3α-[bis(4′-fluorophenyl)methoxy]tropane, DAT Ki = 2340 nM)23 to an affinity virtually identical to that of (±)-1, and (2) the same order of halogen effect on the amine analogues described herein on decreasing DAT affinity (F > Cl > Br) was also reported for the benztropines, and this is in direct opposition to the order observed in the cocaine-like 3-phenyltropane analogues (e.g., WIN 35,428, 2β-carbomethoxy-3β-(4-fluorophenyl)tropane).24 This divergence in DAT SAR between the benztropines and 3-phenyltropane analogues formed one of the early foundations for our hypothesis that these compounds bind differently to the DAT, and these differences may be related to their different behavioral profiles. Importantly, as it was shown that TM10 plays a critical role in propagating the conformational changes of the homologous LeuT from the S1 binding site to the intracellular gate, such divergent interactions with TM10 are likely to have an impact on the overall transporter conformation25 and may contribute to the mechanism underlying the unique pharmacology of (±)-1 and its analogues at DAT.
Experimental Methods
Synthesis
Reaction conditions and yields were not optimized. Anhydrous solvents were purchased from Aldrich and were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium benzophenone ketyl. All other chemicals and reagents were purchased from Sigma-Aldrich Co. LLC, Combi-Blocks, TCI America, OChem Incorporation, Acros Organics, Maybridge, and Alfa Aesar. The diphenylmethanols (3a–c, 3e–i) were commercially available, except 3d, which was synthesized as outlined below. Unless otherwise stated, amine final products were converted into oxalate salts, typically by treating the free base in 2-propanol with a 1:1 molar ratio of oxalic acid in acetone. As described, some of the oxalate salts were recrystallized from hot methanol or a methanol–acetone solvent mixture. Spectroscopic data and yields refer to the free base, except for compounds 6b and 6c, which were synthesized as the hydrochloride salts. Flash chromatography was performed using silica gel (EMD Chemicals, Inc., 230–400 mesh, 60 Å). Compounds 4u and 6f were purified using a Teledyne ISCO CombiFlash Rf instrument. 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 and 100 MHz, respectively. Chemical shifts are reported in parts per million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26 ppm, or DMSO-d6, 2.50 ppm) and 13C spectra (CDCl3, 77.2 ppm, or DMSO-d6, 39.5 ppm). Gas chromatography/mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N gas chromatograph equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 μm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min, then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA), and the results agree within ±0.5% of the calculated values. Melting point determination was conducted using a Thomas-Hoover melting point apparatus, and the melting points are uncorrected. On the basis of NMR and combustion data, all final compounds are >95% pure.
Bis(4-bromophenyl)methanol (3d)
Compound 3d was synthesized by adapting a literature method26 from bis(4-bromophenyl)methanone (10.2 g, 30.0 mmol) and NaBH4 (2.55 g, 67.4 mmol) in anhydrous ethanol (65 mL) at 0 °C under argon. The product 3d (9.8 g, 95% yield) was recovered as a white solid. Mp: 109–111 °C. 1H NMR (CDCl3): δ 7.46 (d, J = 8.6 Hz, 4H), 7.22 (d, J = 8.6 Hz, 4H), 5.76 (sd, J = 3.5 Hz, 1H), 2.21 (sd, J = 3.5 Hz, 1H). 13C NMR (CDCl3): δ 142.4, 131.9, 128.3, 121.9, 75.2.
Thioacetamides
General Thioacetamide Synthesis Procedures
Procedure A
A solution of 2-mercapto-N-methylacetamide (10 mmol) and diphenylmethanol, 3a, or the appropriate substituted diphenylmethanol, 3c or 3d (10 mmol), in trifluoroacetic acid (TFA; 200 mmol) was stirred at room temperature (60 °C for substituted analogues) for 20 h. The solvent was removed in vacuo, and the thick oily residue was washed with water (30 mL). After the water was decanted, a crude solid product was isolated by addition of diisopropyl ether (20 mL) to the oily residue and vigourous mixing. The crude solid was filtered and purified by flash column chromatography using 5% MeOH/CH2Cl2 to give the pure, desired product.
Procedure B
Thioacetamides 4b–4f were synthesized10 in three steps. Step 1: Thioglycolic acid (1 mmol) was reacted with the appropriate substituted diphenylmethanol, 3e–3i (1 mmol), in TFA (14 mmol) overnight at room temperature. After solvent removal in vacuo, the residue obtained was washed with water (5 mL) and hexanes (15 mL) to give the carboxylic acid product, which was carried to the next step without further purification. Step 2: The acid product (3 mmol) from step 1 was reacted with K2CO3 (4.5 mmol) and iodomethane (CH3I; 4.5 mmol) in acetone (50 mL) overnight under reflux conditions. After solvent removal in vacuo, the residue was suspended in water (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was dried over MgSO4 and concentrated to give the methyl ester, which was carried to the next step without further purification. Step 3: A mixture of the ester (3 mmol), NH4Cl (4.2 mmol), concentrated NH4OH (28.0–30.0%, 20 mL), and MeOH (5.7 mL) was stirred at 50 °C for 72 h. MeOH was removed in vacuo, and the reaction mixture was diluted with water (50 mL), extracted with ethyl acetate (3 × 50 mL), and dried over Na2SO4. The solvent was evaporated, and the recovered crude product was purified by flash column chromatography using 1:1 ethyl acetate/hexanes to afford the pure product.
Procedure C
Thioacetamides 4l, 4p, 4s, 4v, and 4w were synthesized in two steps according to a published procedure,10 while compounds 4j, 4k, 4m–4o, 4q, 4r, 4t, 4u, and 4x–4z were synthesized in two steps with slight modifications to the published procedure in the second step. Step 1 is the same as step 1 for procedure B. Step 2: CDI (11 mmol) was added to a solution of the carboxylic acid product (10 mmol) from step 1 in anhydrous THF (25 mL). The reaction mixture was stirred at room temperature for 2 h and then cooled to 0 °C. Water (0.1–0.2 mL) was added to the reaction mixture (to quench excess CDI), followed by the dropwise addition of the appropriate amine (10 mmol, dissolved in THF). The reaction mixture was left to warm to room temperature and stir overnight. The solvent was removed under vacuum to give a crude residue, which was dissolved in diethyl ether or ethyl acetate. The organic solution was washed with aqueous 1.0 M HCl solution (55 mL), water (80 mL), dilute aqueous NaHCO3 solution (36 mL, 1:6 dilution of saturated NaHCO3 solution), and water (2 × 30 mL). The organic layer was dried over MgSO4 and concentrated in vacuo to give the pure product. The bromo-substituted analogues 4q, 4t, 4x, and 4z required further purification by flash column chromatography as indicated.
2-(Benzhydrylthio)acetamide (4a)
Compound 4a was synthesized by stirring a solution of 2-mercaptoacetamide (0.63 g, 6.9 mmol; recovered from the 10% (w/v) methanol/NH3 solution) and diphenylmethanol, 3a (1.3 g, 7.1 mmol), in TFA (11.9 g, 104 mmol) at room temperature for 4 h. The solvent was removed in vacuo, and the brown oily residue was dissolved in CHCl3 (30 mL) and washed with water (30 mL), followed by a dilute NaHCO3 solution (30 mL, 1:3 dilution of saturated NaHCO3 solution) and water (30 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash chromatography using 1:1 ethyl acetate/hexanes to give pure 4a (0.31 g, 17% yield) as a white solid. Mp: 105–106 °C (lit.3 109–110 °C). 1H NMR (CDCl3): δ 7.41 (d, J = 7.6 Hz, 4H), 7.33 (t, J = 7.4 Hz, 4H), 7.25 (tt, J = 7.2, 1.4 Hz, 2H), 6.50 (br s, 1H), 5.57 (br s, 1H), 5.17 (s, 1H), 3.09 (s, 2H). 13C NMR (CDCl3): δ 171.2, 140.3, 128.9, 128.4, 127.8, 54.9, 35.7. Anal. (C15H15NOS) C, H, N.
2-((Di-p-tolylmethyl)thio)acetamide (4b)
Compound 4b was synthesized according to general procedure B to give 4b (450 mg, 52% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.26 −7.30 (m, 4H), 7.12 (d, J = 7.6 Hz, 4H), 6.54 (br s, 1H), 5.53 (br s, 1H), 5.11 (s, 1H), 3.07 (s, 2H), 2.31 (s, 6H). GC/MS (EI): m/z 285 (M+).
2-((Bis(4-(trifluoromethyl)phenyl)methyl)thio)acetamide (4c)
Compound 4c was synthesized according to general procedure B to give 4c (680 mg, 58% yield) as a white foam. 1H NMR (CDCl3): δ 7.61 (d, J = 8.0 Hz, 4H), 7.53 (d, J = 8.0 Hz, 4H), 6.29 (br s, 1H), 5.72 (br s, 1H), 5.34 (s, 1H), 3.08 (s, 2H). GC/MS (EI): m/z 393 (M+).
2-((Bis(3-fluorophenyl)methyl)thio)acetamide (4d)
Compound 4d was synthesized according to general procedure B to give 4d (810 mg, 61% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.27–7.33 (m, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.12 (dt, J = 10.0, 2.0 Hz, 2H), 6.97 (td, J = 8.0, 2.4 Hz, 2H), 6.43 (br s 1H), 6.09 (br s, 1H), 5.19 (s, 1H), 3.09 (s, 2H). GC/MS (EI): m/z 293 (M+).
2-((Bis(3-chlorophenyl)methyl)thio)acetamide (4e)
Compound 4e was synthesized according to general procedure B to give 4e (800 mg, 65% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.38–7.39 (m, 2H), 7.25–7.28 (m, 6H), 6.42 (br s, 1H), 6.05 (br s, 1H), 5.15 (s, 1H), 3.09 (s, 2H). 13C NMR (CDCl3): δ 170.9, 141.6, 134.8, 130.1, 128.3, 128.1, 126.5, 53.4, 35.4. Anal. (C15H13Cl2NOS) C, H, N.
2-(((3-Bromophenyl)phenylmethyl)thio)acetamide (4f)
Compound 4f was synthesized according to general procedure B to give 4f (750 mg, 58% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.57–7.58 (m, 1H), 7.31–7.39 (m, 6H), 7.25–7.29 (m, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.49 (br s, 1H), 6.32 (br s, 1H), 5.16 (s, 1H), 3.07 (s, 2H). GC/MS (EI): m/z 337 (M+).
2-(Benzhydrylthio)-N-methylacetamide (4g)
Compound 4g was synthesized using 2-mercapto-N-methylacetamide and diphenylmethanol, 3a, according to general procedure A. The product 4g (3.5 g, 59% yield) was obtained as a white solid. Mp: 101–102 °C. 1H NMR (DMSO-d6): δ 7.86 (br s, 1H), 7.42 (d, J = 8.2 Hz, 4H), 7.33 (t, J = 7.6 Hz, 4H), 7.23 (t, J = 7.2 Hz, 2H), 5.40 (s, 1H), 2.96 (s, 2H), 2.54 (sd, J = 4.7 Hz, 3H). 13C NMR (DMSO-d6): δ 169.6, 142.2, 129.5, 128.9, 128.1, 54.0, 35.8, 26.7. Anal. (C16H17NOS) C, H, N.
2-((Bis(4-chlorophenyl)methyl)thio)-N-methylacetamide (4h)
Compound 4h was synthesized using 2-mercapto-N-methylacetamide and bis(4-chlorophenyl)methanol, 3c, at 60 °C according to general procedure A. The product 4h (2.12 g, 79% yield) was obtained as a white solid. Mp: 156–158 °C. 1H NMR (DMSO-d6): δ 7.87 (br s, 1H), 7.38–7.44 (m, 8H), 5.45 (s, 1H), 2.99 (s, 2H), 2.53 (sd, J = 4.7 Hz, 3H). 13C NMR (DMSO-d6): δ 169.3, 140.8, 132.8, 130.8, 129.6, 52.4, 35.8, 26.6. Anal. (C16H15Cl2NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-methylacetamide (4i)
Compound 4i was synthesized from 2-mercapto-N-methylacetamide and bis(4-bromophenyl)methanol, 3d, at 60 °C according to general procedure A. The product 4i (1.86 g, 74% yield) was obtained as a white solid. Mp: 149–151 °C. 1H NMR (DMSO-d6): δ 7.86 (br s, 1H), 7.53 (dt, J = 8.4, 2.2 Hz, 4H), 7.35 (dt, J = 8.4, 2.2 Hz, 4H), 5.42 (s, 1H), 2.99 (s, 2H), 2.53 (sd, J = 4.8 Hz, 3H). 13C NMR (DMSO-d6): δ 168.3, 140.2, 131.5, 130.2, 120.4, 51.6, 34.9, 25.7. Anal. (C16H15Br2NOS) C, H, N.
N-Allyl-2-(benzhydrylthio)acetamide (4j)
Compound 4j was synthesized from 2-(benzhydrylthio)acetic acid and allylamine according to the modified general procedure C. The product 4j (1.97 g, 86% yield) was obtained as a viscous yellow oil that solidified over time. Mp: 45–47 °C. 1H NMR (CDCl3): δ 7.40 (d, J = 7.2 Hz, 4H), 7.32 (t, J = 7.4 Hz, 4H), 7.25 (t, J = 8.0 Hz, 2H), 6.67 (br s, 1H), 5.77–5.86 (m, 1H), 5.19 (d, Jtrans = 17.6 Hz, 1H), 5.15 (d, Jcis = 10.6 Hz, 1H), 5.13 (s, 1H), 3.84 (tt, J = 5.6, 1.6 Hz), 3.14 (s, 2H). 13C NMR (CDCl3): δ 168.0, 140.3, 133.8, 128.8, 128.2, 127.6, 116.8, 55.1, 42.1, 36.1. Anal. (C18H19NOS) C, H, N.
2-(Benzhydrylthio)-N-propylacetamide (4k)
Compound 4k was synthesized from 2-(benzhydrylthio)acetic acid and propylamine according to the modified general procedure C. The product 4k (1.05 g, 91% yield) was obtained as a yellow oil that solidified over time. Mp: 57–58 °C. 1H NMR (CDCl3): δ 7.39 (d, J = 7.6 Hz, 4H), 7.32 (tt, J = 7.2, 1.6 Hz, 4H), 7.24 (t, J = 7.2 Hz, 2H), 6.64 (br s, 1H), 5.11 (s, 1H), 3.18 (q, J = 6.8 Hz, 2H), 3.11 (s, 2H), 1.47–1.56 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 168.2, 140.5, 128.9, 128.4, 127.7, 55.2, 41.6, 36.3, 22.9, 11.5. Anal. (C18H21NOS) C, H, N.
2-((Bis(4-fluorophenyl)methyl)thio)-N-propylacetamide (4l)
Compound 4l was synthesized from 2-((bis(4-fluorophenyl)methyl)thio)acetic acid and propylamine according to general procedure C. The product 4l (320 mg, 95% yield) was obtained as a white solid. Mp: 83–85 °C. 1H NMR (CDCl3): δ 7.32–7.37 (m, 4H), 6.99 −7.05 (m, 4H), 6.55 (br s, 1H), 5.14 (s, 1H), 3.20 (q, J = 6.6 Hz, 2H), 3.07 (s, 2H), 1.48–1.58 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 168.0, 162.1 (1JCF = 247 Hz), 136.0 (4JCF = 3.7 Hz), 129.8 (3JCF = 8.1 Hz), 115.7 (2JCF = 21.4 Hz), 53.2, 41.5, 36.0, 22.8, 11.4. Anal. (C18H19F2NOS) C, H, N.
2-((Bis(4-chlorophenyl)methyl)thio)-N-propylacetamide (4m)
Compound 4m was synthesized from 2-((bis(4-chlorophenyl)methyl)thio)acetic acid and propylamine according to the modified general procedure C. The product 4m (2.06 g, 91% yield) was obtained as a viscous yellow oil that solidified over time. Mp: 57–59 °C. 1H NMR (CDCl3): δ 7.30 (s, 8H), 6.34 (br s, 1H), 5.11 (s, 1H), 3.20 (q, J = 6.8 Hz, 2H), 3.07 (s, 2H), 1.48–1.57 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 168.0, 138.6, 133.8, 129.7, 129.2, 53.6, 41.7, 36.1, 22.9, 11.5. Anal. (C18H19Cl2NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-propylacetamide (4n)
Compound 4n was synthesized from 2-((bis(4-bromophenyl)methyl)thio)acetic acid and propylamine according to the modified general procedure C. The product 4n (1.75 g, 80% yield) was obtained as a light yellow solid. Mp: 92–94 °C. 1H NMR (CDCl3): δ 7.45 (d, J = 8.8 Hz, 4H), 7.24 (d, J = 8.8 Hz, 4H), 6.41 (br s, 1H), 5.08 (s, 1H), 3.19 (q, J = 6.8 Hz, 2H), 3.07 (s, 2H), 1.47–1.56 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 168.0, 139.1, 132.1, 130.0, 121.9, 53.7, 41.7, 36.1, 22.9, 11.5. Anal. (C18H19Br2NOS) C, H, N.
2-(Benzhydrylthio)-N-(cyclopropylmethyl)acetamide (4o)
Compound 4o was synthesized from 2-(benzhydrylthio)acetic acid and cyclopropylmethylamine according to the modified general procedure C. The product 4o (0.25 g, 94% yield) was obtained as a yellow oil. 1H NMR (CDCl3): δ 7.41 (d, J = 7.6 Hz, 4H), 7.33 (tt, J = 7.4, 1.9 Hz, 4H), 7.25 (tt, J = 7.4, 1.7 Hz, 2H), 6.73 (br s, 1H), 5.14 (s, 1H), 3.12 (s, 2H), 3.09 (dd, J = 7.2, 5.6 Hz, 2H), 0.90–1.00 (m, 1H), 0.53 (q, J = 6.4 Hz, 2H), 0.22 (q, J = 5.2 Hz, 2H). 13C NMR (CDCl3): δ 168.1, 140.5, 128.9, 128.4, 127.7, 55.1, 44.7, 36.3, 10.8, 3.6. Anal. (C19H21NOS) C, H, N.
2-((Bis(4-fluorophenyl)methyl)thio)-N-(cyclopropylmethyl)acetamide (4p)
Compound 4p was synthesized from 2-((bis(4-fluorophenyl)methyl)thio)acetic acid and cyclopropylmethylamine according to general procedure C. The product 4p (320 mg, 92% yield) was obtained as a white solid. Mp: 103–105 °C. 1H NMR (CDCl3): δ 7.35 (dd, J = 8.8, 5.2 Hz, 4H), 6.99–7.05 (m, 4H), 6.58 (br s, 1H), 5.16 (s, 1H), 3.11 (dd, J = 7.0, 5.4 Hz, 2H), 3.08 (s, 2H), 0.91–1.01 (m, 1H), 0.52–0.56 (m, 2H), 0.23 (q, J = 5.0 Hz, 2H). 13C NMR (CDCl3): δ 167.9, 162.1 (1JCF = 248 Hz), 136.0 (4JCF = 3.0 Hz), 129.8 (3JCF = 8.1 Hz), 115.7 (2JCF = 21.4 Hz), 53.3, 44.6, 36.0, 10.7, 3.4. Anal. (C19H19F2NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-(cyclopropylmethyl)acetamide (4q)
Compound 4q was synthesized from 2-((bis(4-bromophenyl)methyl)thio)acetic acid and cyclopropylmethylamine according to the modified general procedure C. Purification by flash column chromatography using 1:1 ethyl acetate/hexanes gave the pure product 4q (1.08 g, 94% yield) as a white solid. Mp: 84–85 °C. 1H NMR (CDCl3): δ 7.49 (dt, J = 8.8, 2.2 Hz, 4H), 7.25 (dt, J = 8.4, 2.4 Hz, 4H), 6.56 (br s, 1H), 5.12 (s, 1H), 3.09 (dd, J = 7.2, 5.6 Hz, 2H), 3.07 (s, 2H), 0.88–0.97 (m, 1H), 0.53 (q, J = 6.6 Hz, 2H), 0.21 (q, J = 5.2 Hz, 2H). 13C NMR (CDCl3): δ 167.9, 139.0, 132.0, 130.0, 121.8, 53.5, 44.6, 35.9, 10.7, 3.5. Anal. (C19H19Br2NOS·1/4C4H8O2) C, H, N.
2-(Benzhydrylthio)-N-butylacetamide (4r)
Compound 4r was synthesized from 2-(benzhydrylthio)acetic acid and n-butylamine according to the modified general procedure C. The product 4r (264 mg, 87% yield) was obtained as a yellow oil. 1H NMR (CDCl3): δ 7.39 (d, J = 7.2 Hz, 4H), 7.32 (t, J = 7.4 Hz, 4H), 7.24 (tt, J = 7.2, 1.7 Hz, 2H), 6.64 (br s, 1H), 5.11 (s, 1H), 3.21 (q, J = 6.7 Hz, 2H), 3.10 (s, 2H), 1.43–1.51 (m, 2H), 1.30–1.39 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 168.2, 140.5, 128.9, 128.3, 127.7, 55.1, 39.6, 31.7, 20.2, 13.9. Anal. (C19H23NOS) C, H, N.
2-((Bis(4-fluorophenyl)methyl)thio)-N-butylacetamide (4s)
Compound 4s was synthesized from 2-((bis(4-fluorophenyl)methyl)thio)acetic acid and n-butylamine according to general procedure C. The product 4s (350 mg, 100%) was obtained as a white solid. Mp: 63–64 °C. 1H NMR (CDCl3): δ 7.34 (dd, J = 8.8, 5.2 Hz, 4H), 6.99–7.05 (m, 4H), 6.49 (br s, 1H), 5.13 (s, 1H), 3.24 (q, J = 6.6 Hz, 2H), 3.07 (s, 2H), 1.45–1.52 (m, 2H), 1.31–1.40 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 167.9, 162.1 (1JCF = 247 Hz), 136.0 (4JCF = 3.7 Hz), 129.8 (3JCF = 8.1 Hz), 115.7 (2JCF = 21.4 Hz, 4C), 53.3, 39.5, 36.0, 31.6, 20.1, 13.7. Anal. (C19H21F2NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-butylacetamide (4t)
Compound 4t was synthesized from 2-((bis(4-bromophenyl)methyl)thio)acetic acid and n-butylamine according to the modified general procedure C. Purification by flash column chromatography using 10% MeOH/CHCl3 gave the pure product 4t (0.50 g, 88% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.45 (dt, J = 8.4, 2.0 Hz, 4H), 7.24 (dt, J = 8.8, 2.4 Hz, 4H), 6.40 (br s, 1H), 5.07 (s, 1H), 3.22 (q, J = 6.8 Hz, 2H), 3.07 (s, 2H), 1.43–1.51 (m, 2H), 1.30–1.39 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 167.9, 139.1, 132.1, 130.0, 121.9, 53.7, 39.7, 36.1 31.7, 20.2, 13.9. Anal. (C19H21Br2NOS) C, H, N.
2-(Benzhydrylthio)-N-(3-phenylpropyl)acetamide (4u)
Compound 4u was synthesized as previously described10 from 2-(benzhydrylthio)acetic acid and 3-phenyl-1-propylamine according to the modified general procedure C. Purification on a Teledyne ISCO CombiFlash Rf instrument using 1:1 ethyl acetate/hexanes gave the pure product 4u (1.66 g, 94% yield) as a white solid. Mp: 63–65 °C. 1H NMR (CDCl3): δ 7.37–7.39 (m, 4H), 7.28–7.33 (m, 6H), 7.16–7.27 (m, 5H), 6.61 (br s, 1H), 5.10 (s, 1H), 3.24 (q, J = 6.8 Hz, 2H), 3.09 (s, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.78–1.86 (m, 2H). 13C NMR (CDCl3): δ 168.3, 141.3, 140.5, 128.9, 128.6, 128.5, 128.3, 127.7, 126.2, 55.2, 39.5, 36.3, 33.4, 31.2. Anal. (C24H25NOS) C, H, N.
2-((Bis(4-fluorophenyl)methyl)thio)-N-(3-phenylpropyl)acetamide (4v)
Compound 4v was synthesized from 2-((bis(4-fluorophenyl)methyl)thio)acetic acid and 3-phenyl-1-propylamine according to general procedure C. The product 4v (1.2 g, 100%) was obtained as a yellow oil. 1H NMR (CDCl3): δ 7.26–7.35 (m, 6H), 7.16–7.22 (m, 3H), 6.98–7.04 (m, 4H), 6.48 (br s, 1H), 5.12 (s, 1H), 3.27 (q, J = 6.8 Hz, 2H), 3.04 (s, 2H), 2.66 (t, J = 7.8 Hz, 2H), 1.81–1.88 (m, 2H). 13C NMR (CDCl3): δ 168.0, 162.1 (1JCF = 247 Hz), 141.1, 135.9 (4JCF = 2.9 Hz), 129.8 (3JCF = 8.1 Hz), 128.4 (2JCF = 21.4 Hz), 126.1, 115.8, 115.6, 53.3, 39.4, 36.0, 33.2, 31.1. Anal. (C24H23F2NOS) C, H, N.
2-((Bis(4-chlorophenyl)methyl)thio)-N-(3-phenylpropyl)acetamide (4w)
Compound 4w was synthesized as previously described10 from 2-((bis(4-chlorophenyl)methyl)thio)acetic acid and 3-phenyl-1-propylamine according to general procedure C. The product 4w (1 g, 75%) was obtained as a yellow oil. 1H NMR(CDCl3): δ 7.15–7.31 (m, 13H), 6.38 (br s, 1H), 5.09 (s, 1H), 3.26 (q, J = 6.6 Hz, 2H), 3.04 (s, 2H), 2.65 (t, J = 7.6 Hz, 2H), 1.80–1.87 (m, 2H). 13C NMR (CDCl3): δ 167.9, 141.1, 138.4, 133.6, 129.5, 129.0, 128.5, 128.3, 126.1, 53.4, 39.4, 35.9, 33.2, 31.0. Anal. (C24H23Cl2NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-(3-phenylpropyl)acetamide (4x)
Compound 4x was synthesized as previously described10 from 2-((bis(4-bromophenyl)methyl)thio)acetic acid and 3-phenyl-1-propylamine according to the modified general procedure C. Purification by flash column chromatography using 7:3 ethyl acetate/hexanes and trituration in boiling diisopropyl ether gave the pure product 4x (1.94 g, 76% yield) as a white solid. Mp: 89–91 °C. 1H NMR (DMSO-d6): δ 7.97 (t, J = 5.4 Hz, 1H), 7.52 (dt, J = 8.8, 2.2 Hz, 4H), 7.35 (dt, J = 8.8, 2.3 Hz, 4H), 7.26 (t, J = 7.6 Hz, 2H), 7.17 (d, J = 7.6 Hz, 2H), 7.15–7.18 (m, 1H), 5.42 (s, 1H), 2.99–3.04 (m, 4H), 2.55 (t, J = 7.8 Hz, 2H), 1.62–1.70 (m, 2H). 13C NMR (DMSO-d6): δ 168.9, 142.5, 141.1, 132.5, 131.1, 129.2, 126.7, 121.4, 52.6, 52.5, 39.3, 35.9, 33.4, 31.6. Anal. (C24H23Br2NOS) C, H, N.
2-(Benzhydrylthio)-N-(4-phenylbutyl)acetamide (4y)
Compound 4y was synthesized from 2-(benzhydrylthio)acetic acid and 4-phenyl-1-butylamine according to the modified general procedure C. The product 4y (0.73 g, 96% yield) was obtained as a yellow oil. 1H NMR (CDCl3): δ 7.35–7.38 (m, 4H), 7.28–7.32 (m, 6H), 7.16–7.26 (m, 5H), 6.61 (br s, 1H), 5.08 (s, 1H), 3.22 (q, J = 6.7 Hz, 2H), 3.10 (s, 2H), 2.64 (t, J = 7.4 Hz, 2H), 1.61–1.69 (m, 2H), 1.48–1.55 (m, 2H). 13C NMR (CDCl3): δ 168.2, 142.1, 140.4, 128.9, 128.52, 128.50, 128.3, 127.7, 126.0, 55.2, 39.7, 36.2, 35.6, 29.2, 28.8. Anal. (C25H27NOS) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)-N-(4-phenylbutyl)acetamide (4z)
Compound 4z was synthesized from 2-((bis(4-bromophenyl)methyl)thio)acetic acid and 4-phenyl-1-butylamine according to the modified general procedure C. Purification by flash column chromatography using 1:1 ethyl acetate/hexanes gave the pure product 4z (1.2 g, 90% yield) as a yellow oil. 1H NMR (CDCl3): δ 7.43 (dt, J = 8.0, 2.3 Hz, 4H), 7.28 (t, J = 7.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 4H), 7.15–7.19 (m, 3H), 6.39 (br s, 1H), 5.04 (s, 1H), 3.23 (q, J = 6.7 Hz, 2H), 3.05 (s, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.61–1.69 (m, 2H), 1.48–1.55 (m, 2H). 13C NMR (CDCl3): δ 168.0, 142.0, 139.0, 132.1, 130.0, 128.54, 128.52, 126.1, 121.9, 53.6, 39.8, 36.0, 35.6, 29.2, 28.8. Anal. (C25H25Br2NOS) C, H, N.
Sulfinylacetamides
2-((Di-p-tolylmethyl)sulfinyl)acetamide (5a)
Compound 5a was synthesized following a literature procedure.10 Briefly, H2O2 (0.11 mL, 1.1 mmol, 1 equiv) was added to a solution of compound 4b (310 mg, 1.1 mmol, 1 equiv) in a solvent mixture of acetic acid (1.1 mL) and MeOH (3.3 mL). The reaction mixture was stirred at 40 °C overnight. The solvent was removed in vacuo, and the isolated crude residue was purified by flash column chromatography using a gradient solvent system, viz., 1:1 ethyl acetate/CH2Cl2 to 5% MeOH/CH2Cl2. The pure product 5a (310 mg, 72%) was obtained as a white solid. Mp: 138–139 °C. 1H NMR (CDCl3): δ 7.36 (d, J = 8.2 Hz, 2H), 7.30, (d, J = 8.2 Hz, 2H), 7.20 (sd, J = 3.7 Hz, 4H), 7.12 (br s, 1H), 5.72 (br s, 1H), 5.12 (s, 1H), 3.46 (d, J = 14.8 Hz, 1H), 3.10 (d, J = 14.4 Hz, 1H), 2.34 (s, 6H). 13C NMR (CDCl3): δ 166.4, 138.8, 138.5, 131.3, 131.2, 130.1, 129.6, 129.2, 128.6, 71.2, 51.2, 21.1. Anal. (C17H19NO2S·1/2H2O) C, H, N.
2-((Bis(4-(trifluoromethyl)phenyl)methyl)sulfinyl)acetamide (5b)
Compound 5b was synthesized as described for 5a using compound 4c (680 mg, 1.73 mmol) to give the product 5b (510 mg, 72%) as a white solid. Mp: 75–77 °C. 1H NMR (CDCl3): δ 7.70 (dd, J = 8.0, 6.0 Hz, 4H), 7.60 (dd, J = 8.6, 2.6 Hz, 4H), 6.70 (br s, 1H), 5.71 (br s, 1H), 5.40 (s, 1H), 3.56 (d, J = 14.0 Hz, 1H), 3.12 (d, J = 14.2 Hz, 1H). 13C NMR (CDCl3): δ 165.3, 137.8, 137.0, 131.4 (2JCF = 33.2 Hz), 131.2 (2JCF = 33.2 Hz), 129.9, 129.3, 126.6 (3JCF = 3.7 Hz), 126.0 (3JCF = 3.7 Hz), 123.8 (1JCF = 272 Hz), 123.6 (1JCF = 273 Hz), 69.6, 51.8. Anal. (C17H13F6NO2S·1/2H2O) C, H, N.
2-((Bis(3-fluorophenyl)methyl)sulfinyl)acetamide (5c)
Compound 5c was synthesized as described for 5a using compound 4d (810 mg, 2.76 mmol) to give the product 5c (600 mg, 70%) as a white solid. Mp: 161–162 °C. 1H NMR (CDCl3): δ 7.34–7.42 (m, 2H), 7.14–7.27 (m, 4H), 7.04–7.10 (m, 2H), 6.98 (br s, 1H), 6.18 (br s, 1H), 5.33 (s, 1H), 3.49 (d, J = 13.6 Hz, 1H), 3.23 (d, J = 14.0 Hz, 1H). 13C NMR (CDCl3): δ 166.0, 163.0 (1JCF = 248 Hz), 162.8 (1JCF = 248 Hz), 136.5, 135.8, 131.2 (3JCF = 8.1 Hz), 130.5 (3JCF = 8.1 Hz), 125.3, 124.5 (4JCF = 3.0 Hz), 116.5 (2JCF = 22.8 Hz), 115.9 (2JCF = 22.1 Hz), 69.7, 52.2. Anal. (C15H13F2NO2S) C, H, N.
2-((Bis(3-chlorophenyl)methyl)sulfinyl)acetamide (5d)
Compound 5d was synthesized as described for 5a using compound 4e (800 mg, 2.45 mmol) to give the product 5d (600 mg, 71%) as a white solid. Mp: 115–116 °C. 1H NMR (CDCl3): δ 7.43–7.43 (m, 2H), 7.31–7.37 (m, 6H), 7.05 (br s, 1H), 6.36 (br s, 1H), 5.36 (s, 1H), 3.47 (d, J = 13.6 Hz, 1H), 3.27 (d, J = 13.6 Hz, 1H). 13C NMR (CDCl3): δ 166.3, 136.3, 135.6, 135.4, 134.8, 130.8, 130.2, 129.6, 129.2, 129.0, 128.8, 127.7, 126.9, 69.4, 52.9. Anal. (C15H13Cl2NO2S) C, H, N.
2-(((3-Bromophenyl)phenylmethyl)sulfinyl)acetamide (5e)
Compound 5e was synthesized as described for 5a using compound 4f (750 mg, 2.23 mmol) to give the product 5e (540 mg, 69%) as a white solid. Mp: 149–151 °C. 1H NMR (DMSO-d6): δ 7.65–7.69 (m, 2H), 7.48–7.57 (m, 4H), 7.30–7.43 (m, 5H), 5.36 (s, 1H), 3.39 (d, J = 13.6 Hz, 1H), 3.20 (d, J = 13.6 Hz, 1H). 13C NMR (DMSO-d6): δ 166.2, 137.4, 136.7, 132.2, 130.8, 130.6, 129.7, 129.1, 128.7, 128.6, 128.4, 128.1, 121.6, 67.5, 56.4. Anal. (C15H14BrNO2S) C, H, N.
2-(Benzhydrylsulfinyl)-N-methylacetamide (5f)
Compound 5f was synthesized as described for 5a using compound 4g (500 mg, 1.84 mmol) to give the product 5f (427 mg, 81%) as a yellow oil. 1H NMR (CDCl3): δ 7.35–7.49 (m, 10H), 7.02 (br s, 1H), 5.18 (s, 1H), 3.44 (d, J = 14.0 Hz, 1H), 3.13 (d, J = 14.0 Hz, 1H), 2.82 (sd, J = 4.7 Hz, 3H). 13C NMR (CDCl3): δ 164.9, 134.9, 134.2, 129.7, 129.6, 129.13, 129.08, 129.01, 128.9, 71.7, 52.3, 26.7. Anal. (C16H17NO2S·3/4H2O) C, H, N.
2-((Bis(4-bromophenyl)methyl)sulfinyl)-N-(3-phenylpropyl)acetamide (5g)
Compound 5g was synthesized as described for 5a using compound 4x (220 mg, 0.41 mmol) to give the product 5g (100 mg, 44%) as a colorless oil. 1H NMR (CDCl3): δ 7.51–7.55 (m, 4H), 7.16–7.31 (m, 9H), 6.69 (t, J = 5.4 Hz, 1H), 5.13 (s, 1H), 3.42 (d, J = 14.0 Hz, 1H), 3.33 (q, J = 7.0 Hz, 2H), 3.05 (d, J = 14.0 Hz, 1H), 2.67 (t, J = 7.8 Hz, 2H), 1.85–1.89 (m, 2H). 13C NMR (CDCl3): δ 163.7, 141.3, 133.4, 132.9, 132.5, 132.3, 131.3, 130.7, 128.7, 128.6, 126.3, 123.53, 123.47, 69.7, 52.4, 39.7, 33.4, 31.2. Anal. (C24H23Br2NO2S·1/2H2O) C, H, N.
Thioethanamines
General Thioethanamine Synthesis Procedure
Procedure D
Compounds 6a–6c were synthesized following a literature procedure.12,13 A solution of cysteamine hydrochloride (10 mmol), diphenylmethanol, 3a, or the appropriate halogen-substituted diphenylmethanol, 3c or 3d (10 mmol), and BF3·OEt2 (11 mmol) in glacial acetic acid (40 mL) was stirred at 90–95 °C for 20 min (40–50 min for substituted analogues). The reaction mixture was cooled to room temperature, and diethyl ether (200 mL) was added to precipitate a solid (the hydrochloride salt) from the mixture. The solid was filtered and dried under vacuum for 3 days in the presence of NaOH pellets. The dried solid was dissolved in hot ethanol and filtered and the solvent removed in vacuo. Finally, the solid was triturated in hot (boiling) ethyl acetate to give the pure product as the hydrochloride salt.
2-(Benzhydrylthio)ethan-1-amine (6a)
Compound 6a was synthesized from diphenylmethanol, 3a, according to general procedure D to give the hydrochloride salt in quantitative yield. The hydrochloride salt of 6a (10.1 g, 36.1 mmol) was converted to the free base by being dissolved in saturated aqueous NaHCO3 solution (120 mL) and extracted into CHCl3 (150 mL). The layers were separated, and the organic layer was washed with distilled water (80 mL) and aqueous brine solution (100 mL) and dried over MgSO4. The solvent was evaporated in vacuo to give the free base 6a (7.90 g, 90% yield) as a yellow oil. Some of the isolated free base was converted into the oxalate salt. Mp: 177–179 °C. 1H NMR (CDCl3): δ 7.43 (d, J = 8.0 Hz, 4H), 7.31 (t, J = 7.4 Hz, 4H), 7.22 (tt, J = 7.4, 1.5 Hz, 2H), 5.16 (s, 1H), 2.81 (t, J = 6.2 Hz, 2H), 2.51 (t, J = 6.4 Hz, 2H). 13C NMR (CDCl3): δ 141.5, 128.7, 128.4, 127.4, 54.0, 41.0, 36.7. Anal. (C15H17NS·3/4C2H2O4) C, H, N.
2-((Bis(4-chlorophenyl)methyl)thio)ethan-1-amine (6b)
Compound 6b was synthesized from bis(4-chlorophenyl)methanol, 3c, according to general procedure D with a reaction time of 50 min. The hydrochloride salt product 6b (1.06 g, 62% yield) was obtained as an off-white solid. Mp: 179–181 °C. 1H NMR (HCl salt, DMSO-d6): δ 8.12 (br s, 3H), 7.48 (d, J = 8.4 Hz, 4H), 7.42 (d, J = 8.8 Hz, 4H), 5.56 (s, 1H), 2.94 (t, J = 7.6 Hz, 2H), 2.60 (t, J = 7.2 Hz, 2H). 13C NMR (HCl salt, DMSO-d6): δ 139.9, 132.0, 129.9, 128.7, 50.2, 38.0, 28.6. Anal. (C15H15Cl2NS·3/4HCl·3/4H2O) C, H, N.
2-((Bis(4-bromophenyl)methyl)thio)ethan-1-amine (6c)
Compound 6c was synthesized from bis(4-bromophenyl)methanol, 3d, according to general procedure D with a reaction time of 40 min. The hydrochloride salt product 6c (4.15 g, 72% yield) was obtained as an off-white solid. Mp: 192–194 °C. 1H NMR (HCl salt, DMSO-d6): δ 8.09 (br s, 3H), 7.55 (dt, J = 8.4, 2.3 Hz, 4H), 7.41 (dt, J = 8.8, 2.2 Hz, 4H), 5.53 (s, 1H), 2.94 (t, J = 7.4 Hz, 2H), 2.59 (t, J = 7.4 Hz, 2H). 13C NMR (HCl salt, DMSO-d6): δ 140.2, 131.6, 130.2, 120.5, 50.2, 37.9, 28.5. Anal. (C15H15Br2NS·HCl) C, H, N.
2-(Benzhydrylthio)-N-(cyclopropylmethyl)ethan-1-amine (6d)
Compound 6d was synthesized according to general procedure D starting with compound 6a.27 A suspension of the hydrochloride salt of 6a (1.0 g, 3.6 mmol) and cyclopropanecarboxaldehyde (0.28 g, 4.0 mmol) in 1,2-dichloroethane (62 mL) was stirred at room temperature under an argon atmosphere for 1.3 h. Sodium cyanoborohydride (0.69 g, 11 mmol) dissolved in methanol (2.0 mL) was added to the reaction mixture, and the mixture was stirred at room temperature under an argon atmosphere overnight. After 19 h of reaction time, saturated NaHCO3 solution (30 mL), distilled water (30 mL), and CH2Cl2 (15 mL) were added to the reaction mixture, and the resulting mixure was stirred vigorously for 1 h. The layers were separated, and the aqueous layer was washed with CH2Cl2 (3 × 25 mL). The combined CH2Cl2 extract was washed with water (50 mL), dried over MgSO4, and concentrated in vacuo to give a crude product. The isolated crude was purified by flash column chromatography using an ethyl acetate/hexanes solvent gradient (from 4:1 to 1:4) to give the free base 6d (0.50 g, 47% yield) as a yellow oil. Some of the isolated free base was converted into the hydrochloride salt in CHCl3 using a 1.0 M HCl in ether solution. Mp: 122–124 °C. 1H NMR (CDCl3): δ 7.42 (d, J = 7.4 Hz, 4H), 7.30 (t, J = 7.4 Hz, 4H), 7.22 (tt, J = 7.2, 1.6 Hz, 2H), 5.17 (s, 1H), 2.76 (t, J = 6.4 Hz, 2H), 2.59 (t, J = 6.6 Hz, 2H), 2.40 (d, J = 6.8 Hz, 2H), 0.81–0.97 (m, 1H), 0.44–0.48 (m, 2H), 0.09 (qd, J = 4.8, 1.2 Hz, 2H). 13C NMR (CDCl3): δ 141.6, 128.7, 128.4, 127.3, 54.7, 54.3, 48.1, 32.9, 11.4, 3.5. Anal. (C19H23NS·HCl·1/4H2O) C, H, N.
N-(2-(Benzhydrylthio)ethyl)butan-1-amine (6e)
Compound 6e was synthesized by adapting a literature procedure14 using compound 6a (general procedure D). A mixture of CsOH·H2O (0.29 g, 1.7 mmol) and activated 4 Å molecular sieves (0.52 g) in anhydrous DMF (8.3 mL, freshly distilled and stored over activated 4 Å molecular sieves) was purged of air under vacuum and flushed with argon gas. After the mixture was stirred for 13 min, the free base of compound 6a (0.41 g, 1.7 mmol), dissolved in anhydrous DMF (4.0 mL), was added. The reaction mixture was stirred under vacuum for 25 min and flushed with argon for 5 min, and n-butyl bromide (0.28 g, 2.04 mmol) was added. This was followed by another 10 min of vacuum purging, and the reaction was left to stir overnight at room temperature. The reaction mixture was filtered after 20 h of reaction time, and the undissolved solids were washed with ethyl acetate. The filtrate was evaporated in vacuo to give a liquid residue, which was taken up in aqueous 1 M NaOH (30 mL) and extracted with ethyl acetate (2 × 25 mL). The organic extract was washed with brine (50 mL), dried over a 1:1 Na2SO4/MgSO4 mixture, and concentrated in vacuo. The crude product was purified by flash column chromatography using 5% diethyl ether/hexanes (with 0.5% NEt3) to give the free base 6e (0.22 g, 44% yield) as a yellow oil. Some of the isolated free base was converted into the oxalate salt. Mp: 209–211 °C. 1H NMR (CDCl3): δ 7.42 (d, J = 7.2 Hz, 4H), 7.30 (t, J = 7.6 Hz, 4H), 7.22 (tt, J = 7.2, 1.6 Hz, 2H), 5.17 (s, 1H), 2.74 (t, J = 6.4 Hz, 2H), 2.58 (t, J = 6.2 Hz, 2H), 2.53 (t, J = 7.2 Hz, 2H), 1.40–1.47 (m, 2H), 1.27–1.37 (m, 2H), 0.90 (t, J = 7.6 Hz, 3H). 13C NMR (CDCl3): δ 141.6, 128.7, 128.4, 127.3, 54.2, 49.3, 48.3, 32.8, 32.3, 20.6, 14.1. Anal. (C19H25NS·C2H2O4) C, H, N.
N-(2-(Benzhydrylthio)ethyl)-3-phenylpropan-1-amine (6f)
Compound 6f was synthesized from compound 4u.10 Briefly, sulfuric acid (98%; 305 mg, 3.11 mmol) in THF (8.0 mL) was added dropwise at 0 °C to LiAlH4 (227 mg, 5.99 mmol) in THF (13 mL), and the mixture was stirred for 15 min at room temperature. Compound 4u (563 mg, 1.50 mmol) in THF (11 mL) was added dropwise to the reduction mixture at room temperature and the resulting mixture stirred overnight. The reaction mixture was cooled to 0 °C and quenched with water (5.0 mL) and 10% NaOH (20 mL) successively. The mixture was filtered, the insolubles were washed with THF, and the filtrate was evaporated to dryness. The crude product was purified on a Teledyne ISCO CombiFlash Rf instrument using 97:3:0.03 CHCl3/MeOH/NH4OH to give the pure product 6f (312 mg, 58%) as a yellow oil. The free base was converted to the oxalate salt. Mp: 196–198 °C. 1H NMR (CDCl3): δ 7.42 (d, J = 7.6 Hz, 4H), 7.27–7.32 (m, 6H), 7.16–7.23 (m, 5H), 5.16 (s, 1H), 2.73 (t, J = 6.4 Hz, 2H), 2.63 (t, J = 7.8 Hz, 2H), 2.54–2.58 (m, 4H), 1.74–1.82 (m, 2H). 13C NMR (CDCl3): δ 142.2, 141.6, 128.7, 128.53, 128.47, 128.4, 127.4, 125.9, 54.2, 49.1, 48.3, 33.7, 32.8, 31.8. Anal. (C24H27NS · C2H2O4) C, H, N.
N-(2-((Bis(4-fluorophenyl)methyl)thio)ethyl)-3-phenylpropan-1-amine (6g)
Compound 6g was synthesized as described for compound 6f using compound 4v, except that the reaction mixture was stirred at room temperature for 2 h (instead of overnight) before being quenched with water and NaOH (15% instead of 10%).The crude product was purified by flash column chromatography (95:5:0.5 CHCl3/MeOH/NH4OH) to give the pure product 6g (820 mg, 86.6%) as a yellow oil. The free base was converted to the oxalate salt, which was recrystallized from a methanol/acetone mixture. Mp: 198–200 °C. 1H NMR (CDCl3): δ 7.33–7.37 (m, 4H), 7.16–7.30 (m, 5H), 6.97–7.02 (m, 4H), 5.13 (s, 1H), 2.72 (t, J = 6.4 Hz, 2H), 2.64 (t, J = 7.8 Hz, 2H), 2.52 (m, 4H), 1.75–1.82 (m, 2H). 13C NMR (CDCl3): δ 161.9 (1JCF = 246 Hz), 142.0, 137.0 (4JCF = 3.0 Hz), 129.7 (3JCF = 8.1 Hz), 128.4, 125.8, 115.5 (2JCF = 21.4 Hz, 4C), 52.5, 48.8, 48.0, 33.6, 32.6, 31.6. Anal. (C24H25F2NS·C2H2O4) C, H, N.
N-(2-((Bis(4-chlorophenyl)methyl)thio)ethyl)-3-phenylpropan-1-amine (6h)
Compound 6h was synthesized as described for 6g using compound 4w (1 g, 2.2 mmol). The crude product 6h (850 mg) was obtained as a yellow oil and carried to the next step without further purification.
N-(2-((Bis(4-bromophenyl)methyl)thio)ethyl)-3-phenylpropan-1-amine (6i)
Compound 6i was synthesized by reducing compound 4x with a borane·THF reagent.28 A solution of 1 M BH3·THF complex (14 mL, 14.0 mmol) was added slowly (in two aliquots) to a solution of compound 4x (1.50 g, 2.81 mmol) in freshly distilled THF (15 mL) at 2 °C. The reaction mixture was refluxed for 16 h, cooled to 0 °C, quenched with CH3OH (30 mL), saturated with aqueous HCl (5.0 mL of concentrated HCl (37%)), and refluxed for another 23 h, successively. The solvent was removed in vacuo to give a yellow, oily residue which was taken up in CHCl3 (50 mL) and washed with distilled water (2 × 50 mL). The combined aqueous extract was back-washed with CHCl3 (3 × 30 mL) and then discarded. The combined CHCl3 extract was washed with water (100 mL) and brine (100 mL) and concentrated in vacuo to give the hydrochloride salt of 6i. The salt was suspended in a small amount of water and the suspension made basic to a pH of 13 with 10 M NaOH (20 mL). The basic solution was continuously extracted with CHCl3 for 6 h, and the layers were separated. Solvent was removed from the organic layer to give the crude free base of compound 6i, which was purified by flash column chromatography (5% MeOH/CH2Cl2). The pure product 6i (0.58 g, 40% yield) was obtained as a yellow oil and converted to the oxalate salt. Mp: 187–189 °C. 1H NMR (CDCl3): δ 7.43 (dt, J = 8.4, 2.3 Hz, 4H), 7.23–7.30 (m, 6H), 7.16–7.20 (m, 3H), 5.07 (s, 1H), 2.74 (t, J = 6.6 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.53–2.29 (m, 4H), 1.77–1.83 (m, 2H). 13C NMR (CDCl3): δ 142.1, 140.1, 131.9, 131.6, 130.0, 128.5, 126.0, 121.5, 53.0, 49.0, 48.3, 33.7, 32.7, 31.7. Anal. (C24H25Br2NOS·C2H2O4) C, H, N.
Sulfinylethanamines
2-(Benzhydrylsulfinyl)ethan-1-amine (7a)
Compound 7a was synthesized with slight modifications to a published procedure.29 Briefly, a solution of sodium periodate (NaIO4; 2.25 g, 10.5 mmol) in water (50 mL) was added in a dropwise manner to a solution of the hydrochloride salt of compound 6a (2.80 g, 10.0 mmol) in ethanol (150 mL) at 0 °C. The reaction was allowed to stir and warm to room temperature for ∼20 h under an argon atmosphere. The reaction mixture, which contained a white precipitate, was cooled in an ice bath and filtered. The filtrate was concentrated in vacuo to give a dark yellow, oily residue. The oily residue (the hydrochloride salt) was dissolved in CHCl3, washed with an aqueous NaHCO3 solution (2:3 dilution in water of saturated NaHCO3 solution), distilled water, and aqueous brine, and dried over Na2SO4, successively. After filtration, solvent was removed in vacuo to give the crude, free base of compound 7a. The crude product was purified by flash column chromatography using a MeOH/CHCl3 (with 0.1% NH4OH) gradient (from 0% to 1% MeOH) to give pure 7a (1.12 g, 43% yield) as a yellow oil. Some of the isolated free base was converted to the oxalate salt. Mp: 161–163 °C. 1H NMR (CDCl3): δ 7.50 (d, J = 7.8 Hz, 2H), 7.31–7.44 (m, 8H), 4.90 (s, 1H), 3.10–3.23 (m, 2H), 2.53–2.65 (m, 2H). 13C NMR (CDCl3): δ 135.8, 135.2, 129.4, 128.9, 128.7, 128.5, 128.4, 73.1, 54.4, 36.5. Anal. (C15H17NOS·C2H2O4) C, H, N.
N-(2-(Benzhydrylsulfinyl)ethyl)butan-1-amine (7b)
Compound 7b was synthesized as described for 7a from compound 6e (0.070 g, 0.23 mmol) and NaIO4 (0.053 g, 0.25 mmol) in an ethanol/water (EtOH/H2O) mixture (4.0 mL/1.2 mL, v/v). The pure free base product 7b (0.020 g, 41% yield) was obtained as a yellow oil after purification of the crude by flash column chromatography using a MeOH/CHCl3 (with 0.1% NH4OH) gradient (from 0% to 2% MeOH). The isolated free base was converted to the oxalate salt. Mp: 162–164 °C. 1H NMR (CDCl3): δ 7.49 (d, J = 7.6 Hz, 2H), 7.30–7.44 (m, 8H), 4.92 (s, 1H), 2.99–3.13 (m, 2H), 2.65 (t, J = 5.8 Hz, 2H), 2.56 (t, J = 7.0 Hz, 2H), 1.40–1.47 (m, 2H), 1.27–1.36 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 135.9, 135.2, 129.44, 129.42, 128.9, 128.8, 128.49, 128.44, 73.0, 51.1, 49.6, 43.6, 32.1, 20.5, 14.1. Anal. (C19H25NOS·C2H2O4·1/2H2O) C, H, N.
N-(2-((Bis(4-fluorophenyl)methyl)sulfinyl)ethyl)-3-phenylpropan-1-amine (7c)
Compound 7c was synthesized as described for compound 5a using 6g (900 mg, 2.27 mmol). The free base product 7c (820 mg, 87.5% yield) was obtained as a yellow oil and converted into the oxalate salt, which was recrystallized from a methanol/acetone mixture. Mp: 180–181 °C dec. 1H NMR (CDCl3): δ 7.37–7.44 (m, 4H), 7.05–7.29 (m, 9H), 5.00 (s, 1H), 3.16–3.22 (m, 1H), 3.04–3.11 (m, 1H), 2.60–2.83 (m, 6H), 1.80–1.88 (m, 2H). 13C NMR (CDCl3): δ 162.8 (1JCF = 248 Hz), 162.6 (1JCF = 249 Hz), 141.1, 131.0 (3JCF = 8.1 Hz), 130.3 (3JCF = 8.1 Hz), 130.1 (4JCF = 3.7 Hz), 128.4, 128.3, 126.0, 116.4 (2JCF = 21.4 Hz), 115.8 (2JCF = 21.4 Hz), 70.6, 48.7, 48.4, 43.1, 33.1, 30.1. Anal. (C24H25F2NOS·C2H2O4) C, H, N.
N-(2-((Bis(4-chlorophenyl)methyl)sulfinyl)ethyl)-3-phenylpropan-1-amine (7d)
Compound 7d was synthesized as described for compound 5a using 6h (850 mg, 1.97 mmol). The free base product 7d (640 mg, two steps yield 72.6%) was obtained as a yellow oil and converted into the oxalate salt, which was recrystallized from hot MeOH. Mp: 153–155 °C dec. 1H NMR (CDCl3): δ 7.15–7.39 (m, 13H), 4.88 (s, 1H), 2.99–3.08 (m, 2H), 2.57–2.65 (m, 6H), 1.75–1.82 (m, 2H). 13C NMR (CDCl3): δ 141.9, 134.7, 134.6, 134.0, 132.9, 130.6, 129.9, 129.6, 129.0, 128.4, 125.9, 70.6, 51.2, 49.1, 43.1, 33.5, 31.4. Anal. (C24H25Cl2NOS·C2H2O4·1/2H2O) C, H, N.
N-(2-((Bis(4-bromophenyl)methyl)sulfinyl)ethyl)-3-phenylpropan-1-amine (7e)
Compound 7e was synthesized as described for compound 5a using 6i (230 mg, 0.443 mmol). The free base product 7e (130 mg, 55% yield) was obtained as a yellow oil and converted into the oxalate salt, which was recrystallized from hot MeOH. Mp: 161–162 °C dec. 1H NMR (CDCl3): δ 7.45–7.53 (m, 4H), 7.15–7.32 (m, 7H), 4.83 (s, 1H), 2.99–3.08 (m, 2H), 2.57–2.65 (m, 6H), 1.75–1.82 (m, 2H). 13C NMR (CDCl3): δ 141.9, 134.5, 133.3, 132.5, 132.3, 132.0, 131.4, 130.9, 130.2, 128.4, 125.9, 122.9, 122.7, 70.7, 51.2, 49.1, 43.1, 33.5, 31.4. Anal. (C24H25Br2NOS·C2H2O4) C, H, N.
Radioligand Binding Assays
DAT Binding Assay
Striata were dissected from male Sprague–Dawley rat brains (supplied on ice from Bioreclamation (Hicksville, NY), prepared by homogenizing tissues in 20 volumes (w/v) of ice cold modified sucrose phosphate buffer (0.32 M sucrose, 7.74 mM Na2HPO4, 2.26 mM NaH2PO4, pH adjusted to 7.4) using a Brinkman Polytron (setting 6 for 20 s), and centrifuged at 30000g for 10 min at 4 °C. The resulting pellet was resuspended in buffer, recentrifuged, and suspended in buffer again to a concentration of 10 mg/mL, original wet weight (OWW). Experiments were conducted in assay tubes containing 0.5 mL of sucrose phosphate buffer, 0.5 nM [3H]WIN 35,428 (Kd = 5.53, specific activity 84 Ci/mmol; Perkin-Elmer Life Sciences, Waltham, MA), 1.0 mg of tissue OWW, and various concentrations of inhibitor. The reaction was started with the addition of tissue, and the tubes were incubated for 120 min on ice. Nonspecific binding was determined using 100 μM cocaine hydrochloride.
SERT Binding Assay
Membranes from frozen brain stem dissected from male Sprague–Dawley rat brains (supplied on ice from Bioreclamation) were homogenized in 20 volumes (w/v) of 50 mM Tris buffer (120 mM NaCl and 5 mM KCl, adjusted to pH 7.4) at 25 °C using a Brinkman Polytron (at setting 6 for 20 s). The tissue was centrifuged at 30000g for 10 min at 4 °C. The resulting pellet was suspended in buffer and centrifuged again. The final pellet was resuspended in cold buffer to a concentration of 15 mg/mL OWW. Experiments were conducted in assay tubes containing 0.5 mL of buffer, 1.4 nM [3H]citalopram (Kd = 1.94 nM, specific activity = 83 Ci/mmol; Perkin-Elmer Life Sciences), 1.5 mg of brain stem tissue, and various concentrations of inhibitor. The reaction was started with the addition of the tissue, and the tubes were incubated for 60 min at room temperature. Nonspecific binding was determined using 10 μM fluoxetine.
NET Binding Assay
Membranes from frozen frontal cortex dissected from male Sprague–Dawley rat brains (supplied on ice from Bioreclamation) were homogenized in 20 volumes (w/v) of 50 mM Tris buffer (120 mM NaCl and 5 mM KCl, adjusted to pH 7.4) at 25 °C using a Brinkman Polytron (at setting 6 for 20 s). The tissue was centrifuged at 30000g for 10 min at 4 °C. The resulting pellet was suspended in buffer and centrifuged again. The final pellet was resuspended in cold buffer to a concentration of 80 mg/mL OWW. Experiments were conducted in assay tubes containing 0.5 mL of buffer, 0.5 nM [3H]nisoxetine (Kd = 1.0 nM, specific activity 82 Ci/mmol; Perkin-Elmer Life Sciences), 8 mg of frontal cortex tissue, and various concentrations of inhibitor. The reaction was started with the addition of the tissue, and the tubes were incubated for 180 min at 0–4 °C. Nonspecific binding was determined using 1 μM desipramine.
The solvent used to dissolve the various analogues of modafinil was typically methanol and was present at a final concentration of 5%. Extensive studies previously in this and other laboratories determined that methanol has no effect on binding at the DAT and SERT. However, there is an effect of methanol on binding at the NET, and therefore, the methanol concentration was controlled in all tubes in that assay. When compounds were not soluble in methanol, we used either ethanol or DMSO at final concentrations of no greater than 5% or 6%, respectively. Previous studies found no effect of either of these solvents at these concentrations on binding at any of the sites. For all three MAT binding assays, incubations were terminated by rapid filtration through Whatman GF/B filters, presoaked in 0.3% (SERT) or 0.05% (DAT, NET) polyethylenimine, using a Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed twice with 5 mL of cold buffer and transferred to scintillation vials. Cytoscint (MP Biomedicals, Solon, OH) (3.0 mL) was added, and the vials were counted the next day using a Beckman 6000 liquid scintillation counter (Beckman Coulter Instruments, Fullerton, CA) or a Tri-Carb 2910-B liquid scintillation counter (Perkin-Elmer Life Sciences). The Ki values for the modafinil derivatives were obtained using nonlinear least-squares regression (using GraphPad Prism software, GraphPad Software, Inc., San Diego, CA) of the displacement data, giving IC50 values, from which affinities (Ki values) were calculated using the Cheng–Prusoff equation.30
Molecular Pharmacology
Site-Directed Mutagenesis
Synthetic cDNA encoding the human DAT (synDAT) was subcloned into pcDNA3 (Invitrogen, Carlsbad, CA). cDNA encoding the human SERT (hSERT) was cloned into the pUbi1z expression vector. Mutations herein were generated by the QuickChange method (adapted from Stratagene, La Jolla, CA) and confirmed by restriction enzyme mapping and DNA sequencing. Positive clones were amplified by transformation into XL1 blue competent cells (Stratagene), positive colony picked, and grown in LB media overnight at 37 °C in an orbital incubator (Infors) at 200 rpm. Plasmids were harvested using the maxi prep kit provided by Qiagen according to the manufacturer’s manual.
Cell Culture and Transfection
COS-7 cells were grown in Dulbecco’s modified Eagle’s medium 041 01885 supplemented with 10% fetal calf serum, 2 mM l-glutamine, and 0.01 mg/mL gentamicin at 37 °C in 10% CO2. Wild-type and mutant constructs were transiently transfected into COS-7 cells with Lipo2000 (Invitrogen) according to the manufacturer’s manual using cDNA:Lipo2000 ratios of 3:6 and 2:6 for hDAT and hSERT, respectively.
[3H]Dopamine and [3H]-5-HT Uptake Experiments
Uptake assays were performed essentially as previously described31 using [2,5,6,7,8-3H](dihydroxyphenyl)ethylamine ([3H]DA; 94.4 Ci/mmol, Perkin-Elmer) or 5-[1,2-3H(N)]hydroxytryptamine ([3H]-5-HT; 28 Ci/mmol, Perkin-Elmer) for hDAT- and hSERT-expressing cells, respectively. Transiently transfected COS-7 cells were plated in 12-well (3 × 105 cells/well) or 24-well (105 cells/well) dishes coated with polyornithine to achieve an uptake level of no more than 10% of the total added radioligand. The uptake assays were carried out 2 days after transfection. Prior to the experiment, the cells were washed once in 500 μL of uptake buffer (25 mM HEPES, 130 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM l-ascorbic acid, 5 mM d-glucose, and 1 μM catechol O-methyltransferase inhibitor Ro 41-0960 (Sigma), pH 7.4) at room temperature. All the tested ligands were solubilized in DMSO to obtain a stock concentration of 10 mM. From here the compounds were further diluted 10-fold in H2O, followed by consecutive dilutions in uptake buffer. The trace amounts of DMSO (maximum 1% for the highest added concentration) did not influence the binding affinity (Loland et al., unpublished experiments). The unlabeled ligand (e.g., modafinil [(±)-1] or analogues) was added to the cells in 10 concentrations from 1 nM to 0.1 mM equally distributed around the expected IC50 value, and uptake was initiated by addition of ∼10 nM radioligand in a final volume of 500 μL. After 3 (for the hSERT) or 5 (hDAT) min of incubation, the reaction was stopped by rapid washing with 2 × 500 μL of ice cold uptake buffer, lysed in 250 μL (300 μL for 12-well plates) of 1% SDS, and left for 30 min at 37 °C with gentle shaking. All samples were transferred to 24-well counting plates, and 500 μL (or 600 μL) of Opti-phase Hi Safe 3 scintillation fluid (Perkin-Elmer) was added followed by counting of the plates in a Wallac Tri-Lux β-scintillation counter (Perkin-Elmer). Nonspecific uptake was determined in the presence of 5 μM paroxetine for hSERT-expressing cells and 50 μM nomifensine for hDAT-expressing cells. All determinations were performed in triplicate. Uptake data were analyzed by nonlinear regression analysis using Prism 5.0 from GraphPad Software. The IC50 values used in the estimation of KM for uptake were calculated from the means of pIC50 values and the SE intervals from the pIC50 ± SE. The Ki values were calculated from the IC50 values using the equation Ki = IC50/(1 + (L/KM)) (L = concentration of [3H]DA or [3H]-5-HT).
Molecular Modeling
We docked the modafinil derivative compound 4h in our LeuT-based SERT model. The preparation and MD equilibration of the homology model of SERT were previously described.19 The compound was constructed and prepared for docking using LigPrep (Schrodinger Inc., Portland, OR). Docking of the compound was carried out with Glide (Schrodinger Inc.). The binding modes shown in Figure 2 were chosen on the basis of both the docking scores and the consistency with the (±)-1 pose in the previously modeled DAT–(±)-1 complexes.1
Acknowledgments
Support for this research was provided to A.H.N., O.M.O.-B., J.C., T.K., and J.L.K. by the National Institute on Drug Abuse Intramural Research Program. C.J.L. is supported by the Lundbeck Foundation and the Danish Council for Independent Research Sapere Aude program.
Glossary
Abbreviations Used
- NET
norepinephrine transporter
- MAT
monoamine transporter
- LeuT
leucine transporter
- TM10
transmembrane helix 10
- CDI
N,N′-carbonyldiimidazole
- IA
inactive
- NT
not tested
- S=O
sulfoxide
- C=O
carbonyl
- CDCl3
deuterated chloroform
- DMSO-d6
deuterated dimethyl sulfoxide
Supporting Information Available
Elemental analysis results. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Loland C. J.; Mereu M.; Okunola O. M.; Cao J.; Prisinzano T. E.; Mazier S.; Kopajtic T.; Shi L.; Katz J. L.; Tanda G.; Newman A. H. R-Modafinil (Armodafinil): A Unique Dopamine Uptake Inhibitor and Potential Medication for Psychostimulant Abuse. Biol. Psychiatry 2012, 72, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu M.; Bonci A.; Newman A. H.; Tanda G. The Neurobiology of Modafinil as an Enhancer of Cognitive Performance and a Potential Treatment for Substance Use Disorders. Psychopharmacology 2013, 229, 415–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K. C.; Reith M. E. The Atypical Stimulant and Nootropic Modafinil Interacts with the Dopamine Transporter in a Different Manner Than Classical Cocaine-like Inhibitors. PLoS One 2011, 6, e25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler S. V.; Hensley-Simon M.; Tahsili-Fahadan P.; Lalumiere R. T.; Thomas C.; Fallon R. V.; Kalivas P. W.; Aston-Jones G. Modafinil Attenuates Reinstatement of Cocaine Seeking: Role for Cystine-Glutamate Exchange and Metabotropic Glutamate Receptors. Addict. Biol. 2014, 19, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg M. J.; Carter C. S. Modafinil: A Review of Neurochemical Actions and Effects on Cognition. Neuropsychopharmacology 2008, 33, 1477–1502. [DOI] [PubMed] [Google Scholar]
- Reichel C. M.; See R. E. Modafinil Effects on Reinstatement of Methamphetamine Seeking in a Rat Model of Relapse. Psychopharmacology 2010, 210, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel C. M.; See R. E. Chronic Modafinil Effects on Drug-Seeking Following Methamphetamine Self-Administration in Rats. Int. J. Neuropsychopharmacol. 2012, 15, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoriels L.; Jones P. B.; Sahakian B. J. Modafinil Effects on Cognition and Emotion in Schizophrenia and Its Neurochemical Modulation in the Brain. Neuropharmacology 2013, 64, 168–184. [DOI] [PubMed] [Google Scholar]
- Tahsili-Fahadan P.; Malcolm R.; Aston-Jones G. Modafinil: An Anti-Relapse Medication. Neuropsychopharmacology 2010, 35, 343–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Prisinzano T. E.; Okunola O. M.; Kopajtic T.; Shook M.; Katz J. L.; Newman A. H. Structure–Activity Relationships at the Monoamine Transporters for a Novel Series of Modafinil (2-[(Diphenylmethyl)Sulfinyl]Acetamide) Analogues. ACS Med. Chem. Lett. 2010, 2, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisinzano T.; Podobinski J.; Tidgewell K.; Luo M.; Swenson D. Synthesis and Determination of the Absolute Configuration of the Enantiomers of Modafinil. Tetrahedron: Asymmetry 2004, 15, 1053–1058. [Google Scholar]
- Caldirola P. M.; Vandergoot H.; Timmerman H. New Prenylamine Analogs—Synthesis and Ca-2+-Entry Blocking Activity. Eur. J. Med. Chem. 1992, 27, 571–579. [Google Scholar]
- Hiskey R. G.; Harpold M. A. Chemistry of Aliphatic Disulfides 0.13. Formation of Unsymmetrical Disulfides from S-Benzydryl Thioethers. Tetrahedron 1967, 23, 3923–3929. [Google Scholar]
- Salvatore R. N.; Nagle A. S.; Jung K. W. Cesium Effect: High Chemoselectivity in Direct N-Alkylation of Amines. J. Org. Chem. 2002, 67, 674–683. [DOI] [PubMed] [Google Scholar]
- Zou M. F.; Cao J.; Kopajtic T.; Desai R. I.; Katz J. L.; Newman A. H. Structure–Activity Relationship Studies on a Novel Series of (S)-2β-Substituted 3α-[Bis(4-fluoro- or 4-chlorophenyl)methoxy]tropane Analogues for in Vivo Investigation. J. Med. Chem. 2006, 49, 6391–6399. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- Bisgaard H.; Larsen M. A.; Mazier S.; Beuming T.; Newman A. H.; Weinstein H.; Shi L.; Loland C. J.; Gether U. The Binding Sites for Benztropines and Dopamine in the Dopamine Transporter Overlap. Neuropharmacology 2011, 60, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller P. C. 2nd; Stephan M.; Glomska H.; Rudnick G. Cysteine-Scanning Mutagenesis of the Fifth External Loop of Serotonin Transporter. Biochemistry 2004, 43, 8510–8516. [DOI] [PubMed] [Google Scholar]
- Plenge P.; Shi L.; Beuming T.; Te J.; Newman A. H.; Weinstein H.; Gether U.; Loland C. J. Steric Hindrance Mutagenesis in the Conserved Extracellular Vestibule Impedes Allosteric Binding of Antidepressants to the Serotonin Transporter. J. Biol. Chem. 2012, 287, 39316–39326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen A. S.; Andersen J.; Jorgensen T. N.; Sorensen L.; Eriksen J.; Loland C. J.; Stromgaard K.; Gether U. Slc6 Neurotransmitter Transporters: Structure, Function, and Regulation. Pharmacol. Rev. 2011, 63, 585–640. [DOI] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X.; Olivella M.; Govaerts C.; Ballesteros J. A.; Campillo M.; Pardo L. Ser and Thr Residues Modulate the Conformation of Pro-Kinked Transmembrane α-Helices. Biophys. J. 2004, 86, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoston G. E.; Wu J. H.; Izenwasser S.; George C.; Katz J.; Kline R. H.; Newman A. H. Novel N-Substituted 3α-[Bis(4′-fluorophenyl)methoxy]tropane Analogues: Selective Ligands for the Dopamine Transporter. J. Med. Chem. 1997, 40, 4329–4339. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Kline R. H.; Allen A. C.; Izenwasser S.; George C.; Katz J. L. Novel 4′-Substituted and 4′,4″-Disubstituted 3α-(Diphenylmethoxy)tropane Analogs as Potent and Selective Dopamine Uptake Inhibitors. J. Med. Chem. 1995, 38, 3933–3940. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Terry D. S.; Shi L.; Quick M.; Weinstein H.; Blanchard S. C.; Javitch J. A. Substrate-Modulated Gating Dynamics in a Na+-Coupled Neurotransmitter Transporter Homologue. Nature 2011, 474, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharul R. K.; Goswami A.; Gite A.; Godha A. K.; Jain M.; Patel P. R. Convenient Synthesis of Structurally Novel 1,3-Disubstituted Azetidine Derivatives. Synth. Commun. 2008, 38, 1703–1717. [Google Scholar]
- Ghosh B.; Antonio T.; Zhen J.; Kharkar P.; Reith M. E.; Dutta A. K. Development of (S)-N6-(2-(4-(Isoquinolin-1-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]-thiazole-2,6-diamine and Its Analogue as a D3 Receptor Preferring Agonist: Potent in Vivo Activity in Parkinson’s Disease Animal Models. J. Med. Chem. 2010, 53, 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn O.; Meares C. F. Large-Scale Synthesis of the Bifunctional Chelating Agent 2-(p-Nitrobenzyl)-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-Tetraacetic Acid, and the Determination of Its Enantiomeric Purity by Chiral Chromatography. Bioconjugate Chem. 1992, 3, 563–569. [DOI] [PubMed] [Google Scholar]
- Hiskey R. G.; Harpold M. A. Chemistry of Aliphatic Disulfides. XIV. Preparation of Disulfide Sulfoxides by Selective Oxidation. J. Org. Chem. 1967, 32, 3191–3194. [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Cha J. H.; Zou M. F.; Adkins E. M.; Rasmussen S. G.; Loland C. J.; Schoenenberger B.; Gether U.; Newman A. H. Rhodamine-Labeled 2β-Carbomethoxy-3β-(3,4-dichlorophenyl)tropane Analogues as High-Affinity Fluorescent Probes for the Dopamine Transporter. J. Med. Chem. 2005, 48, 7513–7516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.