

Abstract
Background
Interleukin-32 (IL-32) is a recently discovered proinflammatory cytokine involved in inflammatory diseases. We investigated the expression of IL-32 and its regulation mechanism in the inflammatory response of patients with Helicobacter pylori (H. pylori) infection.
Design and Methods
IL-32 mRNA and protein expression in gastric tissues was detected by quantitative real-time PCR and immunohistochemistry. The regulation of IL-32 in human gastric epithelia cell line AGS was investigated by different cytokine stimulation and different H. pylori strain infection.
Results
Gastric IL-32 mRNA and protein expression were elevated in patients with H. pylori infection and positively correlated with gastritis. In H. pylori-infected patients, the mRNA level of IL-32 was also correlated with that of proinflammatory cytokines IL-1β and TNF-α. In vitro IL-1β and TNF-α could upregulate IL-32 mRNA and protein level in AGS cells, which was dependent on NF-κB signal pathway. The regulation of IL-32 expression in response to H. pylori-infection could be weakened by using neutralizing antibodies to block IL-1β and TNF-α. Moreover, H. pylori-infected AGS cells also induced IL-32 mRNA and protein expression, which was dependent on CagA.
Conclusions
IL-32 level is elevated in patients with H. pylori infection and its expression is regulated by proinflammatory stimuli, suggesting that IL-32 may play a role in the pathogenesis of H. pylori-related gastritis.
Introduction
Helicobacter pylori (H. pylori) is a Gram-negative, microaerophilic bacterium that colonizes the stomach of about 50% of the population worldwide. The persistent infection of H. pylori causes chronic and persistent gastritis and increases the risk of peptic ulcer and gastric cancer.
H. pylori-infected gastric mucosa has been characterized by the infiltration of immune cells and the production of inflammatory factors. Th1 and Th2 response are reported to mediate immune response to H. pylori, and Th1 response is thought to be predominant [1]. Then induction of Th17 response in H. pylori-infected stomach is confirmed [2]. Besides Th1, Th2 and Th17 cells, regulatory T cells (Tregs) also play an important role in H. pylori-related gastritis [3]. Meanwhile, proinflammtory cytokines such as IL-1β, TNF-α and IL-6 have also been shown to be elevated in H. pylori-infected stomach [4], and these cytokines could influence T cell immune response in inflammatory disorders, suggesting that inflammatory factors may be crucial in H. pylori-related gastric inflammation. However, the exact mechanism of the process has not been fully elucidated.
IL-32 is a newly identified proinflammatory cytokine produced by immune cells (NK cells, T cells, monocytes) and non-immune cells (endothelial cells, epithelial cells) [5]–[7]. It was originally cloned as a gene induced by IL-2 and called NK-4, but its function was unknown until 2005 [5], [8]. There are six splice variants including IL-32α, β, γ, δ, ε, and ζ, and diverse roles are potentially played by its different isoforms. However, a specific receptor for IL-32 has not been discovered, although neutrophil proteinase 3 binds to IL-32 with a high affinity [9]. IL-32 plays an important role in various inflammatory disorders and its expression has been shown to correlate with the severity of diseases in rheumatoid arthritis, Crohn’s disease and atopic dermatitis [10]–[12]. Moreover, IL-32 had been implied in some infectious diseases including H. pylori infection [13]–[16]. However, the association between IL-32 expression and H. pylori-induced gastritis and its exact regulation mechanism including whether auto−/paracrine effects on the expression of IL-32 may be involved in the process was remain unknown.
In the present study, we detected IL-32 expression in biopsy specimens from patients with H. pylori infection and analyzed the relationship between gastric IL-32 level and the severity of mucosal inflammation. Subsequently, we explored the impact of proinflammatory stimuli and H. pylori infection on IL-32 expression in human gastric epithelia cell lines. Our results showed that IL-32 might be involved in the pathogenesis of H. pylori-related gastritis.
Materials and Methods
Ethics Statement
Human gastric mucosal biopsies were collected by routine endoscopy at the Xinqiao Hospital of the Third Military Medical University. Blood was obtained from the same subject who underwent endoscopy for H. pylori serology test. The study was approved by the Ethics Committee of Xinqiao Hospital, Third Military Medical University. Written informed consent was obtained from each subject.
Subjects
Gastric tissues and blood were collected from 54 patients (male/female = 27/27; average age 47±1.2 years) with H. pylori infection who underwent endoscopy at the Xinqiao Hospital of the Third Military Medical University. H. pylori infection was confirmed by rapid-Urease test, serology test, 13C-urea breath test and histology. Patients were classified as H. pylori positive if two of the four tests were positive. Normal gastric tissues from 47 subjects (male/female = 23/24; average age 47±1.3 years) who had negative results for all four tests were enrolled as controls.
Biopsy Specimens and Histology Assessment
Biopsy specimens were taken from the subjects at each endoscopy. One was immediately frozen in liquid nitrogen and stored at −80°C for RNA extraction. The rest of biopsy specimens were fixed in formalin and embedded in paraffin. Haematoxylin-Eosin (H&E) stained sections were examined by two experienced histopathologist. The histological severity of gastritis was graded from normal to severe based on the density of infiltrating mononuclear and polymorphnuclear cells according to the established criteria [17], [18].
Cell Culture
The gastric epithelial line AGS (ATCC, American Type Culture Collection) was cultured at 37°C and 5% CO2 in Ham’s F12 (Hyclone, Logan, UT, USA), which contained 10% fetal calf serum (FCS). AGS Cells were seeded in six-well plates at a density of 1×106 cells/well and stimulated with 10 ng/ml TNF-α or/and 10 ng/ml IL-1β (PeproTech, Rocky Hill, NJ, USA); cells were collected at indicated times for analysis of IL-32 mRNA and protein expression. For the signal pathway inhibition assay, NF-κB inhibitor (BAY 11-7082), MEK1/2 inhibitor (U0126), p38/MAPK inhibitor (SB203580), JNK inhibitor (SP600125), JAK inhibitor I (all at 10 µM and all from Calbiochem, San Diego, CA, USA) or the vehicle DMSO (Sigma, Saint Louis, MO, USA) were added to the cell culture 1 hour before cytokine stimulation.
Infection of AGS Cells with H. pylori
H. pylori 11637 strain and its isogenic CagA-negative mutant strain (CagA- strain) were grown on brain-heart infusion plates containing 10% rabbit blood at 37°C under microaerophilic conditions (5% O2, 10% CO2, 85% N2). H. pylori was washed off the culture plates with PBS and centrifuged at 2500×g for 5 min, before being resuspended in PBS for optical density quantification at 600 nm (1 OD600 = 1×109 H. pylori/ml). A multiplicity of infection (MOI) of 1, 10, and 100 was used for infecting AGS cells. For neutralization assays, neutralizing antibodies against TNF-α (nTNF-α, 1 µg/ml, Biolegend) or IL-1β (nIL-1β, 1 µg/ml, eBioscience) was added in the coculture system.
RNA Isolation and Quantitative Real-time PCR
RNA was extracted from biopsy specimens or cells by TRIzol reagent® (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed to cDNA using ReverTra Ace (TOYOBO, Osaka, Japan). Quantitative real-time PCR was carried out by the iQ5 Detection System (Bio-Rad, USA). The PCR primers were designed to cross an exon-intron border and used to detect the mRNA expression of IL-32, TNF-α, IL-1β and β-actin and their sequences are as follows: IL-32, forward, 5′-ACGACTTCAAAGAGGGCTACC-3′; reverse, 5′-GCCTCGGCACCGTAATCCAT-3′; TNF-α, forward, 5′-TCTCTAATCAGCCCTCTGGC-3′; reverse, 5′-ATGAGGTACAGGCCCTCTGA-3′; IL-1β, forward, 5′-GTTCTTTGAAGCTGATGGCC-3′; reverse, 5′-GTGGTCGGAGATTCGTAGCT-3′; β-actin, forward, 5′-TTCCTTCCTGGGCATGGAGTCC-3′; reverse, 5′-TGGCGTACAGGTCTTTGCGG-3′. β-actin was used as an internal control. The relative gene expression was calculated as fold change by the ΔΔCt method.
Immunohistochemistry
Forty paraffin-embedded samples were cut into 5-µm sections. After being deparaffinized and hydrated, the sections in citrate buffer (pH = 6.0) were subjected to heat-induced antigen retrieval in a microwave oven and treated with 3% hydrogen peroxide. Following incubation with rabbit anti-human IL-32 (Abcam, MA, USA) overnight at 4°C, Slides were treated with horseradish peroxidase-conjugated secondary anti-rabbit antibody (Zhongshan Golden Bridge Biotech., Beijing, China) followed by substrate 3,3′-diaminobenzidine tetrahydrochloride (DAB). Isotype matched antibody was used as negative control. Images were acquired on a microscope equipped with a digital camera Nikon Eclipse 80i (Tokyo, Japan). For semi-quantitative analysis of the immunohistochemistry, each section was chosen for evaluating IL-32 immunostaining in gastric tissues and was graded as follows: score 0, no expression; score 1, low expression; score 2, intermediate expression; score 3, high expression.
Western Blot
Cells were washed in ice-cold PBS and then disrupted in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin and protease inhibitor). The protein concentration was measured with a BCA protein assay kit (boster, Wuhan, China). Cell lysates were separated by 12% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membranes were blocked for 1 h with 3% bovine serum albumin in Tris-buffered saline–Tween at room temperature and then incubated overnight at 4°C with rabbit anti-human IL-32 (Abcam, MA, USA) or mouse anti-human β-actin ((Tianjin Sungene Biotech Co., Ltd, China). Horseradish peroxidase-conjugated secondary antibody was used according to the manufacturer’s instructures. The proteins of interest were visualized by using Supersignal® West Dura Duration substrate reagent (Thermo, IL, USA).
Statistical Analysis
All results were summarized as mean ± standard error of the mean (SEM), and statistical analysis was performed using the GraphPad Prism 5.0 Software. The differences between two groups were analyzed by the Mann-Whitney U test and multiple groups were analyzed by a one-way analysis of variance (ANOVA). When variances were detected, Spearman’s correlation was used to evaluate the degree of association between variables. P<0.05 was considered statistically significant.
Results
Elevated IL-32 mRNA Level Detected in Patients with H. pylori Infection
To study whether IL-32 is involved in the pathogenesis of H. pylori induced gastritis, we first determined the IL-32 mRNA expression in gastric biopsy specimens from subjects with and without H. pylori infection. As shown in Fig. 1A, IL-32 expression was significantly higher in H. pylori-positive samples than that in H. pylori-negative samples (P<0.001). To evaluate whether the expression of IL-32 was related to the degree of inflammation in H. pylori-infected gastric samples, we divided the samples into normal and mild, moderate and severe gastritis groups based on histology assessment as described in Materials and Methods. The results showed that IL-32 mRNA levels in H. pylori-positive samples were positively correlated with the degree of gastric inflammation (Fig. 1B). Moreover, the level of IL-32 mRNA also showed a positive correlation with TNF-α and IL-1β mRNA levels (Fig. 1C).
Figure 1. Gastric IL-32 mRNA in patients with H. pylori infection correlated with inflammation and inflammatory cytokine gene expression.

A. IL-32 mRNA expression was detected by real-time PCR in gastric mucosa tissues from 54 H. pylori-positive patients and 47 H. pylori-negative healthy individuals; B. Analysis of the relationship between IL-32 mRNA levels and the degree of gastric inflammation in H. pylori-positive patients; C. IL-32 mRNA showed a positively correlation with those of TNF-α and IL-1β. *P<0.05; ***P<0.001.
Increased IL-32 Protein Expression in Patients with H. pylori Infection
To visualize IL-32 expression in gastric biopsy specimens, immunohistochemical staining of IL-32 was performed on paraffin-embedded tissues. As shown in Fig. 2B, a few of IL-32-producing cells were detected in H. pylori–negative gastric tissues, whereas in H. pylori–positive gastric tissues, we observed that IL-32 protein expression was significantly increased (Fig. 2C). In addition, the semi-quantitative evaluation of IL-32 immunoreactivity confirmed that the expression of IL-32 in gastric tissues of H. pylori-infected patients at protein level was also significantly increased compared with those in H. pylori–negative gastric tissues and in mild gastritis.
Figure 2. IL-32 protein was detected in the gastric biopsy tissues.
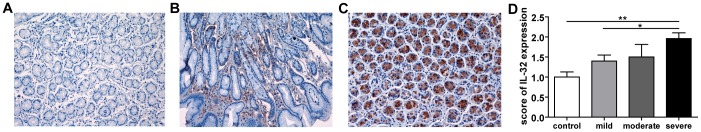
A. control isotype IgG (200×); B. A representative microphotograph showing biopsy sections from H. pylori-negative normal controls stained for IL-32 (200×); C. A representative microphotograph showing IL-32 immunostaining in biopsy sections from H. pylori-positive patients (200×) ; D. The immunohistochemistry of IL-32 in the gastric biopsy tissues was semi-quantitatively graded as scores (0 = no expression; 1 = low expression; 2 = intermediate expression; 3 = high expression). *P<0.05; **P<0.01.
IL-32 mRNA and Protein Level were Upregulated by TNF-α and IL-1β
Because we found a positive relationship between IL-32 mRNA levels and the degree of gastric inflammation in gastric tissues and correlation between IL-32 mRNA and IL-1β and TNF-α mRNAs, the regulation of proinflammatory cytokines on IL-32 mRNA expression was investigated in AGS cells. As shown in Fig. 3A, after being treated with cytokines for 24 hours, IL-32 mRNA level was significantly upregulated: 5.0±0.5-fold by TNF-α, 6.9±0.5-fold by IL-1β, and 11.2±1.4-fold by IL-1β and TNF-α. This effect was completely dependent on NF-κB signaling pathway as pretreatment with NF-κB inhibitor BAY 11-7082, but not JNK, p38/MAPK, MEK1/2 or JAK/STAT signaling inhibitors inhibited the induction of IL-32 mRNA after stimulation by TNF-α or IL-1β (Fig. 3C). Furthermore, Western blot confirmed that IL-32 protein level was also induced by TNF-α and/or IL-1β stimulation which depended on NF-κB signaling pathway (Fig. 3B and D). We next studied whether TNF-α or IL-1β was involved in H. pylori-induced upregulation of IL-32 expression. AGS cells were infected with H. pylori and simultaneously treated with neutralizing antibodies to block TNF-α or IL-1β. As shown in Fig. 3E, H . pylori-induced IL-32 protein expression was significantly decreased by using indicated cytokines-blocking antibodies.
Figure 3. Induction and regulation of IL-32 mRNA in AGS cells.

A. IL-32 mRNA expression was analyzed in AGS cells after stimulation with TNF-α and/or IL-1β for 24 hours. *P<0.05; B. IL-32 protein expression was detected by Western blot from (A); C. AGS cells were pretreated with 10 µM NF-κB inhibitor (BAY 11-7082), MEK1/2 inhibitor (U0126), p38/MAPK inhibitor (SB203580), JNK inhibitor (SP600125), JAK inhibitor I or the vehicle DMSO for 1 hour prior to IL-1β or TNF-α stimulation. IL-32 mRNA level was determined by real-time PCR. *P<0.05 versus the vehicle DMSO treated cells; D. The level of IL-32 protein was detected after AGS cells pretreation with 10 µM NF-κB inhibitor (BAY 11-7082) or not and then stimulation with TNF-α or IL-1β; E. AGS cells were infected by H. pylori at a MOI = 100 for 24 hours and simultaneously treated with neutralizing antibodies to block TNF-α or IL-1β.
H. pylori-induced IL-32 Expression
To further study the direct effect of H. pylori on the expression of IL-32 in gastric epithelial cells, AGS cells were infected with H. pylori at a MOI of 1, 10, and 100. As shown in Fig. 4A and B, IL-32 mRNA and protein levels were increased after H. pylori infection in a dose-dependent manner. We next analyzed whether H. pylori strain differences would contribute to different expression of IL-32. Cells were infected with H. pylori 11637 strain and CagA- strain. Although CagA- strain infection slightly increased the expression of IL-32 in AGS cells, the induction of IL-32 mRNA and protein level by CagA- strain infection was significantly lower than that by H. pylori 11637 strain infection (Fig. 4C and D).
Figure 4. IL-32 induction in response to H. pylori infection.

A. IL-32 mRNA level were assessed in AGS cells after H. pylori 11637 strain infection for 24 hours at a MOI of 1, 10, 100; B. IL-32 protein level were detected in (A); C. AGS cells were stimulated with either H. pylori 11637 strain or its isogenic CagA-negative mutant strain for 24 hours, and then the mRNA level of IL-32 was analyzed. Data are mean ± SEM of three separate experiments. *P<0.05; ***P<0.001; D. The protein level of IL-32 was detected and one representative blot was shown from (C).
Discussion
In the present study, we observed that gastric IL-32 was significantly elevated in patients with H. pylori infection at both mRNA and protein levels. The increased IL-32 mRNA level correlated with the severity of gastric inflammation. In addition, IL-32 mRNA levels were also correlated with TNF-α and IL-1β mRNA levels in H. pylori-positive gastric biopsy specimens, and either of the two cytokines could upregulate the IL-32 mRNA and protein level. Furthermore, we found that H. pylori infection of gastric epithelial cell lines also induced IL-32 mRNA and protein expression. These results indicate that IL-32 is likely involved in gastric inflammation from patients with H. pylori infection.
IL-32 was recently described as a proinflammtory factor involved in many inflammatory disorders such as chronic rhinosinusitis, a condition mostly caused by gram positive bacteria infection [19], [20]. Elevated IL-32 was then reported in HCV and HBV-infected liver and to correlate with the severity of hepatic inflammation and liver fibrosis [15], [21]. In this study, we showed that IL-32 mRNA level was significantly elevated in patients with H. pylori infection, and a high correlation between IL-32 mRNA and gastric inflammation was also observed, suggesting that IL-32 may play an important role in H. pylori-infected stomach. In addition, immunohistochemistry was used to detect the source of IL-32 and results showed that IL-32 was expressed in gastric epithelial cells and its highly expression was observed in H. pylori-positive gastric tissues. Because IL-32 was found to induce the production of IL-8 in gastric epithelial cells and inhibited the proangiogenic factor VEGF secretion by bronchial epithelial cells [16], [22], and we also observed IL-32 mRNA level was positively correlated with IL-8 expression (data not shown). It is reasonable that IL-32 influence the function of gastric epithelial cells in H. pylori-infected stomach.
The classic gastric inflammation with H. pylori infection could be influenced by infiltrating immune cells and inflammatory cytokines, the later may include the recently discovered IL-32 [19]–[22]. In our study, we found that in vitro TNF-α and/or IL-1β stimulated AGS cells to upregulate IL-32 mRNA and protein expression, which supported a positive relationship between IL-32 and TNF-α and IL-1β in vivo. However, Th17 cytokine (IL-17A, IL-17F, IL-6), Th2 cytokine IL-4, Tregs cytokine TGF-β1 and Th1 cytokine IFN-γ all failed to induce IL-32 mRNA and protein expression in our system, although all the relevant immune cell types and cytokines were reported to infiltrate H. pylori-infected stomach (see Figure S1). These results suggested that IL-32 is likely produced before the infiltration of these cells, which is supported by the report that IL-32 induces dendritic cell maturation and promotes the Th1 and Th17 polarization [23]. Moreover, the induction of IL-32 mRNA and protein by TNF-α or IL-1β was blocked by NF-κB inhibitor BY 11-7082, indicating that the molecular mechanism underlying this process is likely NF-κB dependent. Interestingly, using neutralizing antibodies to block TNF-α or IL-1β in parallel with H. pylori infection, we observed that IL-32 protein level was significantly decreased. Therefore, our results indicated that epithelial cytokines (TNF-α and IL-1β) response to H. pylori infection were important for the regulation of IL-32 expression.
Our results also showed that the IL-32 mRNA and protein level were increased in a dose-dependent manner following H. pylori infection, suggesting that the effect of H. pylori infection on IL32 expression is directly physiological. In addition, CagA mutations in H. pylori could significantly weaken the expression of IL-32 in AGS cells, indicating that H. pylori–induced IL-32 upregulation was dependent on CagA. H. pylori express many proteins to facilitate its pathogenesis [24]. UreB is also an important virulence protein for the colonization of H. pylori and mediated H. pylori-induced dysfunction of gastric barrier [25], [26]. We found that UreB mutations also attenuated H. pylori–induced IL-32 mRNA expression (see Figure S2). Therefore, IL-32 expression was predominantly regulated by translocation of CagA into epithelial cells and other virulence proteins might also influence the pathogenic response of H. pylori to gastric epithelial cells.
In conclusion, our data demonstrated that IL-32 expression was elevated in patients with H. pylori infection and correlated with the severity of gastric inflammation. The regulation of IL-32 expression in response to H. pylori infection depends on different interacting factors. These data together suggest that IL-32 may play an important role in the pathogenesis of gastritis caused by H. pylori infection.
Supporting Information
AGS cells were seeded in six-well plates at a density of 1×106 cells/well and stimulated with 10 ng/ml Th17 cytokine (IL-17A, IL-17F, IL-6), Th2 cytokine IL-4, Tregs cytokine TGF-β1 and Th1 cytokine IFN-γ for 24 hours, and cells were collected for analysis of IL-32 mRNA and protein expression. Data are mean ± SEM of three separate experiments and one representative blot was shown.
(TIF)
H. pylori strain 26695 were grown on brain-heart infusion plates containing 10% rabbit blood at 37°C under microaerophilic conditions (5% O2, 10% CO2, 85% N2), and its isogenic Urease subunit B-negative mutant strain (UreB- strain) was obtained as before described [27] . A multiplicity of infection (MOI) of 100 was used for infecting AGS and GES-1 cells. Cells were collected for analysis of IL-32 mRNA expression. Data are mean ± SEM of three separate experiments. *P<0.05; **P<0.01; ***P<0.001.
(TIF)
Funding Statement
This work was supported by grants from the Medical Science Youth Training Project of Chinese People’s Liberation Army (13QNP108) and National Basic Research Program of China (973 Program, No. 2009CB522606). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, et al. (1998) Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 114: 482–492. [DOI] [PubMed] [Google Scholar]
- 2. Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, et al. (2008) IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. European Journal of Immunology 38: 470–478. [DOI] [PubMed] [Google Scholar]
- 3. Harris PR, Wright SW, Serrano C, Riera F, Duarte I, et al. (2008) Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134: 491–499. [DOI] [PubMed] [Google Scholar]
- 4. Lindholm C, Quiding-Järbrink M, Lönroth H, Hamlet A, Svennerholm AM (1998) Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun 66: 5964–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, et al. (2011) Interleukins, from 1 to 37, and interferon-gamma: receptors, functions, and roles in diseases. J Allergy Clin Immunol 127: 701–721. [DOI] [PubMed] [Google Scholar]
- 6. Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA (2005) Interleukin-32: a cytokine and inducer of TNFalpha. Immunity 22: 131–142. [DOI] [PubMed] [Google Scholar]
- 7. Dinarello CA, Kim SH (2006) IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis 65: iii61–iii64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dahl CA, Schall RP, He HL, Cairns JS (1992) Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol 148: 597–603. [PubMed] [Google Scholar]
- 9. Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, et al. (2006) Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci 103: 3316–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, et al. (2006) IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci 103: 3298–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shioya M, Nishida A, Yagi Y, Ogawa A, Tsujikawa T, et al. (2007) Epithelial overexpression of interleukin-32α in inflammatory bowel disease. Clin Exp Immunol 149: 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer N, Zimmermann M, Bürgler S, Bassin C, Woehrl S, et al. (2010) IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol 125: 858–865. [DOI] [PubMed] [Google Scholar]
- 13. Bai X, Kim SH, Azam T, McGibney MT, Huang H, et al. (2010) IL-32 Is a Host Protective Cytokine against Mycobacterium tuberculosis in Differentiated THP-1 Human Macrophages. J Immunol 184: 3830–3840. [DOI] [PubMed] [Google Scholar]
- 14. Nold MF, Nold-Petry CA, Pott GB, Zepp JA, Saavedra MT, et al. (2008) Endogenous IL-32 Controls Cytokine and HIV-1 Production. J Immunol 181: 557–565. [DOI] [PubMed] [Google Scholar]
- 15. Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, et al. (2011) Interleukin-32: a new proinflammatory cytokine involved in hepatitis C virus-related liver inflammation and fibrosis. Hepatology 53: 1819–1829. [DOI] [PubMed] [Google Scholar]
- 16. Sakitani K, Hirata Y, Hayakawa Y, Serizawa T, Nakata W, et al. (2012) Role of interleukin-32 in Helicobacter pylori-induced gastric inflammation. Infect Immun 80: 3795–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dixon MF, Genta RM, Yardley JH, Correa P (1996) Classification and grading of gastritis: the updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 20: 1161–1181. [DOI] [PubMed] [Google Scholar]
- 18. Andrew A, Wyatt JI, Dixon MF (1994) Observer variation in the assessment of chronic gastritis according to the Sydney system. Histopathology 25: 317–322. [DOI] [PubMed] [Google Scholar]
- 19. Keswani A, Chustz RT, Suh L, Carter R, Peters AT, et al. (2012) Differential expression of interleukin-32 in chronic rhinosinusitis with and without nasal polyps. Allergy 67: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soyka MB, Treis A, Eiwegger T, Menz G, Zhang S, et al. (2012) Regulation and expression of IL-32 in chronic rhinosinusitis. Allergy 67: 790–798. [DOI] [PubMed] [Google Scholar]
- 21. Xu Q, Pan X, Shu X, Li X, Zhang K, et al. (2012) Increased interleukin-32 expression in chronic hepatitis B virus-infected liver. J Infect 65: 336–342. [DOI] [PubMed] [Google Scholar]
- 22. Meyer N, Christoph J, Makrinioti H, Indermitte P, Rhyner C, et al. (2012) Inhibition of angiogenesis by IL-32: possible role in asthma. J Allergy Clin Immunol 129: 964–973. [DOI] [PubMed] [Google Scholar]
- 23. Jung MY, Son MH, Kim SH, Cho D, Kim TS (2011) IL-32γ Induces the Maturation of Dendritic Cells with Th1- and Th17-Polarizing Ability through Enhanced IL-12 and IL-6 Production. J Immunol 186: 6848–6859. [DOI] [PubMed] [Google Scholar]
- 24. Andersen LP (2007) Colonization and infection by Helicobacter pylori in humans. Helicobacter 12 Suppl 212–15. [DOI] [PubMed] [Google Scholar]
- 25. Tan S, Berg DE (2004) Motility of urease-deficient derivatives of Helicobacter pylori. J Bacteriol 186: 885–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wroblewski LE, Shen L, Ogden S, Romero-Gallo J, Lapierre LA, et al. (2009) Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 136: 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang JY, Liu T, Guo H, Liu XF, Zhuang Y, et al. (2011) Induction of a Th17 cell response by Helicobacter pylori Urease subunit B. Immunobiology. 216: 803–810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AGS cells were seeded in six-well plates at a density of 1×106 cells/well and stimulated with 10 ng/ml Th17 cytokine (IL-17A, IL-17F, IL-6), Th2 cytokine IL-4, Tregs cytokine TGF-β1 and Th1 cytokine IFN-γ for 24 hours, and cells were collected for analysis of IL-32 mRNA and protein expression. Data are mean ± SEM of three separate experiments and one representative blot was shown.
(TIF)
H. pylori strain 26695 were grown on brain-heart infusion plates containing 10% rabbit blood at 37°C under microaerophilic conditions (5% O2, 10% CO2, 85% N2), and its isogenic Urease subunit B-negative mutant strain (UreB- strain) was obtained as before described [27] . A multiplicity of infection (MOI) of 100 was used for infecting AGS and GES-1 cells. Cells were collected for analysis of IL-32 mRNA expression. Data are mean ± SEM of three separate experiments. *P<0.05; **P<0.01; ***P<0.001.
(TIF)