

Abstract
While conventional MAP kinase pathways are one of the most highly studied signal transduction molecules, less is known about the MEK5 signaling pathway. This pathway has been shown to play a role in normal cell growth cycles, survival and differentiation. This MEK5 pathway is also believed to mediate the effects of a number of oncogenes. MEK5 is the upstream activator of ERK5 in many epithelial cells. Activation of the MEK-MAPK pathway is a frequent event in malignant tumor formation and contributes to chemoresistance and anti-apoptotic signaling. This pathway may be involved in a number of more aggressive, metastatic varieties of cancer due to its role in cell survival, proliferation and EMT transitioning. Further study of this pathway may lead to new prognostic factors and new drug targets to combat more aggressive forms of cancer.
Keywords: mitogen-activated protein kinase, big-mitogen activated protein kinase, Erk5, cellular signaling, epithelial-to-mesenchymal transition, kinase inhibitors
Introduction
The MEK5/ERK5 pathway is one of the lesser studied members of the mitogen-activated protein kinase (MAPK) family of protein kinases. This pathway has been implicated in cell survival, anti-apoptotic signaling, angiogenesis, cell motility, differentiation and cell proliferation(1–3). However, there is still much to learn regarding the potential activators of this pathway and many of the intermediary molecules involved in signaling both upstream and downstream of MEK5/ERK5 activation. Studies suggest that MEK5/ERK5 is involved in angiogenesis through its critical role in maintaining vascular integrity (4), but the role that MEK5 signaling may play in VEGF-mediated neovascularization is not fully understood. In addition, the specific involvement of MEK5 in EMT has not been well characterized. Also, it has been demonstrated the ERK5 is capable of activating downstream protein targets and functioning as a transcription factor. Yet, the regulation of the dual signaling capabilities of ERK5 is not clear. Understanding the nuances of this signaling cascade in normally functioning cells may provide additional insight into the mechanisms of dysregulation in disease states.
MEK5/ERK5 is over-expressed or constitutively active in a number of cancers relative to expression in healthy cells. Studies have examined MEK5 activation in prostate tumors and found strong expression in better than a third of them (10). Additional research examined the expression of MEK5 and ERK5 in breast tumors. One group found MEK5 expressed in over 50% of tumors and another studies found ERK 5 over expressed in 20% of patients (11–12). Additional tissue analysis is needed to further establish MEK5/ERK5 over-expression in tumors relative to activation in healthy tissue as well as quantification of the expression levels relative to baseline. Furthermore, more data is needed to determine clear associations between the expression of MEK5/ERK5, activation, cancer morphology and prognosis. This understanding will help to tailor treatments to situations where abnormal activation is triggered.
Understanding of the MEK5 pathway relatively to other MAP kinases may also help in tailoring appropriate drug treatments for many cancers. Current studies suggest there may be some redundancies and cross-talk among members of the MAP kinase family (2, 13–16). Future studies or re-examination of existing data with newer pathway specific inhibitors many help to elicit the specific signaling role of MEK5/ERK5 in these tissues (15, 17). This understanding may help with the development of specific treatments and possibly minimize side effects with future treatments.
Many features indicate that the MEK5/ERK5 pathway may provide a novel target for future therapeutics. Structurally, the C-terminus of ERK5 is much larger than other MAP kinases and contains both auto-inhibitory and nuclear shuttling functions (18–22). This structure lends itself to development of specific drug targets which may not interfere with other MAP kinases’ functions. This specificity may allow for targeted therapeutics to inhibit this pathway while minimizing the inhibition of other MAP kinases that may be critical to healthy cell survival.
In addition, the MEK5 pathway plays a novel role in chemoresistance within breast cancer cell lines (12, 23–25). Studies suggest that acquired resistance is a significant problem with all current therapeutic approaches (26). Inhibition of the MEK5/ERK5 pathway may make current therapies more effective and may be an integral component of a multi-drug therapy regimen (23, 25). However, additional work needs to be done to support these studies and determine chemotherapeutics that are enhanced through MEK5/ERK5 inhibition.
Further understanding of the MEK5/ERK5 pathway and the effectors of dysregulation and matching the therapy to these disease mechanisms may improve the success rates for the treatment of these more aggressive varieties of cancer (27). Given what has been learned to date, the MEK5/ERK5 pathway offers a promising target as a possible prognostic factor and potential therapeutic drug target due to its role in dysregulation of cell survival, proliferation and metastatic transitioning in a number of more aggressive varieties of cancer (5).
Overview of MAPKs
Mitogen-activated protein kinase (MAPKs) pathways are a family of related and sometimes interconnected pathways that play a critical role in the regulation of many cellular processes such as growth, differentiation and apoptosis. MAPKs are activated by signaling molecules such as growth factors, cytokines, neurotransmitters, hormones or various cell stressors which generally transmit their signaling through tyrosine kinase, G-coupled protein or hormone receptors(28). In MAPKs pathways, signals are transduced through a three-tiered kinase signaling cascade which begins with the phosphorylation of a mitogen-activated kinase kinase kinase (MAPKKK) that phosphorylates a second mitogen-activated kinase kinase (MAPKK) which subsequently phosphorylates a third mitogen-activated kinase (MAPK). Activation of MAPKs regulates critical cellular processes such as gene expression, proliferation and cell death [Figure 1](5). In mammals, there are many distinct subfamilies of MAPKs; the ERK 1/2, JNK 1/2/3, p38 proteins, ERK3/4, ERK5 (also known as Big Map Kinase), ERK7 and ERK8. All MAPKs are part of an evolutionary conserved family of kinases which predominantly phosphorylate serine and threonine residues which are preceded by a proline rich residue (29). The specificity of each MAP kinase is determined by the docking domains of the targeted substrate. The MAP kinases tether the specific substrate to their docking domain which aids in the efficiency of these reactions. These phosphorylation reactions are regulated by numerous transcription factors and enzymes and further controlled through binding partners, subcellular localizations, conformational changes and protein stability (30–31). Conventional members of the MAPK family have known activators. For example, MEK1 and MEK2 activate ERK1/2; MKK4 and MKK7 for JNKs; MKK3 and MKK6 for p38; and MEK5 for ERK5 (5–6). However, the activators of the atypical MAPKs such as ERK3/4, ERK7 and ERK8 are still not fully understood (7–9). These atypical MAPKs may also have different phosphorylation motifs and it is not clear if they conform to the classical three-tiered kinase activation cascade (6–8).
Fig. 1.
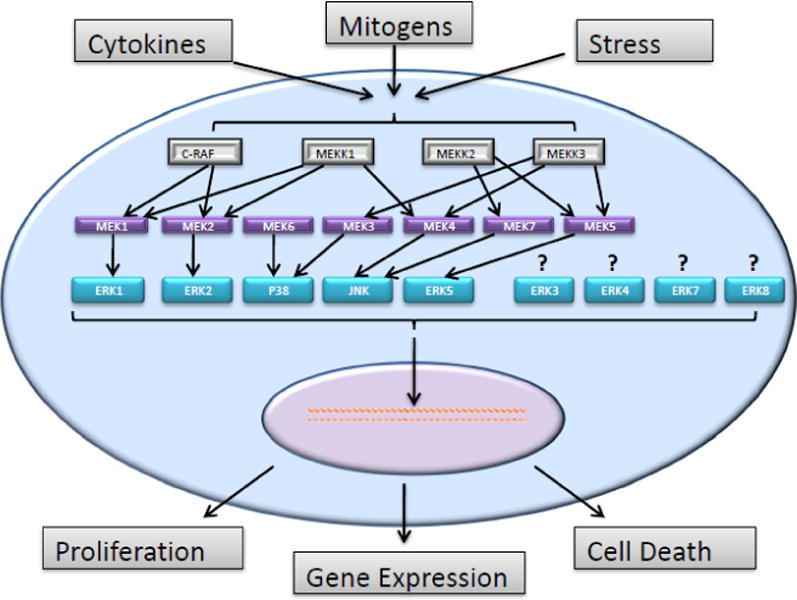
Mitogen-activated protein kinase (MAPK) pathways are a family of related and sometimes interconnected pathways. Signals are transduced through a three-tiered kinase cascade; MAPKKK through MAPKK to MAPK (5). Conventional members of the MAPK family have known activators. For example, MEK1 and MEK2 activate ERK1/2; MKK4 and MKK7 for JNKs; MEK3 and MEK6 for p38; and MEK5 for ERK5 (5–6). However, the activators of the atypical MAPKs such as ERK3/4, ERK7 and ERK8 are still not fully understood (7–9).
Initially, experimental data suggest that each MAPK subfamily is associated with different biological reactions. For example, it was believed that the ERK subfamily played a larger role in cell survival and proliferation, while the JNK and p38 MAKs were predominately activated by cytokines or extracellular stress and had a greater involvement in apoptosis (3, 5, 32). However, many of these distinctions have been blurred. It now seems clear that there are considerable tissue and developmental variations which affect signaling inputs and there may also be points of crosstalk or redundancy between these MAPK pathways (2, 6, 14, 33–34).
Upstream Activators of MEK5/ERK5 Signaling
MEK5/ERK5 interaction has been established as a unique MAP Kinase signaling pathway. MEK5 can be activated by both MEKK2 and MEKK3. However, it has been shown that MEK5 was not activated by MEKK1, an upstream activator of the JNK pathway (5). The precise pathway of activation varies based on the difference in the stimuli and the cell type. (35–37). MEKK2 and MEKK3 kinase domains are very similar, but their regulatory domains, located in the N-terminus, are significantly different. When compared with MEKK3, MEKK2 has a higher binding affinity for MEK5 (37). It has been shown that MEKK3 induces activation of the MEK5/ERK5 pathways through growth factor-induced cellular stimulation and oxidative stress (35).
Subsequent studies have shown that epidermal growth factor (EGF) mediates proliferation through ERK5 signaling(18). However, the role Ras plays in this activation is unclear (38). One study indicated that strong activation of ERK5 occurred through both EGF and nerve growth factor (NGF) signaling and this activation was induced by an oncogenic mutant of Ras (Ras Val12) in PC12 cells, but blocked by a dominant negative form of Ras. This study suggested that Ras is required for activation(34). Yet, conflicting studies demonstrated that RasVal12 was not sufficient to activate ERK5 and a dominant negative form of Ras did not block activation in PC12 cells (38). Additional work indicated RasVal12 was not sufficient to activate ERK5 in Cos7 cells(39). Other studies also support that EGF activation of ERK5 is independent of Ras(18).
The leukemia inhibitory factor (LIF), a cytokine, has been demonstrated to activate MEK5 through a LIF-induced tyrosine activated phosphorylation of Grb2-associated binder (Gab 1) and protein tyrosine phosphatase (SHP2) in cardiomyocytes. The activated Gab 1 and SHP2 form a complex that signals through ERK5 to cause elongation of cardiomyocyte cells (40). This LIF signaling pathway is independent of MEKK2 or MEKK3 signaling. Stat-3 has also been implicated as an upstream activator of MEK5 in both breast cancer cell lines and in breast cancer tissues (14).
MEK5 Structure and Activation
Binding of MEKK proteins occurs on the N-terminus extension of MEK5 (36). Two N-terminus splice variant isoforms of MEK5 have been identified, MEK5α and MEK5ß (36, 41). Experimental results indicate that MEK5α is a stronger activator of ERK5, relative to MEK5ß, due to its higher affinity for ERK5. This led to the discovery of a novel MAPK docking site on the N-terminus of MEK5Aα which is different from the consensus motifs identified in other MEKs (3). The N-terminus of MEK5 α includes a phox and Bem1p (PB1) domain which regulates the binding of MEKK2/3 with MEK5. Inhibiting the PB1 dependent formation of the MEKK/MEK5 complex has been shown to inhibit activation of MEK5α (42–43). It has been proposed that the N-terminus of MEK5 also contains an auto-inhibitory domain that can affect the interaction of MEK5 with other kinases (43). Binding of MEKK2 with MEK5 affects the overall confirmation of the MEK5α protein so that Ser311 and Thr315 become accessible for phosphorylation (44). Both MEKKs and ERK5 bind to the N-terminus extension of MEK5α. It is hypothesized that MEKK dissociates from the MEK5 complex to allow interaction with its ERK5 substrate (43, 45).
MEK5-Mediated ERK5 Activation
Once activated, MEK5 transmits signaling through ERK5. MEK5 has been identified as a binding partner for ERK5 in several studies (20, 22). It has been shown that ERK5 will not interact with either MEK1 or MEK2, indicating that MEK5/ERK5 interaction represents a unique signaling pathway (22). Similar to other MAPKs, ERK5 is present in a wide variety of cell types and is believed to be ubiquitously expressed. Presumably, it serves to regulate diverse functions depending on the cellular context and circumstances (19). During signal transmission, the N-terminus of the activated form of MEK5 binds to the functional domain of ERK5 (aa 78–139) to convey the activation signal. ERK5 has dual phosphorylation sites characterized by a TEY sequence which are similar to the binding sites of ERK1 and ERK2. However, ERK5 demonstrates a larger molecular mass than other ERK enzymes due to a unique C terminus extension (20, 34). The large C-terminus domain regulates activation, autophosphorylation, subcellular localization and nuclear shuttling. It is hypothesized that the N and C terminal halves are bound to one another in the cytosol and, once phosphorylated by MEK5, this binding is disrupted. This kinase includes a nuclear localization signaling (NLS) domain from aa 505–539(46). The open conformation of ERK5 promotes activation and allows exposure of the nuclear localization signaling (NLS) element which facilitates translocation to the nucleus (19, 21, 47). Once ERK5 has been imported into the nucleus, it associates with, phosphorylates and activates many transcription factors including Sap1, c-FOS, c-Myc and MEF2 (31, 34, 46, 48–50). The auto-phosphorylation of the C-terminus may also be required for transcriptional activation (19, 21) and the terminus contains two proline rich domains that serve as binding sites for nuclear transcription factors with SH3 domains(46). ERK 5 is exported back to the cytosol when the N and C-terminal halves renew their interaction and shuttling is accomplished through a nuclear export receptor, the Chromosome Region Maintenance 1(CRM1), dependent nuclear export signal (NES) (21, 47).
Adaptor/Scaffold Proteins Involved in the MEK5/ERK5 Pathway
Based on physiological stimuli that initiate the reaction, different adaptor/scaffold proteins facilitate and fine tune specific MAPK activation through formation of multi-enzyme complexes (52). These adaptor/scaffold proteins provide spatial and temporal regulation by binding multiple members of the MAPK pathway to bring them into close proximity and facilitate efficient phosphorylation and propagation of signaling (53–54). Many scaffold proteins have been identified for the MEK1/2, ERK 1/2 signaling cascade, such as the KSR family, MP-1, IQGAP, β-Arrestins, CNK, Sef and Paxillin which facilitate MEK/ERK interactions (55–57). However, there is much less information regarding scaffolding proteins for the MEK5/ERK5 cascade. It is unclear if any of MEK1/2 proteins also provide a scaffold function in the MEK5 pathway. Yet, a functional interaction has been shown between MEKK2 and Lck-associated adapter (LAD) which is required for EGF activation of ERK through Src (37). Also, muscle specific protein kinase A (PKA) anchoring protein mAKAP is involved in ERK5 activation which induces hypertrophic signaling in cardiomyocytes (58).
Furthermore, MEK5’s PB1 domain has developed as an internal platform or scaffold to facilitate MEKK2 (or MEKK3)-MEK5-ERK5 signaling (51, 59). The MEK5 PB1 domain organizes the interaction to ensure rapid phosphorylation of ERK5 upon upstream signaling from MEKK2 or MEKK3 (43, 51, 59).
In addition, all MAPKs have an evolutionarily conserved, common docking (CD) domain that consists of a cluster of negatively charged amino acids in the C-terminus. This CD domain is used to bind the MAPK to their activators (MAPKKs), phosphatases, substrates and scaffold proteins (59–60). In the case of ERK5, this negatively charged domain binds the positively charged lysine and arginine residues in the MEK5 docking site. These docking interactions are pathway specific and may provide a molecular basis for the sequential activation and inactivation of a given pathway. The docking interactions enhance the efficiency of all the enzymatic reactions and may help to regulate the specificity of protein recognition (60).
Regulation of Cell Differentiation and Survival
ERK5 has been implicated in the survival response of PC12 cells to oxidative stress (61). Further work demonstrated that ERK5 contributes to the survival response in neuronal dorsal root ganglia cells through a unique retrograde signaling system mediate by nerve growth factor (NGR)(3, 62). In this pathway, activated ERK5 initiates a phosphorylation cascade through activation of p90 ribosomal S6 kinase (RSK) which ultimately results in activation of Ca2+/cAMP response element binding protein (CREB). CREB regulates the transcription of pro-apoptotic and survival genes (17, 62). Further studies indicate ERK5 contribute to survival in neurons via activation on transcription factor MEF2; a pro-survival, anti-apoptotic transcription factor (63–64). Additional in vivo studies showed ERK5 deleted mice were genetically lethal around embryonic day 10 and these mice displayed severe growth retardation in the head region (3, 65). These studies indicate that the defect is not caused by apoptosis, but by the inhibition of neuronal differentiation due to the suppression of some specific neural markers (3, 17). Under physiological conditions, it remains highly likely that ERK5 participates in normal brain development due to its role in neuron cell differentiation (3, 66–67). Yet, when neuron-specific ERK5 knock down was develop, mice develop normally with a life span similar to their control littermates. The survival of these mice indicates that there may be other intercellular pathways which exist to compensate for the loss of a ERK5 gene in neurons.(17)
ERK5 is needed to mediate the survival response in endothelial cells(ECs) that are subjected to sheer stress. Overexpression of constitutively active MEK5 in blocks caspase-3 activity and prevents apoptosis. However, inhibition of ERK5 activity in these cells by using a dominant negative ERK5 mutation or a gene deletion stimulates EC cell death. The survival mechanism is believed to be PKB independent phosphorylation of Bad which sequesters Bad to the cytosol to prevent apoptotic signaling (17, 68–69).
The serum and glucocorticoid-induced kinase (SGK) have very similar sequence homology with PKB and shares its mechanism of activation. Similar to PKB, SGK is able to block Foxo3, a member of the forkhead family of transcription factors that acts as a tumor suppressor, activity by phosphorylation (70). SGK is a direct target of ERK5 and a key component in the cell’s survival response to environment stress stimuli (71–72). However, it is not yet clear if ERK5 regulation of SGK is responsible for mediating SGK’s survival function (3).
The Role of MEK5/ERK5 in Cell Proliferation
It has been demonstrated that ERK5 signaling dramatically increases the transcription of MEF2 which in turn induces transcription of c-jun, a gene required for cell proliferation (49). Upstream of this signaling, mitogens, including EGF and granulocyte colony stimulating factor (G-CSF), have been found to transmit pro-growth signaling through the ERK5 pathway (18, 73). However, fibroblasts showed no marked difference in progression to S phase when comparing dominant negative ERK5 and MEK5 cells to wild type (4, 65). This suggests that ERK5 signaling pathways may play a central role in promoting or regulating proliferation in given cell types under different conditions(3). Potentially, proliferation signaling may be transduced through SGK; this protein kinase is also closely linked to the progression from G1 to S phase in the cell cycle (74). Cyclin D1 signaling may be another possible pathway for ERK5 mediated proliferation. The cyclin D1 gene is a key proliferation checkpoint and ERK5 has also been shown to regulate this protein. De-regulation of the expression of cyclin D1 is frequently associated with neoplastic transformation. In a number of breast cancer cell lines, blocking the activation of ERK5 has been shown to block synthesis of endogenous cyclin D1 protein (75).
The Role of MEK5/ERK5 in Angiogenesis
Angiogenesis is the process of developing new blood vessels. It plays a crucial role in development, reproduction and wound healing, but it has also been implicated in tumorigenesis of many cancers (76). Work has been done to examine vascular integrity and endothelial failures in animal studies. ERK5 deletion was embryonically lethal in genetic knock-out mice and ERK5 deletion in adult mice lead to lethality within 2–4 weeks. Physiological analysis of the adult mice demonstrated abnormally leaky blood vessels. Histologically these mice demonstrated multiorgan hemorrhage and architectural irregularities in the endothelial lining of their blood vessels. This evidence suggests that ERK5 is critical for endothelial function and preserves the integrity of blood vessels (4). Further studies on mice bearing xerographic tumors found that ERK5 was required for tumor growth due to its role in tumor vascular development. These studies suggested ERK5 is involved in the development of tumor vasculature through its action on rpS6; phosphorylated rpS6 is a ribosomal protein correlated with tumor-associated endothelial cell of blood vessels (1).
The role of ERK5 in vascular endothelial growth factor (VEGF)-induced angiogenesis is unclear. Work has been done on human umbilical vein endothelial cells to look at synergies between VEGF and fibroblast growth factor 2 (FGF2) and to examine some of the intracellular molecules involved in this signaling cascade. These studies showed upregulation of phosphorylated ERK1/2 in response to stimulation of VEGF receptor. The downstream targets of this pathway were strongly inhibited with the use of the protein kinase C (PKC) inhibitor, GF109203X, and the use of a mitogen-activated protein kinase/ERK kinase (MEK) kinase inhibitor, U0126. In addition, ERK activation was reduced by approximately 50% with the PKC inhibitor alone indicating the downstream expression may occur both through and independent of the ERK1/2 pathway (77). These experiments do not preclude MEK5/ERK 5 involvement in this pathway. The MEK inhibitor U0126 has also been shown to inhibit MEK5 (15). Given the demonstrated role of MEK5/ERK5 in endothelial cell survival, proliferation, and angiogenesis (1, 4), it is possible that MEK5/ERK5 plays a role in the VEGF signaling cascade. It is feasible that both MAP kinases are involved in this pathway and inhibition with U0126 has fully blocked signaling cascades to the downstream targets in both pathways (15). The availability of more specific inhibitors would clarify the role of MEK5/ERK5.
MEK5/ERK5 Mediation of Oncogenes
A mutated Ras has been identified in many types of cancer, including pancreatic (90%), thyroid (50%), colon (50%), lung (30%) ovarian (15%), breast, skin, liver, kidney, and some leukemias. The MEK5/ERK5 pathway is believed to be involved in mediating the oncogenic effects of Ras. In some cell types (PC12, C2C12, COS7) ERK5 is activated through a Ras signaling pathway (34, 48). This suggests that an oncogenic form of Ras may induce typical morphological changes, loss of contact inhibition, anchorage-independent growth of fibroblasts and tumorigenicity through the MEK5/ERK5 signaling cascade. However, this hypothesis has not been fully tested (3).
Other oncogenes may activate ERK5, including cancer osaka thyroid (Cot). Experimentally, over-expression of the Cot protein has been shown to potently stimulate the activity of the c-jun promoter utilizing JNK-dependent and also two members of the MAPK family, p38gamma (ERK6) and ERK5. This study also revealed that Cot requires all three protein kinases, JNK, p38s, and ERK5, to stimulate the c-jun promoter fully and to induce neoplastic transformation. In addition, there is a strong suggestion that the transforming ability of Cot results from the coordinated activation of these separate MAPK pathways, which collaborate to regulate the expression and activity of the product of the c-jun proto-oncogene (78).
c-Src has also been shown to mediate activation of ERK5 in response to oxidative stress (79). Studies have demonstrated that the oncogene, Src, can mediate ERK5 signaling which may lead to disruption of the actin cytoskeleton through a loss of actin stress fibers. The mechanism that causes these cytoskeletal disruptions is distinct from ERK1/2 signaling and inhibition of ERK 1/2 signaling is not sufficient to restore actin fibers in the cytoskeleton. This indicates that multiple MAP kinase pathways downstream of Src oncogenes may play a role in cytoskeletal modifications (80–81). A further study concluded that ERK5 promotes increased Src activity which in turn induces the formation of invasive adhesions called podosomes (82).
The Role of MEK5 in Epithelial-Mesenchymal Transitions (EMT)
EMT occurs at critical points during embryonic development. During EMT, epithelial cells will undergo remodeling: increased plasticity, loss of polarity, redistribution of tight junctions and adherins and development of migratory capacity. After development, this process may occur during normal physiological functions such as wound healing and tissue repair, but it can also be associated with hyperplasia, adenomas, tumor progression and metastatic disease(83). One hallmark of EMT is the loss of E-cadherin expression, a central protein component of cell to cell adhesion junctions that is required for all epithelium. This loss increases tumor cell invasiveness and contributes to the transition to carcinomas (84). In order for EMT to occur, there must also be a gain in other mesenchymal cell markers such as fibronectin, vimentin and N-cadherin. The loss of cell adhesion proteins coupled with the cell’s ability to express mesenchymal markers allows cells to morphologically transition from an epithelial to a fibroblastic morphology (85). Studies indicate that the process is mediated through a number of signaling pathways, including activation by several tyrosine kinase receptors, such as EGFR (Epidermal Growth Factor Receptor) and FGFR (Fibroblast Growth Factor Receptor), that act through a RAS dependent/MAPK signaling pathway (83).
A study examined the role of ERK5 in re-epithelialization during cutaneous wound healing of human keratinocytes. The cells were treated with EGF to stimulate the EGF receptor. Treated cells showed increased levels of phosphorylated ERK5 which coincided with increased levels of Snai2 mRNA (86); Snai2 is a transcription factor that plays a role in cell migration during normal tissue turnover and it is also associated with EMT in human melanoma, oral squamous cell and some breast carcinomas (87). An ERK5 knockdown was also used in HaCaT cells, an immortalized human keratinocyte cell line. The treated HaCaT cells demonstrated decreased motility response and reduced Snai2 mRNA expression. The knockdown cells demonstrated a more compact morphology, disruption in desmosome organization and an altered ability to aggregate. This work suggests that ERK5 plays a role in controlling cytoskeleton organization and motility of keratinocytes during cutaneous wound healing (86).
Transforming growth factor-ß receptor (TGFßR)-SMAD has also been demonstrated to act in conjunction with the Ras pathway to induce EMT in culture and in metastatic mouse models (88–89). It has also been demonstrated that EMT is dependent on overexpression of activated forms of the Raf-MAPK pathway in NBT-II rat carcinoma cell line in vitro. In this study overexpression of activated forms of c-Raf and MEK1 led to cell dissociation through the loss of desmosomes from the cell periphery. In addition, the MEK1 inhibitor, PD098059, repressed EGF and Ras-induced cell dispersion. The authors concluded that EMT depends on activation of a Raf-MEK1 pathway (90). However, this does not preclude signaling through MEK5 rather than MEK1; overexpression of RAF can also lead to increased MEK5 signaling (48) and PD09859 can inhibit activation of both MEK1 and MEK5 (34).
EMT signaling has been shown to induce transcription of several developmentally important genes; Twist, Snai1 and ZEB1/ZEB2 (84). When over expressed, Snai1 and ZEB1/ZEB2 are involved in the disruption of E-cadherin-mediated intercellular adhesions and Twist has been shown to confer the ability to enter into the circulation to seed metastases (85). ZEB1/ZEB2 can be activated through TGF-beta signaling and shows high expression in colon and several breast cancer cell lines that under express E-cadherins (89, 91). Alternately, it has been demonstrated that Twist is the downstream target of NF-κB in vertebrates (92). NF-κB is a known transcription factor that is activated in response to inflammatory cytokines and growth factors (93). It is involved in multiple cellular processes, including cytokine gene expression, cellular adhesion, cell cycle activation, apoptosis and oncogenesis. Constitutive activation of NF-κB has been demonstrated in a number of solid tumors which leads to chemoresistance and cancer cell survival (94). Work done in our lab has demonstrated that inhibition of the NF-κB pathway blocked cell survival of MCF-7 breast cancer cells (95). NF-κB activation has also been shown to be an essential step for EMT and metastasis in breast cancer (93). Snai1 expression is regulated by several upstream targets. A TGF-beta-SMAD pathway converges with Ras/MAPK signaling pathway to induce EMT and metastasis progression in mouse models (88). In addition, activation of LIV1 has been reported to control Stat3 (signal transducer and activator or transcription 3)-dependant activation of EMT during gastrulation in zebra fish (96). Prior to this work, it had been demonstrated that LIV1 was implicated in metastatic progression in breast cancer (96–97). Both studies show that LIV1-induced Stat3 signaling mediated through SNAI1 plays a role in EMT and metastasis. Conversely, ER receptor signaling mediated through MTA3 (metastasis-associated gene 3) represses Snai1 signaling. In estrogen receptor negative cancers, loss of MTA3 may lead to EMT and metastatic progression (84).
The role of MEK5/ERK5 in Cancer
Hepatocyte growth factor (HGF) is highly expressed in most malignant mesotheliomas. HGF has been shown to stimulate proliferation through a phosphatidylinositol 3-kinase (PIK3)/ERK5/fos-related antigen 1(fra-1) pathway in some malignant mesothelioma cells. These studies used human mesothelial LP9/TERT-1 and seven other malignant mesothelioma cell lines to demonstrate increased levels of phosphorylated ERK5 after the addition of HGF. Furthermore, it was shown that increased cell viability induced by HGF was blocked by knocking down MEK5 using a siRNA construct (siMEK5). Increased expression of Fra-1 was noted in 3 of the 7 cell lines. Fra-1, a member of the FOS gene family, dimerizes with proteins of the JUN family to form the transcriptional factor complex AP-1 which has been implicated as a regulator of cell proliferation, differentiation, and transformation (98). Fra-1 is up-regulated in several tumor types. Previous studies had demonstrated that Fra-1 expression is regulated by the ERK 1/2 pathway (99–102), but this work suggests that Fra-1 may also be regulated through the MEK5 pathway. Moreover, Fra-l could modulate it own pathway through a negative feedback loop blocking activation of MEK5(103).
ERK5 may also play a role in the regulation of apoptosis in multiple myeloma cells. Studies have shown that the myeloma growth factor, interleukin 6 (IL-6), activated ERK5 independent of the Ras and Src signaling. These studies examined ERK5 level in cell lines MM1S, MM1R MM144, OPM2, U266 and RPMI8226 as well as bone marrow plasma cells from three multiple myeloma patients. ERK5 expression was demonstrated in all cell lines and tissue cultures. After treatment with IL6, dual phosphorylation of ERK5 was shown in both the MM1S and MM144 cell lines. RT-PCR analysis of these two cell lines also demonstrated expression of MEF2 transcription factor, a known downstream target of ERK5. In addition, treatment with a dominant-negative form of ERK5 restricted proliferation of multiple myeloma cells and sensitized these cells to treatment with PS341 and dexamethasone to induce apoptosis. Furthermore, overexpression of a wild type ERK5 was shown to convey resistance to PS341-induced cell death. (23)
More recent experiments have examined the role of ERK5 in the FAK (focal adhesion kinase) signaling pathway (104). FAK is over expressed in a variety of human cancers and this overexpression has been associated with metastasis and invasion (105). This study found that intergrin-mediated activation of FAK showed up-regulation of the dual phosphorylated ERK5. Cells were then treated with dominant negative FAK and showed a significant reduction in phosphorylated ERK5 levels as well as a reduction in cell adhesion and cell motility. These studies suggest that integrin-mediated FAK signaling promotes cancer metastasis through activation of the ERK5 signaling pathway thus this pathway offers an opportunity to develop an anti-metastatic inhibitor to fight a variety of tumors associated with increased levels of FAK and ERK5 (104).
Over-expression of MEK5 has been demonstrated in many cancers. One study looked at MEK5 expression in 127 prostate tumors and cases of benign prostatic hyperplasia. MEK5 was moderately expressed in 44% of tumors and strongly expressed in 37% of tumors. In this study, high grade prostate cancers, those with Gleason scores of 8 or above, generally had increased levels of MEK5 expression. Among those with Gleason scores of 7 or greater, MEK5 levels could be used to define a subgroup with the worst prognosis. Although MEK5 expression was not significantly associated with tumor grade, it was strongly associated with the presence of bony metastases and reduced disease-specific survival (10).
Activation of ERK5 is also associated with metastases in oral squamous cell carcinoma (OSCC). Gene expression profiling was performed on 35 primary OSCC tumors. In total, 7390 genes were found with differential expression in the OSCC tumors relative to healthy oral mucosal samples. Dysregulation was found in several cancer related pathways; transforming growth factor beta, apoptosis and MAPK signaling. Concentrating on the MAPK pathway, additional microarray analysis using immunohistochemistry (IHC) uncovered increased protein expression of ERK1 in 22.8% of the samples and an increase of ERK5 in 27.4% of the samples. However, there was an association of high ERK5, but not ERK1 expression, with the presence of advanced staged tumors and lymph node metastases (106). Furthermore, ERK5, but not ERK1, was shown to contribute to the Src-mediated structural disruption of the actin cytoskeleton. This disruption may be the first phase in development of metastatic disease (21, 106). These results suggest that ERK5, as a downstream target of EGFR, may be a novel molecular target and potential interference point in future therapeutic approaches given its association with advanced tumor stage (106).
Studies completed in our laboratory examined the role of MEK5 in chemoresistance in MCF-7 breast cancer cells. Microarray analysis indicated a 22-fold increase in the levels of MEK5 in apoptically resistant cells (APO- cells). Transfection of these cells with dominant negative forms of ERK5 and ERK2 and subsequent treatment with and without several apoptotic inducing agents in addition to one cell survival compound revealed the anti-apoptotic characteristics of MEK5. This demonstrated that the MEK5 pathway plays a novel role in chemoresistance and this pathway is a central mediator of cell survival and apoptotic regulation in MCF-7 cells (25).
Within the same MCF-7 cell line model, others demonstrated that ERK5 was activated through the HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4 receptors through the binding of neuregulins (NRGs); Neuregulins are part of the family of epidermal growth factor proteins which have diverse effects on neuronal development. These NRG-activated cells affected ERK5 in a time and dose-dependent fashion. When a dominant negative form of ERK5 was introduced into these cell lines, there was a reduction in cell proliferation. The BT474 and SKBR3 human breast tumor cell lines also express a constitutively active form of ERK5 in activated ErbB2, ErbB3 and ErbB4 cells. When the cells were treated to inhibit ErbB receptor activity, ERK5 was inactivated. In addition, when a dominant negative form of ERK5 was introduced into these cells, the proliferation rates fell indicating ERK5 may be involved in proliferation of these breast cancer cells (107).
In 2005, a study was performed to evaluate the potential role of Stat3 in regulating human breast cancer. This study examined tissue from 136 invasive breast tumors. Microarray analysis and immunohistochemistry (IHC) were done to determine expression and phosphorylation of Stat3. Increased levels of phosphorylated Stat3 were detected in 35% of the tumors (relative to Stat3 phosphorylation levels in normal breast tissue) and the area of increased Stat3 activation was linked to the metastasis in the regional lymph nodes. Also, phosphorylation of Stat3 was associated with increased expression of many downstream target proteins involved in metastasis and proliferation of breast cancer such as MEK5, c-Myc and c-Fos. IHC tests indicated that positive staining for MEK5 was found in 55% of the tissues that demonstrated increased Stat3 activation. The authors suggest that MEK5 may be a downstream target for Stat3 signaling due to the high expression in tumors and previous reports of MEK5/ERK5 role in malignancies and tumor formation (11). Other studies have also reported that MEK5 levels are significantly higher in breast carcinoma cell lines with constitutively activated Stat3 when compared with non-malignant breast epithelial cells or normal tissue with activated Stat3 signaling. Based on this work, it is clear that Stat3 signaling may be involved with MEK5 in inducing malignancies. However, it is not entirely clear if each pathway plays its own independent role or if there is crosstalk among these different EMT, proliferative and anti-apoptotic pathways that participate in a network of system regulation in the development of breast cancers (14).
Other studies looked at downstream targets of Breast Tumor Kinase (Brk). Brk is a protein tyrosine kinase found to be over expressed in 86% of invasive ductal breast tumors. It is co-expressed with ErbB family members and has been shown to phosphorylate STAT3. In these experiments, Brk expression was knocked down in the T447D cell line and found there was a reduction in Rac, p38, ERK5, cyclin D1 expression and cell migration. This demonstrated that Brk expression enhances MEF2-luciferase activity. Brk enhances EGF and heregulin-induced ERK5 activation. The activation of ERK5 signaling contributes to breast cancer progression. However, this work suggests that p38 MAPK may have a dominant role in this pathway. Further work is needed to understand Brk activation of ERK5 and to examine the relative role of ERK5 in inducing cyclin D1 expression, MEF2 transcription activity and breast cancer cell migration independent of p38 signaling. This additional data is necessary to determine areas of possible cross-talk as well as potential signaling specificity between the two MAPK pathways (13).
Additional research examined the expression of ERK5 in 84 human breast tumors relative to normal breast tissue. This study found ERK 5 was expressed in most patients and over expressed in 20% of patients. The data showed an inverse relationship between ERK5 overexpression and disease free survival rates independent of other clinical-pathological parameters of prognosis. HER2 overexpression and phospho-HER2 cells also over expressed pERK5, but other tissue samples demonstrated pERK5 expression with no overexpression of HER2. This suggested that activation of ERK5 may also occur through a HER2-independent route. Other experiments looked at an animal tumor model using the BT474 cell lines. ERK5 function was reduced either through a knockdown or the use of a stable dominant negative form of ERK5 cells. The cells with reduced ERK5 function demonstrated lower proliferation rates and a greater sensitivity to HER2 inhibitors. These studies suggest that ERK5 overexpression may help to predict patient outcomes and may be a potential therapeutic target and, when ERK5 activity is inhibited, it may enhance the action of other known chemotherapeutic agents (12).
Our laboratory completed novel studies looking at the role of MEK5/ERK5 signaling in a tumor necrosis factor (TNF)-α chemo-sensitized breast cancer cell line to examine this pathway’s involvement in mechanisms of resistance and EMT. The proteome of TNF-α sensitive MCF-7 cells was compared to TNF-α resistant MCF-7-MEK5 cells and the results were confirmed with gene expression levels using RT-PCR assays. Prior work done by the lab demonstrated that stable anti-apoptotic phenotype of MCF-7 cells could be derived from prolonged exposure to increasing concentrations of TNF-α and this resistance was in part dependent on mitogen-activated protein kinase and nuclear factor-κβ signaling (NF- κβ) (25, 108). For these experiments, a DNA expression construct, pCMV-HA-CA-MEK5, was used to derive a MEK5 over-expressing variant of the TNF-α resistant cell and a stable vector was introduced in to TNF-α sensitive MCF-7 cells as a control, MCF-7-VEC. The TNF-α resistant cell line showed increases in vimentin (VIM), an member of the intermediate filament family involved in attachment, migration and signaling; creatine kinase, brain (CKB), a cytoplasm enzyme which catalysis phosphate transfer; heat shock protein 4 (HSPA4), a cellular chaperone protein; and glutathione S-transferase pi1(GSTP1), an enzyme which plays an important role in detoxification. Other proteins were downregulated in the TNF-α resistant cells: keratin 19 (KRT19), an intermediate filament protein responsible for the structural integrity of epithelial cells; keratin 8 (KRT8), a intermediate filament protein which maintains structural integrity; and glutathione S-transferase mu 3 (GSTM3), an enzymes which function in the detoxification of electrophilic compounds including carcinogens (24, 109). In addition, the MCF-7-MEK5 cells demonstrated an EMT phenotype under immunofluorescence staining for E-cadherin and vimentin. Moreover, the MEK5 cells demonstrated downregulation of E-cadherin with upregulation of the EMT regulatory genes SNA12, ZEB1 and N-cadherin consistent with an EMT phenotype relative the MCF-7-VEC cells. These results suggest that the MEK5 pathway may promote both chemoresistance and the EMT phenotype in breast cancer cells (24).
Inhibitors of MEK5/ERK5
PD98059 and U0126, two drugs thought to solely inhibit the MEK1/ERK 1 and ERK 2 pathways, were studied to determine their effects on MEK5/ERK5. Both drugs were also shown to inhibit MEK5 and ERK5 activity which seems plausible given that the TEY sequences of ERK5 are similar to those in ERK1 and ERK2 and MEK5 shares a 48% identity relative to MEK1. U0126 suppressed MEK5 activity in a dose dependent fashion, while PD98059 only partially blocked activity. These results call into question previous data generated using these inhibitors and suggest that more specific blocking agents should be used to determine the exact role for each of these MAP kinase pathways (34).
In a later study, PD98059 and U0126 were found to inhibit activation of endogenous ERK5 in Hela cells, but at a higher concentration than required to block activation of ERK1/ERK2. PD184352 was also found to block activation of MEK5/ERK5 and MEK1/ERK 1/2 pathways at high concentrations. At lower concentrations, this drug blocked MEK1/ERK1/2, but it actually prolonged activation of MEK5/ERK5 suggesting that MEK1/ERK1/2 activation may suppress activation the MEK5/ERK5 pathway (15).
Recently, BIX02188 and BIX02189 were identified as two pharmacological inhibitors specific to the MEK5/ERK5 pathway. These inhibitors suppressed MEK5 catalytic activity in a dose dependent fashion with IC50s of 4.3 and 1.5 nM, respectively. Both compounds also inhibited ERK5 phosphorylation in a dose dependent manner, yet BIX02189 had a greater potency. Both drugs were shown to inhibit MEF2 driven gene expression, a downstream target of MEK5/ERK5. Cells were incubated for a 24 hour period with these drugs and there were no cytotoxic effects during that period. In addition, these compounds did not inhibit other closely related MAP kinases: MEK1, MEK2, ERK2 and JNK2. Due to their efficacy and specificity, these inhibitors may provide an additional tool for understanding the role of the MEK5/ERK5 pathway relative to other closely related MAP kinases and a starting point for potential therapeutic treatments (110).
Another class of MEK5 inhibitors is novel benxamidazole compounds that were originally designed as CDK5 inhibitors. Investigators looked at the ability of these compounds to inhibit EGF-initiated MEK5-mediated ERK5 phosphorylation in human embryonic kidney cells. One of the compounds examined was shown to block 12.35% of ERK5 phosphorylation. This compound also demonstrated a modest preferential inhibition of ERK5 phosphorylation relative to inhibition of ERK1/2 phosphorylation. NCI 60 cell line screen showed that a single dose of this compound selectively inhibits MCF-7 cell line growth, but this compound was not selective for long dose response analysis. Long dose response analysis, the process of evaluating the drug’s efficacy at five concentration levels, is completed for compounds that exhibit significant growth inhibition during single dose analysis. Investigators are continuing studies on variations of this compound for their potential to inhibit EGF-mediated activation of the MEK5/ERK5 signaling pathway (111).
Most recently, XMD8-92 has been identified as an ERK5 (BMK1) inhibitor. XMD8-92 has been shown to reduce ERK5 transactivation of MEF2C, a known substrate of ERK5. Researchers also demonstrated that the inhibition of ERK5 activation by XMD8-92 treatment was indistinguishable from cells treated with a dominant negative form of ERK5 in HeLa cells. This inhibitor also exhibits a high selectivity for ERK5. When XMD8-92 was profiled against all kinases expressed in HeLa cells, it was found to be at least 10-fold more selective for ERK5 than any other kinase studied with an IC50 of 1.5 μM. In addition, it did not block growth factor induced activation of ERK 1/2 and did not significantly inhibit MEK5. Researchers also examined the interaction of ERK5 with the tumor suppressor promyelocytic leukemia protein (PML) and the impact that this interaction had on the induction of p21; a known downstream effector of PML which modulates cell proliferation (112–114). It was shown that ERK5 interacts to forms a complex with PML which suppresses expression of p21 thus blocking PML’s anti-proliferative function. Treatment with XMD8-92 induced the expression of p21 which in turn suppressed cancer cell proliferation.
Future Perspective
The discovery of the mitogen-activated protein kinase family has provided significant insight into major signaling pathways that have a significant role in human development normal physiology and disease. While the majority of research effort has focused on the Erk1/2, Jnk and P38 family members, emerging evidence has now demonstrated an important role for the less well known MAPK family members such as ERK5 and ERK8. With a greater understanding of the mechanisms and biological targets of the MEK5-ERK5 pathway and the recent development of effective pharmacological and molecular targeting strategies, we are beginning to delineate the true importance of this pathway. As our understanding of the MEK5-ERK5 pathway continues to evolve we expect to define its unique role in human disease as well as the signaling crosstalk and interconnection between the individual MAPK family member pathways. Ultimately we expect that this will reveal a critical role for ERK5 as a viable target for therapeutic intervention particularly in targeting cancer.
Fig. 2.

MEK5 regulation and downstream targets. MEK5/ERK5 pathway activation can occur through extracellular stressors, mitogens or cytokine activation. Activation of this pathway may lead to epithelial mesenchymal transitions (EMT), cell survival, anti-apoptotic signaling and an increase in cell proliferation.
Fig. 3.

MEK5/ERK5 pathway is activated by cell stressors, mitogens or cytokines. Once stimulated, MEKK2 or MEKK3 will phosphorylate MEK5 on its serine 311 and threonine 315 residues(51). MEK5 then phosphorylates the threonine 218 and tyrosine 220 residues of ERK5(2). Upon activation, ERK5 can phosphorylate downstream target molecules such as Sap1, cFOS, c-Myc and MEF2 or autophosphorylate its carboxyl-terminal region which contains a NLS region (nuclear localization signal) allowing ERK5 to shuttle from the cytosol to the nucleus. This gives ERK5 two possible mechanisms for signaling; either by activating downstream molecules or potentially acting directly as a transcription factor (2, 21).
Fig. 4.

Pathway for MEK5/ERK5- mediated cell survival: When cell stress occurs, a nonphosphorylated form of Bad translocates to the mitochondria to induce cytochrome c release and a nonphosphorylated Foxo3a translocates to the nucleus to induces transcription of FasL receptor. Both processes initiate apoptosis. Activated ERK5 phosphorylates Bad and Foxo3a either directly or through a PKB dependent mechanism. Phosphorylation sequesters Bad and Foxo3a in the cytoplasm thereby blocking their apoptotic effects. In neurons, ERK5 also mediates cell survival in response to growth factors by activation of the transcriptional factors CREB and MEF2 (3).
Fig. 5.

Proposed MEK5 regulation in breast cancer: Similarly to MEK5/ERK5 pathway activation in physiologically healthy tissue, activation may occur through extracellular stressors, mitogens or cytokine activation to promote survival, anti-apoptotic signaling and proliferation of cancer cells. Some of the microarray analysis of breast cancer tissue samples as well as experiments completed by our laboratory suggests that activation of this pathway may lead to epithelial mesenchymal transitions (EMT), metastasis and invasion especially in more aggressive phenotypes of breast cancer.
Table 1.
Some of the known MEK5/ERK5 pathway inhibitors and their reported IC50 values in various cell lines.
| Inhibitor | Cell Line | MEK 5 specificity | IC50 [nM] | Reference |
|---|---|---|---|---|
| PD98059 | SK-Br-3 | No | 27 | Normanno et al., 2006 (115) |
| MDA-MB-361 | No | 52 | Normanno et al., 2006 (115) | |
| MDA-MB-468 | No | 40 | Normanno et al., 2006 (115) | |
| U0126 | AP-1-bla ME-180 | No | 543 | Grady et al., 2005 (116) |
| BIX02188 | HeLa | Yes | 1.15 | Takate et al., 2008 (117) |
| HEK293 Cells | Yes | 0.82 | Takate et al., 2008 (117) | |
| BIX02189 | HeLa | Yes | 0.53 | Takate et al., 2008 (117) |
| HEK293 Cells | Yes | 0.26 | Takate et al., 2008 (117) | |
| XMD8-92 | HeLa | Yes | 1.5×103 | Yang et al., 2008 (112) |
Acknowledgments
This work is supported by a grant from NIH (CA125806 to M.E.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005 Sep 1;65(17):7699–706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- 2.Roberts OL, Holmes K, Muller J, Cross DA, Cross MJ. ERK5 and the regulation of endothelial cell function. Biochem Soc Trans. 2009 Dec;37(Pt 6):1254–9. doi: 10.1042/BST0371254. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006 Jun;18(6):753–60. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004 Apr;113(8):1138–48. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001 Mar 1;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 6.Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007 Aug;1773(8):1150–60. doi: 10.1016/j.bbamcr.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. 2007 Aug;1773(8):1376–87. doi: 10.1016/j.bbamcr.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Bogoyevitch MA, Court NW. Counting on mitogen-activated protein kinases--ERKs 3, 4, 5, 6, 7 and 8. Cell Signal. 2004 Dec;16(12):1345–54. doi: 10.1016/j.cellsig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Rousseau J, Klinger S, Rachalski A, Turgeon B, Deleris P, Vigneault E, Poirier-Heon JF, Davoli MA, Mechawar N, El Mestikawy S, Cermakian N, Meloche S. Targeted Inactivation of Mapk4 in Mice Reveals Specific Non-Redundant Functions of Erk3/Erk4 Subfamily MAP Kinases. Mol Cell Biol. 2010 Oct 18; doi: 10.1128/MCB.01147-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003 Mar 6;22(9):1381–9. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh FC, Cheng G, Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Res Commun. 2005 Sep 23;335(2):292–9. doi: 10.1016/j.bbrc.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 12.Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009;4(5):e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res. 2007 May 1;67(9):4199–209. doi: 10.1158/0008-5472.CAN-06-3409. [DOI] [PubMed] [Google Scholar]
- 14.Song H, Jin X, Lin J. Stat3 upregulates MEK5 expression in human breast cancer cells. Oncogene. 2004 Oct 28;23(50):8301. doi: 10.1038/sj.onc.1208026. [DOI] [PubMed] [Google Scholar]
- 15.Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001 Jul 27;502(1–2):21–4. doi: 10.1016/s0014-5793(01)02651-5. [DOI] [PubMed] [Google Scholar]
- 16.Ramos-Nino ME, Blumen SR, Sabo-Attwood T, Pass H, Carbone M, Testa JR, Altomare BA, Mossman BT. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am J Resp Cell Mol. 2008 Feb;38(2):209–17. doi: 10.1165/rcmb.2007-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi M, Lee JD. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J Mol Med. 2004 Dec;82(12):800–8. doi: 10.1007/s00109-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 18.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998 Oct 15;395(6703):713. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 19.Buschbeck M, Ullrich A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J Biol Chem. 2005 Jan 28;280(4):2659–67. doi: 10.1074/jbc.M412599200. [DOI] [PubMed] [Google Scholar]
- 20.Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995 Aug 15;213(2):715–24. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 21.Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006 Aug;7(8):782–6. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995 May 26;270(21):12665. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 23.Carvajal-Vergara X, Tabera S, Montero JC, Esparis-Ogando A, Lopez-Perez R, Mateo G, Gutierrez N, Parmo-Cabanas M, Teixido J, San Miguel JF, Pandiella A. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005 Jun 1;105(11):4492–9. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- 24.Zhou C, Nitschke AM, Xiong W, Zhang Q, Tang Y, Bloch M, Elliott S, Zhu Y, Bazzone L, Yu D, Weldon CB, Schiff R, McLachlan JA, Beckman BS, Wiese TE, Nephew KP, Shan B, Burow ME, Wang G. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008;10(6):R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, Beckman BS, Burow ME. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002 Aug;132(2):293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 26.Gee JM, Howell A, Gullick WJ, Benz CC, Sutherland RL, Santen RJ, Martin LA, Ciardiello F, Miller WR, Dowsett M, Barrett-Lee P, Robertson JF, Johnston SR, Jones HE, Wakeling AE, Duncan R, Nicholson RI. Consensus statement. Workshop on therapeutic resistance in breast cancer: impact of growth factor signalling pathways and implications for future treatment. Endocr Relat Cancer. 2005 Jul;12(Suppl 1):S1–7. doi: 10.1677/erc.1.01054. [DOI] [PubMed] [Google Scholar]
- 27.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Jr, Marks JR, Dressman HK, West M, Nevins JR. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006 Jan 19;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 28.Flaherty PT, Chopra I, Jain P, Yi S, Allen E, Cavanaugh J. Identification of benzimidazole-based inhibitors of the mitogen activated kinase-5 signaling pathway. Bioorg Med Chem Lett. 2010 May 1;20(9):2892–6. doi: 10.1016/j.bmcl.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 29.Tanoue T, Nishida E. Molecular recognitions in the MAP kinase cascades. Cell Signal. 2003 May;15(5):455–62. doi: 10.1016/s0898-6568(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 30.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999 Jan;79(1):143–80. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 31.Yang SH, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene. 2003 Nov 27;320:3–21. doi: 10.1016/s0378-1119(03)00816-3. [DOI] [PubMed] [Google Scholar]
- 32.Kuida K, Boucher DM. Functions of MAP kinases: insights from gene-targeting studies. J Biochem. 2004 Jun;135(6):653–6. doi: 10.1093/jb/mvh078. [DOI] [PubMed] [Google Scholar]
- 33.Charlson AT, Zeliadt NA, Wattenberg EV. Extracellular signal regulated kinase 5 mediates signals triggered by the novel tumor promoter palytoxin. Toxicol Appl Pharmacol. 2009 Dec 1;241(2):143–53. doi: 10.1016/j.taap.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999 Sep 10;274(37):26563. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 35.Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999 Dec 17;274(51):36035. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 36.English JM, Vanderbilt CA, Xu S, Marcus S, Cobb MH. Isolation of MEK5 and differential expression of alternatively spliced forms. J Biol Chem. 1995 Dec 1;270(48):28897. doi: 10.1074/jbc.270.48.28897. [DOI] [PubMed] [Google Scholar]
- 37.Sun W, Kesavan K, Schaefer BC, Garrington TP, Ware M, Johnson NL, Gelfand EW, Johnson GL. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001 Feb 16;276(7):5093. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- 38.Obara Y, Nakahata N. The signaling pathway leading to extracellular signal-regulated kinase 5 (ERK5) activation via G-proteins and ERK5-dependent neurotrophic effects. Mol Pharmacol. 2010 Jan;77(1):10–6. doi: 10.1124/mol.109.060236. [DOI] [PubMed] [Google Scholar]
- 39.Fukuhara S, Marinissen MJ, Chiariello M, Gutkind JS. Signaling from G protein-coupled receptors to ERK5/Big MAPK 1 involves Galpha q and Galpha 12/13 families of heterotrimeric G proteins. Evidence for the existence of a novel Ras AND Rho-independent pathway. J Biol Chem. 2000 Jul 14;275(28):21730–6. doi: 10.1074/jbc.M002410200. [DOI] [PubMed] [Google Scholar]
- 40.Nakaoka Y, Nishida K, Fujio Y, Izumi M, Terai K, Oshima Y, Sugiyama S, Matsuda S, Koyasu S, Yamauchi-Takihara K, Hirano T, Kawase I, Hirota H. Activation of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ Res. 2003 Aug 8;93(3):221–9. doi: 10.1161/01.RES.0000085562.48906.4A. [DOI] [PubMed] [Google Scholar]
- 41.Pearson G, English JM, White MA, Cobb MH. ERK5 and ERK2 cooperate to regulate NF-kappaB and cell transformation. J Biol Chem. 2001 Mar 16;276(11):7927. doi: 10.1074/jbc.M009764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura K, Johnson GL. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J Biol Chem. 2003 Sep 26;278(39):36989. doi: 10.1074/jbc.C300313200. [DOI] [PubMed] [Google Scholar]
- 43.Seyfried J, Wang X, Kharebava G, Tournier C. A novel mitogen-activated protein kinase docking site in the N terminus of MEK5alpha organizes the components of the extracellular signal-regulated kinase 5 signaling pathway. Mol Cell Biol. 2005 Nov;25(22):9820. doi: 10.1128/MCB.25.22.9820-9828.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Thakur A, Sun Y, Wu J, Biliran H, Bollig A, Liao DJ. Synergistic effect of cyclin D1 and c-Myc leads to more aggressive and invasive mammary tumors in severe combined immunodeficient mice. Cancer Res. 2007 Apr 15;67(8):3698–707. doi: 10.1158/0008-5472.CAN-06-4000. [DOI] [PubMed] [Google Scholar]
- 45.Xia Y, Wu Z, Su B, Murray B, Karin M. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 1998 Nov 1;12(21):3369–81. doi: 10.1101/gad.12.21.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan C, Luo H, Lee JD, Abe J, Berk BC. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001 Apr 6;276(14):10870. doi: 10.1074/jbc.M009286200. [DOI] [PubMed] [Google Scholar]
- 47.Kondoh K, Terasawa K, Morimoto H, Nishida E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol Cell Biol. 2006 Mar;26(5):1679–90. doi: 10.1128/MCB.26.5.1679-1690.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.English JM, Pearson G, Hockenberry T, Shivakumar L, White MA, Cobb MH. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem. 1999 Oct 29;274(44):31588. doi: 10.1074/jbc.274.44.31588. [DOI] [PubMed] [Google Scholar]
- 49.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. Embo J. 1997 Dec 1;16(23):7054. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terasawa K, Okazaki K, Nishida E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells. 2003 Mar;8(3):263. doi: 10.1046/j.1365-2443.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura K, Uhlik MT, Johnson NL, Hahn KM, Johnson GL. PB1 domain-dependent signaling complex is required for extracellular signal-regulated kinase 5 activation. Mol Cell Biol. 2006 Mar;26(6):2065–79. doi: 10.1128/MCB.26.6.2065-2079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitmarsh AJ, Davis RJ. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci. 1998 Dec;23(12):481–5. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- 53.Brown MD, Sacks DB. Compartmentalised MAPK pathways. Handb Exp Pharmacol. 2008186:205–35. doi: 10.1007/978-3-540-72843-6_9. [DOI] [PubMed] [Google Scholar]
- 54.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009 Apr;21(4):462–9. doi: 10.1016/j.cellsig.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci USA. 2009 Jul 7;106(27):11022–7. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev. Mol Cell Biol. 2005 Nov;6(11):827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 57.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 58.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005 Sep 22;437(7058):574–8. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Johnson GL. Noncanonical function of MEKK2 and MEK5 PB1 domains for coordinated extracellular signal-regulated kinase 5 and c-Jun N-terminal kinase signaling. Mol Cell Biol. 2007 Jun;27(12):4566–77. doi: 10.1128/MCB.00125-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000 Feb;2(2):110–6. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 61.Suzaki Y, Yoshizumi M, Kagami S, Koyama AH, Taketani Y, Houchi H, Tsuchiya K, Takeda E, Tamaki T. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: potential role in cell survival following oxidative insults. J Biol Chem. 2002 Mar 15;277(11):9614. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- 62.Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci. 2001 Oct;4(10):981–8. doi: 10.1038/nn720. [DOI] [PubMed] [Google Scholar]
- 63.Liu L, Cavanaugh JE, Wang Y, Sakagami H, Mao Z, Xia Z. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Natl Acad Sci USA. 2003 Jul 8;100(14):8532–7. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci. 2001 Jan 15;21(2):434. doi: 10.1523/JNEUROSCI.21-02-00434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang X, Merritt AJ, Seyfried J, Guo C, Papadakis ES, Finegan KG, Kayahara M, Dixon J, Boot-Handford RP, Cartwright EJ, Mayer U, Tournier C. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol Cell Biol. 2005 Jan;25(1):336. doi: 10.1128/MCB.25.1.336-345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishimoto S, Kusakabe M, Nishida E. Requirement of the MEK5-ERK5 pathway for neural differentiation in Xenopus embryonic development. EMBO Rep. 2005 Nov;6(11):1064. doi: 10.1038/sj.embor.7400515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Obara Y, Yamauchi A, Takehara S, Nemoto W, Takahashi M, Stork PJ, Nakahata N. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J Biol Chem. 2009 Aug 28;284(35):23564–73. doi: 10.1074/jbc.M109.027821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pi X, Garin G, Xie L, Zheng Q, Wei H, Abe J, Yan C, Berk BC. BMK1/ERK5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ Res. 2005 Jun 10;96(11):1145. doi: 10.1161/01.RES.0000168802.43528.e1. [DOI] [PubMed] [Google Scholar]
- 69.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004 Feb 20;94(3):362. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 70.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001 Feb;21(3):952–65. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001 Mar 23;276(12):8631–4. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 72.Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum-and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003 Feb 21;278(8):5871–82. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 73.Dong F, Gutkind JS, Larner AC. Granulocyte colony-stimulating factor induces ERK5 activation, which is differentially regulated by protein-tyrosine kinases and protein kinase C. Regulation of cell proliferation and survival. J Biol Chem. 2001 Apr 6;276(14):10811. doi: 10.1074/jbc.M008748200. [DOI] [PubMed] [Google Scholar]
- 74.Buse P, Tran SH, Luther E, Phu PT, Aponte GW, Firestone GL. Cell cycle and hormonal control of nuclear-cytoplasmic localization of the serum- and glucocorticoid-inducible protein kinase, Sgk, in mammary tumor cells. A novel convergence point of anti-proliferative and proliferative cell signaling pathways. J Biol Chem. 1999 Mar 12;274(11):7253–63. doi: 10.1074/jbc.274.11.7253. [DOI] [PubMed] [Google Scholar]
- 75.Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003 Aug 21;22(35):5387. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]
- 76.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007 Apr;6(4):273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 77.Holmes DI, Zachary IC. Vascular endothelial growth factor regulates stanniocalcin-1 expression via neuropilin-1-dependent regulation of KDR and synergism with fibroblast growth factor-2. Cell Signal. 2008 Mar;20(3):569–79. doi: 10.1016/j.cellsig.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 78.Chiariello M, Marinissen MJ, Gutkind JS. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol Cell Biol. 2000 Mar;20(5):1747–58. doi: 10.1128/mcb.20.5.1747-1758.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stressmediated activation of big mitogen-activated protein kinase 1. J Biol Chem. 1997 Aug 15;272(33):20389–94. doi: 10.1074/jbc.272.33.20389. [DOI] [PubMed] [Google Scholar]
- 80.Barros JC, Marshall CJ. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J Cell Sci. 2005 Apr 15;118(Pt 8):1663. doi: 10.1242/jcs.02308. [DOI] [PubMed] [Google Scholar]
- 81.Scapoli L, Ramos-Nino ME, Martinelli M, Mossman BT. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004 Jan 22;23(3):805–13. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- 82.Schramp M, Ying O, Kim TY, Martin GS. ERK5 promotes Src-induced podosome formation by limiting Rho activation. J Cell Biol. 2008 Jun 30;181(7):1195–210. doi: 10.1083/jcb.200801078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003 Aug;4(8):657–65. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 84.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004 Aug 6;118(3):277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 85.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004 Jun 25;117(7):927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 86.Arnoux V, Nassour M, L’Helgoualc’h A, Hipskind RA, Savagner P. Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell. 2008 Nov;19(11):4738–49. doi: 10.1091/mbc.E07-10-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parent AE, Choi C, Caudy K, Gridley T, Kusewitt DF. The developmental transcription factor slug is widely expressed in tissues of adult mice. J Histochem Cytochem. 2004 Jul;52(7):959–65. doi: 10.1369/jhc.4A6277.2004. [DOI] [PubMed] [Google Scholar]
- 88.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002 Mar;3(3):155–6. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 89.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002 Jun;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 90.Edme N, Downward J, Thiery JP, Boyer B. Ras induces NBT-II epithelial cell scattering through the coordinate activities of Rac and MAPK pathways. J Cell Sci. 2002 Jun 15;115(Pt 12):2591–601. doi: 10.1242/jcs.115.12.2591. [DOI] [PubMed] [Google Scholar]
- 91.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001 Jun;7(6):1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 92.Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003 Jan 24;112(2):169–80. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 93.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004 Aug;114(4):569–81. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwartz SA, Hernandez A, Mark Evers B. The role of NF-kappaB/IkappaB proteins in cancer: implications for novel treatment strategies. Surg Oncol. 1999 Nov;8(3):143–53. doi: 10.1016/s0960-7404(00)00012-8. [DOI] [PubMed] [Google Scholar]
- 95.Weldon CB, Burow ME, Rolfe KW, Clayton JL, Jaffe BM, Beckman BS. NF-kappa B-mediated chemoresistance in breast cancer cells. Surgery. 2001 Aug;130(2):143–50. doi: 10.1067/msy.2001.115512. [DOI] [PubMed] [Google Scholar]
- 96.Yamashita S, Miyagi C, Fukada T, Kagara N, Che YS, Hirano T. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature. 2004 May 20;429(6989):298–302. doi: 10.1038/nature02545. [DOI] [PubMed] [Google Scholar]
- 97.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003 Apr 18;113(2):207–19. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 98.NCBI FOSL1 FOS-like antigen 1 [ Homo sapiens ] 2010 [updated updated 14-Jul-2010]; Available from: http://www.ncbi.nlm.nih.gov/gene/8061.
- 99.Ramos-Nino ME, Timblin CR, Mossman BT. Mesothelial cell transformation requires increased AP-1 binding activity and ERK-dependent Fra-1 expression. Cancer Res. 2002 Nov 1;62(21):6065–9. [PubMed] [Google Scholar]
- 100.Hoffmann E, Thiefes A, Buhrow D, Dittrich-Breiholz O, Schneider H, Resch K, Kracht M. MEK1-dependent delayed expression of Fos-related antigen-1 counteracts c-Fos and p65 NF-kappaB-mediated interleukin-8 transcription in response to cytokines or growth factors. J Biol Chem. 2005 Mar 11;280(10):9706–18. doi: 10.1074/jbc.M407071200. [DOI] [PubMed] [Google Scholar]
- 101.Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003 Jul;4(1):67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 102.Young MR, Nair R, Bucheimer N, Tulsian P, Brown N, Chapp C, Hsu TC, Colburn NH. Transactivation of Fra-1 and consequent activation of AP-1 occur extracellular signal-regulated kinase dependently. Mol Cell Biol. 2002 Jan;22(2):587–98. doi: 10.1128/MCB.22.2.587-598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ramos-Nino ME, Blumen SR, Sabo-Attwood T, Pass H, Carbone M, Testa JR, Altomare DA, Mossman BT. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am J Respir Cell Mol Biol. 2008 Feb;38(2):209–17. doi: 10.1165/rcmb.2007-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J Cell Physiol. 2009 Apr;219(1):152–61. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008 Feb 1;14(3):627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 106.Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele CM, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008 May;10(5):462–U26. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002 Jan;22(1):270–85. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weldon CB, Parker AP, Patten D, Elliott S, Tang Y, Frigo DE, Dugan CM, Coakley EL, Butler NN, Clayton JL, Alam J, Curiel TJ, Beckman BS, Jaffe BM, Burow ME. Sensitization of apoptotically-resistant breast carcinoma cells to TNF and TRAIL by inhibition of p38 mitogen-activated protein kinase signaling. Int J Oncol. 2004 Jun;24(6):1473–80. [PubMed] [Google Scholar]
- 109.NCBI Entrez Gene. National Institute of Health. 2010 [updated September 1, 2010]; Available from: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene.
- 110.Takahashi N, Saito Y, Kuwahara K, Harada M, Tanimoto K, Nakagawa Y, Kawakami R, Nakanishi M, Yasuno S, Usami S, Yoshimura A, Nakao K. Hypertrophic responses to cardiotrophin-1 are not mediated by STAT3, but via a MEK5-ERK5 pathway in cultured cardiomyocytes. J Mol Cell Cardiol. 2005 Jan;38(1):185–92. doi: 10.1016/j.yjmcc.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 111.Flaherty PT, Chopra I, Jain P, Yi SY, Allen E, Cavanaugh J. Identification of benzimidazole-based inhibitors of the mitogen activated kinase-5 signaling pathway. Bioorganic & Medicinal Chemistry Letters. 2010 May 1;20(9):2892–6. doi: 10.1016/j.bmcl.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 112.Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, 3rd, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010 Sep 14;18(3):258–67. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007 Dec;8(12):1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 114.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002 Jan 25;108(2):165–70. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 115.Normanno N, De Luca A, Maiello MR, Campiglio M, Napolitano M, Mancino M, Carotenuto A, Viglietto G, Menard S. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol. 2006 May;207(2):420–7. doi: 10.1002/jcp.20588. [DOI] [PubMed] [Google Scholar]
- 116.O’Grady M, Raha D, Hanson BJ, Bunting M, Hanson GT. Combining RNA interference and kinase inhibitors against cell signalling components involved in cancer. BMC Cancer. 2005 Oct 3;5 doi: 10.1186/1471-2407-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tatake RJ, O’Neill MM, Kennedy CA, Wayne AL, Jakes S, Wu D, Kugler SZ, Jr, Kashem MA, Kaplita P, Snow RJ. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008 Dec 5;377(1):120–5. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]