

Summary
The high rate of clinical response to protein kinase-targeting drugs matched to cancer patients with specific genomic alterations has prompted efforts to use cancer cell-line (CCL) profiling to identify additional biomarkers of small-molecule sensitivities. We have quantitatively measured the sensitivity of 242 genomically characterized CCLs to an Informer Set of 354 small molecules that target many nodes in cell circuitry, uncovering protein dependencies that: 1) associate with specific cancer-genomic alterations and 2) can be targeted by small molecules. We have created the Cancer Therapeutics Response Portal (www.broadinstitute.org/ctrp) to enable users to correlate genetic features to sensitivity in individual lineages and control for confounding factors of CCL profiling. We report a candidate dependency, associating activating mutations in the oncogene β-catenin with sensitivity to the Bcl2-family antagonist, navitoclax. The resource can be used to develop novel therapeutic hypotheses and accelerate discovery of drugs matched to patients by their cancer genotype and lineage.
Introduction
Insights into cancer genomes and advances in small-molecule science are providing a foundation for future cancer therapeutics—ones linked to genomic alterations present in patients’ cancers. Several drugs that target dependencies acquired by cancers as a result of somatic mutations or translocations are yielding high clinical response rates, although beneficial responses are observed in a fraction of cancer patients and not always durable (Gonzalez de Castro et al., 2013). Current targeted drugs inhibit protein kinases encoded by driver oncogenes or their wild-type alleles directly (‘oncogene dependencies’). It is not known whether similar clinical responses can result from drugs targeting non-oncogenes that become essential for cancer survival or progression in the context of specific genetic features (‘oncogene-induced dependencies’). To accelerate discovery of patient-matched therapies, systematic approaches are needed to identify: 1) the dependencies cancers acquire as a result of specific genetic features, and 2) small-molecule drugs that target the dependencies.
Cancer cell-line profiling has been used to reveal patterns of small-molecule sensitivities across diverse cancer cell lines (CCLs). These efforts initially focused on relating sensitivity to CCL lineage (Shoemaker, 2006), but now increasingly relate sensitivity to genetic and epigenetic features (Barretina et al., 2012; Garnett et al., 2012; Heiser et al., 2012; Larsen et al., 2011; Sharma et al., 2010; Sun et al., 2007). This approach identified dependencies on oncogenic alleles of EGFR and BRAF that are now exploited by targeted cancer therapeutics (McDermott et al., 2007). Manifestation of genetic dependencies in a lineage-restricted manner, for example sensitivity of V600E BRAF melanoma but not colorectal cancers to BRAF-targeting vemurafenib (Prahallad et al., 2012), highlights the need to integrate genetic and lineage features in CCL profiling.
CCL profiling studies have historically been limited in the quantity, diversity, or level of characterization of CCLs and small molecules used. One of the earliest CCL profiling efforts, the NCI-60, probed a set of 59 CCLs from various lineages with now >105 diverse small molecules. While this approach has been valuable for identifying lineage-selective small-molecule sensitivities, the relatively small number of CCLs and limited genomic characterization restricted the usefulness of these data. More recent studies have aimed to address this limitation. One recent study profiled 479 CCLs with significant genomic characterization against 24 anti-cancer drugs (Barretina et al., 2012). A second study profiled 350 CCLs against 130 pre-clinical or clinical anti-cancer agents, though the genomic alterations correlated to sensitivity were limited to ~70 genes (Garnett et al., 2012). In order for genomic and lineage CCL profiling to link cancer genetic alterations systematically with potential drug-targetable dependencies, we need to obtain sensitivity measurements for extensively characterized CCLs against a larger set of small molecules that span a broad array of cell processes.
Here, we provide a resource, the Cancer Therapeutics Response Portal (CTRP; www.broadinstitute.org/ctrp), that enables researchers to analyze relationships between genetic and lineage features of cancer and small-molecule sensitivity. We profiled the sensitivity of 242 CCLs to an Informer Set of small molecules with well-annotated targets and activities that collectively modulate a broader range of cellular processes than is currently being investigated in cancer drug discovery. We correlated the compound-sensitivity measurements of CCLs to their genomic alterations, identifying significant correlations involving 60% of the compounds tested and suggesting candidate dependencies on their targets. We intend the CTRP to be a living resource, incorporating new data over time involving additional CCLs and compound treatments (single agent and combination), and new analyses linking sensitivity to additional types of cellular features.
Results
Creating an interactive resource
Profiling the sensitivity of CCLs to an Informer Set of small-molecule probes
The two main considerations for inclusion of small molecules in the Informer Set were high selectivity for their targets (e.g., rapamycin (Brown et al., 1995)), and/or collective targeting of many distinct nodes in cell circuitry. Compounds having different structures but targeting the same protein (e.g., cyclosporin A and tacrolimus targeting calcineurin (Liu et al., 1991)), and compounds having differential selectivity towards distinct members of a protein family (e.g., histone deacetylases (Pan et al., 2012)), were included to validate that genetic feature/sensitivity correlations can be attributed to dependency on a defined protein target. Compounds in clinical development, with strong selectivity data, or with pharmacokinetic data, were prioritized to enable rapid drug development. The current Informer Set comprises 35 FDA-approved drugs, 54 clinical candidates, and 266 probes (~30% prepared for this project by synthesis, Table S1).
The 242 CCLs (Table S2j), chosen to align with lineages studied by The Cancer Genome Atlas and in published genome-wide RNAi screens (Cheung et al., 2011), are a subset of the Cancer Cell Line Encyclopedia collection of ~1,000 genetically characterized CCLs. Data regarding gene expression, amplifications/deletions, somatic mutations in 1,645 cancer genes, and lineage/histological subtypes are freely available (www.broadinstitute.org/ccle). Each CCL was grown in its preferred media, plated at a density optimized during assay development (Table S2j) and treated with compound at 8 concentrations for 72h. Sensitivity was assayed using CellTiter-Glo to measure cellular ATP levels as a surrogate for cell number and growth. The area under percent-viability curves (AUC) was computed as a metric of sensitivity (Extended Experimental Procedures) since AUC reflects both relative potency and total level of inhibition observed for a compound across CCLs (Table S2g and Figure S1).
Analysis of sensitivity data
The ability of genomic CCL profiling to identify clinically relevant biomarkers of drug response depends on the ability of CCLs to model tumor responses, which cannot be confirmed without patient-response data to the same perturbations. To evaluate the performance of CCLs in this study, we analyzed distributions of AUCs across all compounds to identify trends among various subpopulations (Figure 1A). While most CCLs respond differentially across our Informer Set, we observed that CCLs within specific tissue lineages and suspension CCLs were often more sensitive to many compounds tested (Figure 1B). These observations motivated us to perform analyses of AUC distributions that include all CCLs, as well analyses that exclude specific context-dependent subsets of CCLs to control for potential “confounding factors” (Figure 1C and Figure S1; Extended Experimental Procedures).
Figure 1. Response of CCLs to Informer Set.
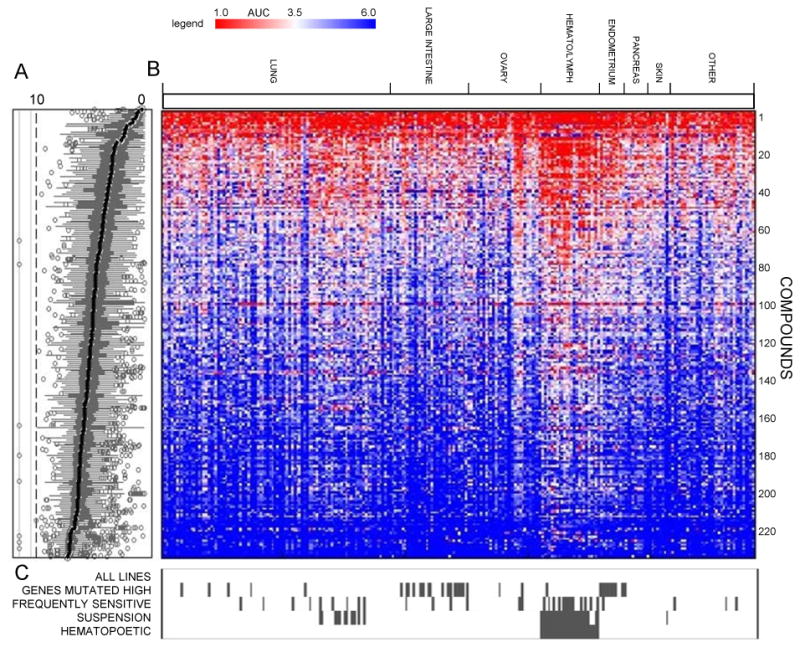
Sensitivity of 242 CCLs to small-molecule probes/drugs was assessed at dose (CellTiterGlo) and areas under the concentration-response curve (AUC) were computed. Data are shown as box plots indicating distributions of AUC values for each compound (A) and a heatmap of AUC values (scale represents AUC values ranging between 1 (sensitive; red) and 6 (unresponsive; blue)) (B) for single CCLs (columns) treated with single compounds (rows). Missing numerical values in heatmap were imputed using a k-nearest neighbors approach. AUC distributions were analyzed by incorporating context-dependent exclusions (C) of cell lines (grey bars represent excluded cell lines). See also Figure S1 and Table S1.
We performed statistics-based enrichment analyses that combined rank-based and parametric tests (Experimental Procedures) to identify genetic alterations and cellular features that are significantly enriched among sensitive (AUC < 3.5) or unresponsive (AUC > 5.5) CCLs. Analyses were performed for each compound across all CCLs and relevant subsets. These correlations are available as a table for download (Table S2 and User Guide S1), and those exceeding a specific threshold of statistical significance are visualized in the CTRP (Extended Experimental Procedures).
Querying the CTRP resource
Validating known dependencies
The resource identified several known mutation/sensitivity relationships, such as the increased sensitivity of BRAF-mutant CCLs to P-0850, an analog of the FDA-approved BRAF-V600E inhibitor, vemurafenib (Smalley, 2010) (Figure 2A). Inspecting P-0850-unresponsive V600E CCLs identified additional features previously associated with resistance to vemurafenib: 1) the unresponsive colorectal CCL, RKO, is reported to produce high levels of hepatocyte growth factor, which activates CRAF via MET in an autocrine fashion and circumvents dependence on BRAF (Corcoran et al., 2011; Straussman et al., 2012); and 2) the unresponsive CCL, SKMEL28, contains an activating mutation in EGFR, and enhanced EGFR signaling has been linked to resistance in vemurafenib-treated colon cancers (Prahallad et al., 2012). These data show the resource can identify candidate resistance mechanisms that may suggest rational combination therapies.
Figure 2. Genetic dependencies targeted by small molecules.
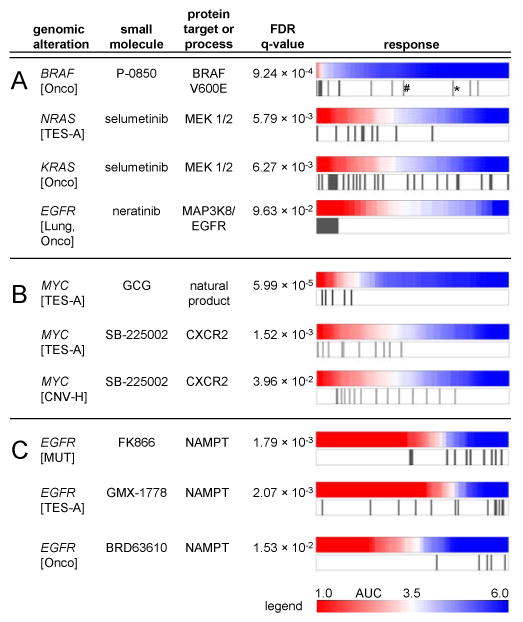
The distribution of CCL response (AUC values) to compound treatment is represented as a heatmap denoting sensitivity (red) or unresponsiveness (blue) aligned with genomic alterations for corresponding CCLs (gray bars). The resource identified known clinically drug-targeted genetic dependencies (A) and known drug-resistance mechanisms (BRAF V600E outlier cell lines: *RKO; #SKMEL28). The resource also suggests dependencies with both mutation and copy number variation in MYC (B). Global analysis of the resource showed EGFR-mutated CCLs are unresponsive to NAMPT inhibitors (C). CNV-H: high-copy number (≥8 copies), TES: all targeted-exome sequencing mutant calls, TES-A: targeted-exome sequencing, non-neutral missense mutations; Onco: Oncomap mutant calls, MUT: any mutation call. See also Figure S2, Table S2, User Guide S1, and Table S3.
The resource identified increased sensitivity of NRAS- and KRAS-mutant CCLs to the MEK1/2 inhibitor, selumetinib, which has shown preliminary moderate activity in clinical trials with KRAS-mutant patients (Janne et al., 2012; National Cancer Institute, 2000; Yoon et al., 2011) (Figure 2A). Several mutant CCLs are unresponsive to selumetinib, suggesting that KRAS/NRAS may be one of several factors determining the response. Analysis of unresponsive outliers may reveal additional features modifying the response to selumetinib.
In some cases, genetic features correlated better with small-molecule sensitivity in the context of specific lineages. We observed that EGFR-mutant lung CCLs were highly sensitive to neratinib, a dual ERBB2/EGFR inhibitor (Figure 2A) (Arteaga, 2006), currently in Phase II trials for advanced non-small cell lung cancer.
Mining for new dependencies
The CTRP suggests dependencies involving oncogenes for which targeted therapies are lacking. We observed that CCLs with MYC mutations, including those interfering with MYC protein degradation (Vervoorts et al., 2006), had increased sensitivity to (-)-gallocatechin-3-monogallate (GCG), a green tea-derived natural product (Figure 2B). Previous studies report that treatment of digestive tract-derived CCLs or mouse tumor models with epigallocatechin-3-monogallate, a GCG analog, led to decreased MYC expression (Ju et al., 2005; Ran et al., 2005). We also observed that mutations in MYC and, to a lesser degree, amplifications of MYC (Figure 2B) correlated with sensitivity to SB-225002, an inhibitor of a chemokine receptor (CXCR2) implicated in promoting oncogene-induced senescence (Acosta et al., 2008). Though the relationship of MYC and SB-225002-targeted biology is not understood, the correlation of sensitivity to SB-225002 with two different types of genomic alterations in MYC supports a potential connection.
We also identified small molecules with strong potency against CCLs of a specific lineage. Although they display a range of sensitivity, ovarian CCLs were among the most sensitive to two probes (ML210 and RSL3; Figure 3A) identified for their ability to kill oncogenically engineered cell lines selectively, BJeLR (HRasG12V, SV40 large T and small T antigens) and DRD (HRasG12V, hTERT, SV40 small T oncoprotein, dominant negative p53, cyclin D1, and mutant CDK4), relative to untransformed controls (Weiwer et al., 2012). Mutations in HRAS did not correlate with sensitivity to RSL3 or ML210 in CCLs used in our study. We confirmed the potency of ML210 against five ovarian CCLs (IC50 ~10 nM), including three not previously profiled, using sulforhodamine B to detect cellular protein content as an assay for cytotoxicity (Skehan et al., 1990) (Figure 3B). Treatment of SKOV3 cells with ML210 also increased expression of the DNA-damage marker phospho-H2AX (Figure S3B) and levels of cleaved caspase-3 (Figure S3C), a marker of apoptosis, suggesting that ML210 is cytotoxic. Similar results were obtained with ML162, another probe identified in the phenotypic screen that yielded ML210 (Figures S3A,B,C).
Figure 3. Lineage dependencies targeted by small molecules.
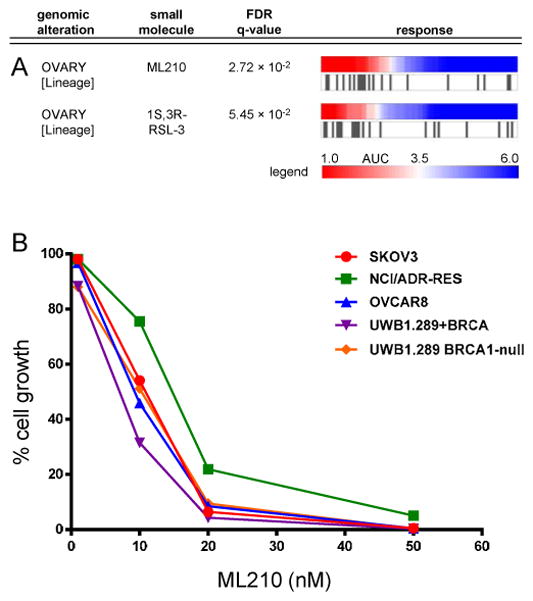
Ovarian CCLs are highly sensitive to ML210 and RSL3 (A). An expanded panel of ovarian CCLs showed sensivity to ML210 (IC50 of ~10 nM) independent of the BRCA1 status of the CCLs (B). See also Figure S3.
RSL3 and related compounds are thought to induce cell death via ferroptosis (Dixon et al., 2012), though they appear to promote markers of apoptosis in this context. It is possible that the sensitive CCLs, whether of ovarian or other lineages, have features in common with the engineered cells described above that render them sensitive to ferroptosis modulators. Testing these compounds in more CCLs and performing multi-feature correlation analyses may help uncover these features.
Global analyses
We determined whether studying gene/compound connections as sets rather than individually could yield insights about dependencies. We limited these analyses to connections having greater statistical significance than those included in Table S2 (Extended Experimental Procedures) to ensure clustering experiments were not dominated by relatively weaker connections. Hierarchical clustering of compounds based on their profile of connections to genetic features (Table S3i) yielded several clusters of compounds that share similar mechanisms of action, including: PRIMA-1 and PRIMA-1-Met (re-activators of mutant p53 signaling), FK866 and GMX-1778 (NAMPT inhibitors), neopeltolide and leucascandrolide A (modulators of respiration), and teniposide and etoposide (topoisomerase inhibitors) (Figure S2).
We also analyzed the frequency with which mutated genes correlate with sensitivity or unresponsiveness to different compounds (Table S3). We found several genes (STK11, EGFR, BRAF) correlated with unresponsiveness to many compounds. EGFR-mutated CCLs were unresponsive to different compounds with the same mechanism of action (NAMPT inhibition; Figure 2C). For example, the top-ranked gene, STK11, has been implicated in resistance to docetaxel in a murine model of KRAS-mutant lung cancer (Chen et al., 2012). Similarly, retrospective clinical analyses indicate patients with BRAF-mutant metastatic colon cancers tend to be non-responsive to EGFR-targeted therapy (Sartore-Bianchi et al., 2009). Our CCL data suggest a possibility that mutations driving certain cancers may lead to unresponsiveness to a wide range of small molecules.
CCLs with activating mutations in β-catenin are more sensitive to navitoclax
The resource suggests oncogene-induced dependencies involving oncogenic alleles of the transcription factor β-catenin (CTNNB1) and alterations in genes encoding proteins that regulate β-catenin stability. CTNNB1 is mutated in several cancer types, yet no targeted treatment has been identified. Activating mutations in the CTNNB1 degradation box (amino acids 32–45) are known to interfere with its phosphorylation and proteasomal degradation, leading to aberrant increases in protein levels (Sparks et al., 1998).
We found that CTNNB1-mutant CCLs were among those most sensitive to navitoclax (Figure 4A), an inhibitor of anti-apoptotic BCL2 family members (BCL-xL, BCL2, BCL-w, but not MCL-1 or BFL-1/A1) previously studied in clinical trials (Gandhi et al., 2011). In studying other proteins regulating β-catenin degradation (APC, AXIN1, CSNK1A1, GSK3B, βTRC), we found that alterations in AXIN1 and CSNK1A1 also correlate with sensitivity to navitoclax (Figure 4A). Collectively, these functionally related alterations account for 37% of the CCLs most sensitive to navitoclax, suggesting alterations increasing β-catenin levels or activity may create a dependency of cancer cells on BCL2 family members for survival (Figure 4B). Our results are consistent with a recent study showing the level of β-catenin pathway activity in CCLs correlates with sensitivity to knockdown of BCL2L1 (encodes BCL-xL) (Rosenbluh et al., 2012). Increased β-catenin activity has also been linked to enhanced expression of BCL2 and BCL2L1 (Kaga et al., 2006; Rosenbluh et al., 2012) and to suppression of BAX-mediated apoptosis (Wang et al., 2009). We do not observe a correlation between CTNNB1 mutation and BCL-2, BCL-xL, or BCL-W protein levels (Figure S4A). We note that in some lineages, unresponsive CCLs lacking CTNNB1 mutations have increased MCL-1 protein levels, which is reported to confer resistance to navitoclax (Tahir et al., 2010). We also observed that correlation between MCL1 gene expression and unresponsiveness to navitoclax ranked highly (top 2%) compared to all other genes (Table S5; Extended Experimental Procedures).
Figure 4. Mutations in β-catenin associate with sensitivity to navitoclax.

Activating mutations in β-catenin (CTNNB1) or mutations in members of its destruction complex (AXIN1; CSNK1A1) correlate with sensitivity to navitoclax (A). Previous studies have linked the Wnt/β-catenin pathway to expression of Bcl2 family members (B). An elastic-net regression model (black circles: observed; red crosses: predicted; weighted root-mean-squared error: 1.45) predicts AUC sensitivity values across CCLs treated with navitoclax (C). Heatmap depicts model features (rows; e.g., mutation, copy number) sorted by descending weight (black bars) across all CCLs tested (columns). Scale represents range of normalized values between -3 and 3 (red: relative higher copy number, presence of mutation; blue: relative lower copy number). All model features (Table S4) were input to Ingenuity Pathway Analysis (Jimenez-Marin et al., 2009) and the highest-scoring network (D) contains β-catenin as a central node (p=10−38). The network contains members of the β-catenin pathway present in the regression model (brown), other genes present in the model (dark grey), and molecular interactions with non-regression-model features (light grey). See also Table S4 and Table S5.
Analytical and experimental confirmation in CCLs
We analyzed the sensitivity data using an orthogonal analytical approach, elastic-net regression (Zhu and Hastie, 2004), that aims to identify a parsimonious model that best predicts response to navitoclax. This analysis used both somatic mutation and copy-number data for each gene as candidate predictive features. Consistent with enrichment analysis, mutation of CTNNB1 is among the top-ranked features in predicting sensitivity to navitoclax (Figure 4C and Table S4). To assess model performance using ten-fold cross validation, we calculated a root-mean-squared error between the predicted and observed sensitivities and compared this value to the day-to-day variability of AUCs in our profiling data (AUC=0.98, 90th percentile). Our estimated prediction error (1.45) is greater but comparable to the biological replicate variability of AUCs. Ingenuity Pathway Analysis (IPA) (Jimenez-Marin et al., 2009) of the full list of predictive features revealed that a majority of the genes in our model are directly linked via interactions annotated in the Ingenuity knowledge base. Our highest-scoring IPA network (p=10−38) identified β-catenin as a centrally connected node in the network, and links it to destruction complex members CSNK1A1 and APC, as well as anti-apoptotic BCL2, a target of navitoclax (Figure 4D).
To confirm the relative sensitivity of CTNNB1-mutant CCLs observed in the large-scale profiling data, we re-tested navitoclax in a subset of CCLs. We first examined a panel of lineage-matched non-mutant and CTNNB1-mutant CCLs and confirmed that mutant CCLs had increased CTNNB1 protein levels (Figure S4A) (Sparks et al., 1998) and AXIN2 expression levels (Figure S4B) (Jho et al., 2002). We then re-tested the sensitivity of seven CTNNB1-mutant and four non-mutant CCLs to 72-hr treatment with navitoclax (CellTiterGlo); the AUCs were similar to those in our profiling data (Figure 5A). We also found that the CCLs most sensitive to navitoclax elicted the largest increase in caspase 3/7 activation, as measured by Caspase-Glo, indicating that loss of viability resulted from apoptosis (Figure 5B) (Tse et al., 2008). We also tested five previously untested CCLs with CTNNB1 mutations in the degradation box and their sensitivity to navitoclax was similar to the original CTNNB1-mutant lines (Figure 5C). These data support our hypothesis that mutations in CTNNB1 and alterations in its destruction complex are biomarkers for sensitivity to navitoclax.
Figure 5. Confirmation experiments for navitoclax/β-catenin.
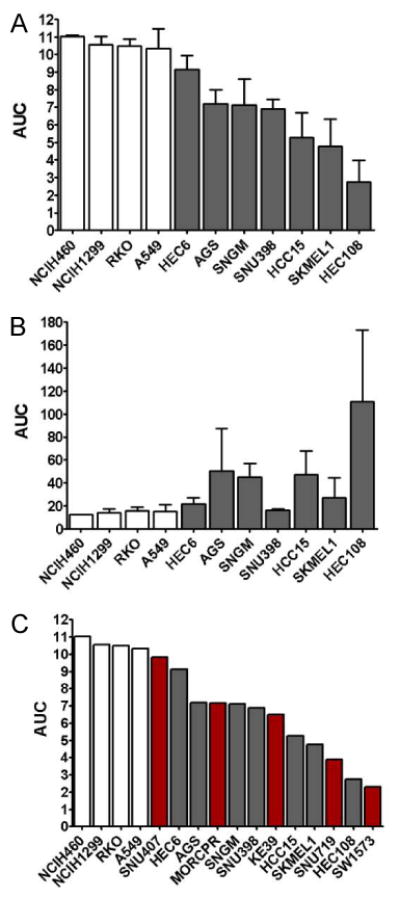
Response to navitoclax observed in large-scale profiling was confirmed in 3 independent experiments (A) with the 7 most sensitive CTNNB1-mutant CCLs (gray bars) and 4 control CCLs lacking mutations in CTNNB1 (white bars). In parallel, caspase 3/7 activation after navitoclax treatment was measured (B), showing that loss of viability was due to induction of apotosis. The response of previously untested CTNNB1-mutant CCLs (red bars) to navitoclax was measured using the same conditions from our initial profiling experiments (C). Data are represented as mean +/- SD. See also Figure S4.
Our data suggest that CTNNB1 mutations that increase β-catenin protein levels sensitize cells to navitoclax. We reasoned that small molecules that increase β-catenin protein levels may also sensitize cells to navitoclax in the absence of CTNNB1 mutations. To explore this hypothesis, we tested CCLs for differential changes in β-catenin levels following a 3d treatment with CHIR-99021, a GSK3β inhibitor that prevents phosphorylation and degradation of β-catenin (Bennett et al., 2002). We identified four CCLs for further testing: RKO and HT29, which lack CTNNB1 mutations and showed significant CHIR-99021-induced increase in β-catenin (Figure 6A); HEC59, which lacks CTNNB1 mutations and showed little change in β-catenin in response to CHIR-99021; and SW48, which contains a mutant GSK3β phosphorylation site in CTNNB1 (S33Y) and did not increase β-catenin in response to CHIR-99021 (Figure 6D).
Figure 6. Small-molecule induction of β-catenin levels and sensitivity to navitoclax.
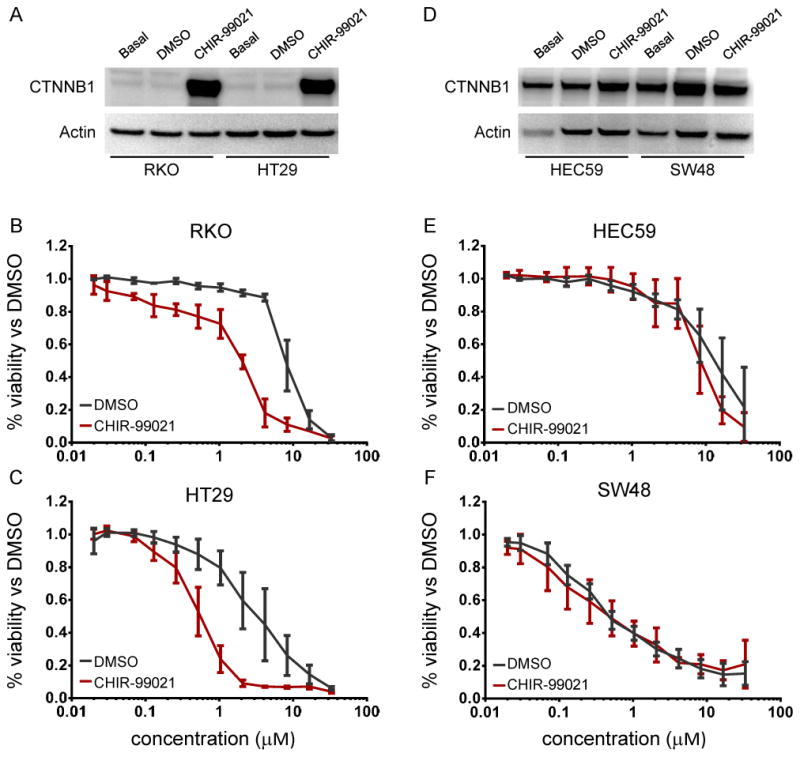
Treatment with the GSK3β inhibitor CHIR-99021 led to increased levels of β-catenin in RKO and HT29 (non-mutant) cells (A) and relatively little change in HEC59 (non-mutant) and SW48 (S33Y CTNNB1 mutant) cells (D). Sensitivity to navitoclax was assessed after pre-treatment with CHIR-99021 (red) and compared to DMSO-pretreated controls (black). RKO (B) and HT29 (C) cells, which had increased levels of β-catenin, showed 4-fold increase in sensitivity to navitoclax, while HEC59 (E) and SW48 (F) cells, which had unchanged levels of β-catenin, demonstrate no significant change in sensitivity. Data are represented as mean +/− SD. See also Figure S5.
Each CCL was pre-treated with CHIR-99021 and then co-treated with both CHIR-99021 and navitoclax. ATP levels were measured as a surrogate for cell viability. The IC50 of both RKO and HT29 response to navitoclax shifted 4-to-8-fold lower (Figures 6B and 6C) after pre-treatment with CHIR-99021 and was more pronounced than after only co-treatment (Figure S5A). Neither CHIR-99021 pre-treatment nor co-treatment significantly increased sensitivity to navitoclax in HEC59 (Figure 6E, Figure S5B) or in SW48 (Figure 6F, Figure S5C). These data suggest that increasing β-catenin levels may correlate with increased sensitivity to navitoclax. We have not determined whether increasing protein levels leads to increased activity, which we observed also correlates to navitoclax sensitivity. Of the 242 CCLs profiled in our study, 41 were tested previously for β-catenin activity in a TCF4 reporter assay (Rosenbluh et al., 2012). Of those 41 CCLs, 8 were sensitive and the remaining 33 unresponsive to navitoclax in our assay. Of the 8 sensitive CCLs, 6 were active in the reporter assay, while only 7 of the 33 unresponsive CCLs were considered active in the reporter assay (p<0.05; data not shown). Thus, compounds that increase β-catenin protein levels may also increase β-catenin activity, rendering them sensitive to navitoclax.
Discussion
Genomic and lineage CCL profiling offers an approach to identify cancer dependencies that are targetable with small molecules, and suggest combinations of compounds that mitigate drug resistance. The Cancer Therapeutic Response Portal (CTRP) suggests candidate dependencies associated with common and medically significant oncogenes. The first version of the CTRP resulted from profiling an Informer Set of small molecules, many of which target non-altered proteins that work in partnership with oncogenes. Exploiting oncogene-induced dependencies contrasts to a related approach based on targeting cell-biological ‘hallmarks’ common to cancers (Hanahan and Weinberg, 2011) without linking these ‘non-oncogene addictions’ to specific genomic alterations (Luo et al., 2009). For example, navitoclax has been tested in phase-I/II clinical trials for small-cell lung cancer (Gandhi et al., 2011); however, our data suggest that navitoclax might best be targeted to patients harboring CTNNB1 mutations, which are present in colorectal, hepatocellular, gastric, and endometrial cancers. We observe that CTNNB1-mutant CCLs are sensitive to navitoclax in several lineages, though more strongly in some (e.g., gastric) than others. The same selectivity was not observed for ABT-199, a BCL-2 specific inhibitor (Souers et al., 2013) (data not shown), suggesting that inhibition of other BCL-2 family members underlies the differential response. Consistently, Rosenbluh et al. recently showed that knockdown of BCL2L1 (BCL-xL) in β-catenin-active CCLs impairs proliferation (Rosenbluh et al., 2012), implicating BCL-xL as a relevant target for navitoclax in CTNNB1-mutant cancers.
Profiling data for single agents may also suggest drug combinations to prevent or overcome drug resistance. By studying the response of BRAF-V600E-mutant CCLs to V600E inhibition, we show how outlier cell lines unresponsive to a small molecule in an otherwise sensitive cohort can reveal additional features that correlate with and confer resistance, in this case upregulation of HGF. Combined treatment with a MET inhibitor to block HGF signaling was sufficient to sensitize these cells to BRAF-V600E inhibition (Corcoran et al., 2011; Straussman et al., 2012). These observations also suggest that correlating small-molecule response to groups of features rather than individual ones may yield biomarkers with greater predictive accuracy.
Since the same oncogene may give rise to different dependencies in different cancer types (e.g., BRAF in melanoma vs. colorectal), the CTRP has been built to allow users to identify dependencies in all CCLs or only in specific lineages. For example, we find that KRAS mutations correlate significantly with sensitivity to navitoclax among colorectal CCLs, but not among all CCLs (data not shown). Interestingly, Corcoran et al. recently showed that navitoclax synergizes with selumetinib to kill KRAS-mutant CCLs in several lineages, but most strongly and consistently in colorectal CCLs (Corcoran et al., 2013).
Corcoran et al. also highlight an important lesson for interpreting CCL profiling data. The authors attribute a lack of efficacy of selumitinib as a single agent in KRAS-mutant tumors to the fact that it is largely cytostatic rather than cytotoxic. Combination with navitoclax, which activates apoptosis, was required for induction of cell death. We note that most CCL profiling data, gathered using a readout for cell growth or proliferation rather than death, may identify gene/sensitivity relationships involving cytostasis; indeed, while KRAS-mutant lines are among those most affected by selumetinib in our study, the compound only leads to partial inhibition of ATP levels, suggestive of cell growth inhibition. Corcoran et al. describe a screening approach for how cytostatic compounds with selectivity for specific cancer genotypes might be exploited in combination strategies to achieve greater efficacy. It also suggests the importance of considering level of inhibition in analyses of existing data, and motivates incorporation of scaleable assays for cell death in future data collection.
The CTRP currently associates small-molecule sensitivity with individual features. In some cases, multiple features associated with sensitivity co-occur in the same CCLs, making it challenging to interpret if an associated feature is causal. For example, we observe that hematopoietic/lymphoid CCLs are more sensitive than those from other lineages to many compounds, including the BRD4 inhibitor JQ-1. MYC-mutant CCLs are among those most sensitive to JQ-1, but they are also frequently of hematopoietic/lymphoid origin, making it difficult to assess whether the genetic or lineage feature is the key determinant of sensitivity. While in this example a larger set of mutant lines will be needed to study MYC mutations separately within hematopoetic/lymphoid and solid tumor CCLs, the CTRP has been built to allow users to perform this analysis in general; for example, MYC amplification associates with sensitivity to SB-225002 whether all CCLs are analyzed or only those from solid cancers.
While CCLs have a long history as models for human cancer, their use in large-scale genomic CCL profiling has emerged more recently. Decisions associated with selection of CCLs, growth conditions, data collection (e.g., assay choice), data filtering (e.g., for possible confounding CCLs), data analysis, and formulation of questions in controlled computational experiments may contribute to differences in results and interpretation of existing profiling studies. For example, this study correlated CTNNB1 mutations with sensitivity to navitoclax, while Garnett et al. correlated sensitivity to navitoclax with NOTCH1 mutations. However, the portal from Garnett et al. suggests that CTNNB1 mutations correlate with sensitivity to TW-37, a pan-Bcl-2 family inhibitor. Despite such differences, both studies identify several similar mutation/sensitivity connections including both established (e.g., KRAS-NRAS/selumetinib, BRAF/V600E inhibitors) and novel associations (e.g., CDKN2A/GW-843682X, a PLK1 inhibitor), as well as features associated with unresponsiveness to compounds (e.g., TP53/nutlin-3). Encouragingly, there are no examples of targeted cancer drugs today that were not predicted by previous genomic CCL profiling studies.
The CTRP is available online (www.broadinstitute.org/ctrp) and the primary sensitivity data underlying the resource can be downloaded from the NCI-CTD2 data portal (http://ctd2.nci.nih.gov). Genetic feature data for CCLs tested can be downloaded from the Broad/Novartis CCLE portal (http://www.broadinstitute.org/ccle).
The CTRP is expected to evolve to include additional data and analyses as they become available. We expect associations identified with the current dataset to change in strength as new lines are examined, and entirely new associations to be uncovered. We are extending this approach to test new probes and drugs, including compounds with novel physical and biological properties, or rationally selected combinations of compounds, across a larger set of CCLs. We are also undertaking systematic analyses to correlate sensitivity to combinations of cellular features, as well as other types of features, including gene expression, signatures of pathway activity (Liberzon et al., 2011), and activity of master regulators inferred computationally (Lefebvre et al., 2010). Further CCL annotations, such as metabolic, proteomic, and epigenetic profiles, will enable additional types of predictive biomarkers to be identified. Our hope is that the cancer biology community will use the CTRP to identify hypotheses for deeper investigation and to accelerate discovery of patient-targeted therapies with better treatment outcomes.
Experimental Procedures
Cancer Cell Line (CCL) Profiling
Frozen cells were obtained from the Broad Institute Biological Samples Platform or ATCC™. CCLs were grown in their specified medium at 37°C/5% CO2. Media were replaced every 2 days. Each CCL was tested for mycoplasma infection (Takara PCR Mycoplasma Detection Set). A list of all CCLs and media conditions is provided (Table S2) and resides on the NCI-CTD2 data portal (http://ctd2.nci.nih.gov).
Cells were plated at a density optimized during assay development (Extended Experimental Procedures) in 384-well opaque, white assay plates and incubated overnight at 37°C/5% CO2. Compound stocks were plated in 384-well format in 8-pt, 2-fold concentration ranges defined by literature review. Compounds were pin-transferred (CyBio Vario) into duplicate assay plates and incubated for 72h. ATP levels were measured using CellTiter-Glo as a surrogate for cell viability.
Assembling the Informer Set
354 small molecules that perturb targets and processes on which cancer cells may become dependent were identified by careful evaluation of the probe-development literature including seminars, journals, NIH Molecular Libraries Initiative Probe Reports, and patents. ~30% of the Informer Set was accessed through organic synthesis. A list of all compounds, with annotated targets and structures, is provided (Table S1) and resides on the NCI-CTD2 data portal (http://ctd2.nci.nih.gov).
Data processing
At each compound concentration, we computed a percent-viability score relative to the effect observed for vehicle-control (DMSO) treatment of the same CCL. Concentration-response curves using percent-viability scores were fit using cubic splines and areas under percent-viability curves (AUC) were computed used as a measure of sensitivity for subsequent analyses (Extended Experimental Procedures).
Genetic Data
Our analyses use publicly available annotations of CCLs, including: gene expression (Affymetrix GeneChip Human Genome U133 Plus 2.0 Array), copy number (Affymetrix Genome-Wide Human SNP Array 6.0), and mutation status from massively parallel sequencing of >1,600 genes and from mass spectrometric genotyping (OncoMap) for 492 mutations in 33 oncogenes/tumor suppressors (Barretina et al., 2012). To illustrate the genetic diversity of the CCLs, we report frequency distributions of the number of mutant genes across the number of CCLs, and the number of unique lesions for each gene (Figure S1).
Enrichment and regression analysis
For each compound, profiling across CCLs yielded a ranked list of sensitivities (AUCs) that could be analyzed for genetic features correlating with the response. For each compound, we used a sorting-based enrichment scoring algorithm (Cormen et al., 2000) to measure how genetic features distribute across the ranked list of sensitivities, followed by a chi-squared test of homogeneity to account for compound potency. The maximum (worst) of the p-values from these two tests was used in subsequent analysis to correct for multiple hypothesis tests, resulting in false-discovery rate (FDR) q-values (Benjamini and Hochberg, 1995). We applied a cutoff of q<0.25 in Table S2, and a more stringent cutoff in the CTRP. For elastic-net regression analysis, we normalized copy number variation, mutation, and lineage features using a z-score (standard normal distribution, with μ=0 and σ=1) for each feature. Elastic net was implemented using Matlab, Python, & R using a core algorithm component from the original authors (Zhu and Hastie, 2004) (Extended Experimental Procedures).
Global analyses of the resource
Global analysis was performed on the subset of connections most robust relative to potential confounding factors. When multiple datasets suggested the same compound-gene connection, the best-scoring connection was retained. Frequency, sum of scores, and average scores for every gene and compound were computed (Table S3). Statistical significance of the number of overlapping genes and compounds (hypergeometric distribution) and a hierarchical clustering of compounds using enrichment scores was performed (Table S3; Extended Experimental Procedures).
Confirming sensitivity of ovarian CCLs to ML210 and ML162
CCLs were plated in 384-well plates at 2000 cells per well in their preferred media and treated with 4 concentrations of ML210 and ML162. Cell number and growth were assayed after 72h treatment using an SRB assay. Results from assays were confirmed using 6 replicates at each compound concentration in each of 3 runs (Extended Experimental Procedures).
Confirming association of CTNNB1 mutation and sensitivity to navitoclax
Four navitoclax-resistant control CCLs and 7 CTNNB1-mutant CCLs were seeded into 384-well plates as during profiling experiments. Caspase 3/7 activity was measured using Caspase-Glo (Promega) after 1.5-h incubation. ATP levels were measured 72h after treatment. Results were confirmed using 8 replicates at each compound concentration in each of 3 runs. Five additional CTNNB1-mutant CCLs were also assayed for sensitivity to navitoclax in a separate run for further comparison (Extended Experimental Procedures).
Induction of β-catenin protein levels and sensitivity to navitoclax
Four CCLs were pre-treated with either DMSO or 4 μM GSK3β inhibitor CHIR-99021, the maximum concentration that did not cause reduction in ATP levels after 3d continuous treatment, as measured by CellTiter-Glo. Cell samples were collected from untreated cells and after 3d of DMSO- or CHIR-99021 treatment, and β-catenin protein levels were assayed by Western blotting (Extended Experimental Procedures).
For pre-treatment experiments, cells were plated overnight, treated with either DMSO or 4 μM CHIR-99021 for 72h, and seeded into 384-well plates with media supplemented with DMSO or CHIR-99021. Cells were incubated overnight, treated with navitoclax in a 12-pt, 2-fold dilution series for 72h, assayed for viability using CellTiter-Glo. All experiments were performed in 8 replicates in each of 2–3 runs. Cells were simultaneously plated for co-treatment experiments under similar conditions (Extended Experimental Procedures).
Supplementary Material
Highlights.
We built a portal to identify cancer genotype/compound sensitivity relationships
We correlated genetic features of cancer cell lines to their response to compounds
We controlled for potential confounding factors of genomic cell-line profiling
We suggest a strategy for treating cancers with mutations in the oncogene β-catenin
Acknowledgments
This work was supported by the NCI’s Cancer Target Discovery and Development Network (RC2-CA148399, awarded to S.L.S.). We acknowledge the following colleagues for contributing compounds and for valuable critique: Drs. D. Adams, A. Beeler, J. Bradner, P. Brown, S. Chattopadhyay, C. Chen, A. Choudhury, J. Clardy, E.J. Corey, M. Dai, K. Hartwell, E. Holson, C. Johannessen, A. Koehler, T. Luo, A. G. Myers, J. Paulk, J. Porco, G. Ramachandran, A. Ramanathan, S. Schaus, K. P. Seiler, M. D. Shair, B. Stockwell, B. Wagner, Q. Wang, and PharmaMar. We thank J. McGrath, G. Wendel and the Broad Compound Management team for handling the Informer Set, D.-K. Jang, A. Li, and M. Reich for supporting web portal development, and the Biological Samples Platform for providing CCLs. The project was enabled by the Broad Institute Chemical Biology Program and Platform. L.A.G. is a consultant for and equity holder in Foundation Medicine, Inc. and received sponsored research from Novartis, Inc. The authors are grateful for the leadership of the CTD2 Network by Daniela Gerhard (Director, Office of Cancer Genomics, NCI). S.L.S. is an Investigator at the Howard Hughes Medical Institute.
Footnotes
Author contributions
Author contributions: C.S.H., B.M., A.M.S., P.A.C., A.F.S., and S.L.S. designed research; A.B., N.E.B., J.H.C., E.V.P., K.L., G.I.S., R.Y.E., M.L.S., D.I., S.W., A.L.B., A.J.W., D.K. and P.A.C. performed research; A.B., N.E.B., J.C.G., T.L., M.W., N.S., G.V.K., J.B., V.D., L.A.G., C.S.H., B.M., and P.A.C. contributed new reagents/analytic tools; A.B., N.E.B, J.H.C., E.V.P., K.L., R.Y.E., G.I.S., M.L.S., D.I., S.W., A.L.B., J.C.G., A.M.S., P.A.C., A.F.S., and S.L.S analyzed data; and A.B., N.E.B, J.H.C., E.V.P., R.Y.E, M.L.S., A.M.S., P.A.C., A.F.S., and S.L.S. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Arteaga CL. EGF receptor mutations in lung cancer: from humans to mice and maybe back to humans. Cancer Cell. 2006;9:421–423. doi: 10.1016/j.ccr.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW, Harrison SD, MacDougald OA. Regulation of Wnt signaling during adipogenesis. The Journal of biological chemistry. 2002;277:30998–31004. doi: 10.1074/jbc.M204527200. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget. 2011;2:336–346. doi: 10.18632/oncotarget.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormen T, Dehne F, Fraigniaud P, Matias Y. ACM Symposium on Parallel Algorithms and Architectures - Guest editors' foreword. Theor Comput Syst. 2000;33:335–335. [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez de Castro D, Clarke PA, Al-Lazikani B, Workman P. Personalized cancer medicine: molecular diagnostics, predictive biomarkers, and drug resistance. Clin Pharmacol Ther. 2013;93:252–259. doi: 10.1038/clpt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heiser LM, Sadanandam A, Kuo WL, Benz SC, Goldstein TC, Ng S, Gibb WJ, Wang NJ, Ziyad S, Tong F, et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A. 2012;109:2724–2729. doi: 10.1073/pnas.1018854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios CH, Franke FA, Grinsted L, Smith PD, Zazulina V, et al. Phase II double-blind, randomized study of selumetinib (SEL) plus docetaxel (DOC) versus DOC plus placebo as second-line treatment for advanced KRAS mutant non-small cell lung cancer (NSCLC) ASCO Meeting Abstracts. 2012;30:7503. [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Molecular and cellular biology. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Marin A, Collado-Romero M, Ramirez-Boo M, Arce C, Garrido J. Biological pathway analysis by ArrayUnlock and Ingenuity Pathway Analysis. BMC Proceedings. 2009;3:S6. doi: 10.1186/1753-6561-3-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J, Hong J, Zhou JN, Pan Z, Bose M, Liao J, Yang GY, Liu YY, Hou Z, Lin Y, et al. Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (−)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer research. 2005;65:10623–10631. doi: 10.1158/0008-5472.CAN-05-1949. [DOI] [PubMed] [Google Scholar]
- Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. Journal of molecular and cellular cardiology. 2006;40:138–147. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Larsen JE, Cascone T, Gerber DE, Heymach JV, Minna JD. Targeted therapies for lung cancer: clinical experience and novel agents. Cancer J. 2011;17:512–527. doi: 10.1097/PPO.0b013e31823e701a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6:377. doi: 10.1038/msb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP- FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Cancer Institute. ClinicalTrials.gov [Internet] Bethesda (MD): National LIbrary of Medicine (US); 2000. Randomized Phase II Study of AZD6244 MEK-Inhibitor With Erlotinib in KRAS Wild Type and KRAS Mutant Advanced Non-Small Cell Lung Cancer. NLM Identifier: NCT01229150. 2000-[cited 2012Jun18] [Google Scholar]
- Pan H, Cao J, Xu W. Selective histone deacetylase inhibitors. Anticancer Agents Med Chem. 2012;12:247–270. doi: 10.2174/187152012800228814. [DOI] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Ran ZH, Zou J, Xiao SD. Experimental study on anti-neoplastic activity of epigallocatechin-3-gallate to digestive tract carcinomas. Chin Med J (Engl) 2005;118:1330–1337. [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani T, Marrapese G, Mazzucchelli L, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS ONE. 2009;4:e7287. doi: 10.1371/journal.pone.0007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nature reviews Cancer. 2010;10:241–253. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nature reviews Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. Journal of the National Cancer Institute. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Smalley KS. PLX-4032, a small-molecule B-Raf inhibitor for the potential treatment of malignant melanoma. Curr Opin Investig Drugs. 2010;11:699–706. [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer research. 1998;58:1130–1134. [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumor microenvironment contributes to innate RAF-inhibitor resistance through HGF secretion. Nature. 2012 doi: 10.1038/nature11183. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. The Journal of clinical investigation. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir SK, Wass J, Joseph MK, Devanarayan V, Hessler P, Zhang H, Elmore SW, Kroeger PE, Tse C, Rosenberg SH, et al. Identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma and leukemia/lymphoma cell lines. Molecular cancer therapeutics. 2010;9:545–557. doi: 10.1158/1535-7163.MCT-09-0651. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. The Journal of biological chemistry. 2006;281:34725–34729. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Havasi A, Gall JM, Mao H, Schwartz JH, Borkan SC. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. J Am Soc Nephrol. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorganic & medicinal chemistry letters. 2012;22:1822–1826. doi: 10.1016/j.bmcl.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer research. 2011;71:445–453. doi: 10.1158/0008-5472.CAN-10-3058. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hastie T. Classification of gene microarrays by penalized logistic regression. Biostatistics. 2004;5:427–443. doi: 10.1093/biostatistics/5.3.427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.