

Abstract
Cancers develop in complex tissue environments, which they depend upon for sustained growth, invasion and metastasis. Unlike tumor cells, stromal cell types within the tumor microenvironment (TME) are genetically stable, and thus represent an attractive therapeutic target with reduced risk of resistance and tumor recurrence. However, specifically disrupting the pro-tumorigenic TME is a challenging undertaking, as the TME has diverse capacities to induce both beneficial and adverse consequences for tumorigenesis. Furthermore, many studies have shown that the microenvironment is capable of normalizing tumor cells, suggesting that reeducation of stromal cells, rather than targeted ablation per se, may be an effective strategy for treating cancer. Here, we will discuss the paradoxical roles of the TME during specific stages of cancer progression and metastasis, and recent therapeutic attempts to re-educate stromal cells within the TME to have anti-tumorigenic effects.
Bidirectional communication between cells and their microenvironment is critical for both normal tissue homeostasis, and for tumor growth. In particular, interactions between tumor cells and the associated stroma represent a powerful relationship that influences disease initiation, progression and patient prognosis1. The link between chronic inflammation and tumorigenesis was first proposed by Rudolf Virchow in 1863, following the observation that infiltrating leukocytes were a hallmark of tumors2. Since then, a plethora of studies have contributed to the characterization of the TME and further complicate the already-challenging task of understanding and treating cancer. Whereas cancer had previously been viewed as a heterogeneous disease involving aberrant mutations in tumor cells, it is now evident that tumors are also diverse by nature of their microenvironmental composition, and stromal cell proportions or activation states3,4. In response to evolving environmental conditions and oncogenic signals from growing tumors, the TME continually changes over the course of cancer progression, underscoring the need to consider TME influences on metastasis as a dynamic process, and to understand how tumor cells drive the construction of their own niche.
Here we discuss current research that demonstrates a crucial role for different stromal compartments during cancer development and metastasis, and recent therapeutic strategies to target the tumor-associated stroma to prevent or regress disease. In light of the breadth and complexity of each step in the invasion-metastasis cascade, and the abundance of microenvironmental influences that play a role during each phase of cancer progression, we have chosen to focus our discussion on specific aspects of the TME during primary tumor growth, survival in the periphery, and secondary outgrowth. We also discuss evidence supporting the extent of interconnectedness within cancers, whereby stromal cells not only signal back and forth to tumor cells, but also to each other, representing the inherent complexity of the TME. For further discussion on topics not covered in extensive detail here, we direct readers to the following recentreviews5–11.
Clinical associations between immune modulation and tumor incidence
One of the most direct pieces of evidence that a deregulated microenvironment impacts tumorigenesis is that tissues subject to chronic inflammation generally exhibit a high cancer incidence12. For instance, hepatocellular carcinoma is the leading cause of death in patients with liver cirrhosis of various etiologies. In a retrospective cohort study of 417 cancer-free patients with cirrhosis, 27% developed liver cancer over ~12 years13. In another large study of 19,486 patients with inflammatory bowel disease, 2841 of which exhibited long-term colitis, the increased risk of colorectal cancer in these two groups was 2.2- and 7.0-fold respectively14. The onset of tumorigenesis in these tumor types is supported by an unresolved inflammatory response, whereby various stromal cell types accumulate, become activated, and their normal function to maintain homeostasis becomes maladaptive, and a pro-tumorigenic niche ensues1,15. Interestingly, a recent meta-analysis found that in ~15% of cancers, tumor initiation can be directly attributed to infection by different etiological agents including viruses, bacteria and parasites16. Moreover, these cancers are associated with chronic inflammation, supporting the growing connection between infection, inflammation and cancer12.
However, it is also critical to note that impaired immune responses can correlate with high cancer incidence. In an analysis of 25,914 female immunosuppressed organ transplant recipients, the observed tumor incidence was higher than predicted for multiple cancers, including lung, GI, reproductive, and skin cancers17. In contrast, breast cancer incidence decreased in this cohort, illustrating the paradoxical nature of immune responses. Furthermore, an analysis of 122,993 individuals with AIDS revealed elevated incidence of not only AIDS-related cancers (e.g. Kaposi’s sarcoma), but also non-AIDS-related cancers (e.g. tongue, skin, lung, CNS, and multiple myeloma)18. Similar retrospective analyses19,20 indicate adequate immune function may be protective against certain cancers, contrasting with evidence supporting pro-tumorigenic functions for inflammation12. This dichotomy underscores the challenges in understanding and therapeutically targeting context-dependent, opposing functions of immune cells in cancer.
Amidst this complexity, therapeutic opportunity lies in the pliancy of the tumor stroma, which imparts strong influences on disease progression. For example, while tumor-associated macrophages exhibit pro-tumorigenic effects in response to stimulation or education by cytokines (e.g. by IL-4, TGF-β, etc.) in many solid tumors, they can also be reprogrammed by various pharmacological agents to exhibit anti-tumor behavior21–26. We can potentially take advantage of this plasticity by reprogramming or re-educating cells in the TME to treat cancer, rather than simply targeting stromal cells for ablation.
Primary growth begins: tissue homeostasis undone
In addition to the clinical association studies discussed above, it is evident that tumorigenesis is indeed modulated by aberrant immune responses and altered homeostasis. In cancer, the coordinated intercellular interactions that are present in normal adult tissues are disrupted as the tumor acquires the capacity to chronically circumvent normalizing cues from the microenvironment, and in turn, the microenvironment evolves to accommodate the growing tumor (Fig. 1)1,27,28. In the following section, we will discuss how the tumor-associated stroma at primary sites is hijacked to support tumor growth, with a focus on the role of macrophages, immune suppressor cells, fibroblasts, the vasculature, and various other components of a tumor-supportive TME.
Figure 1. Multiple stromal cell types converge to support a tumorigenic primary niche.
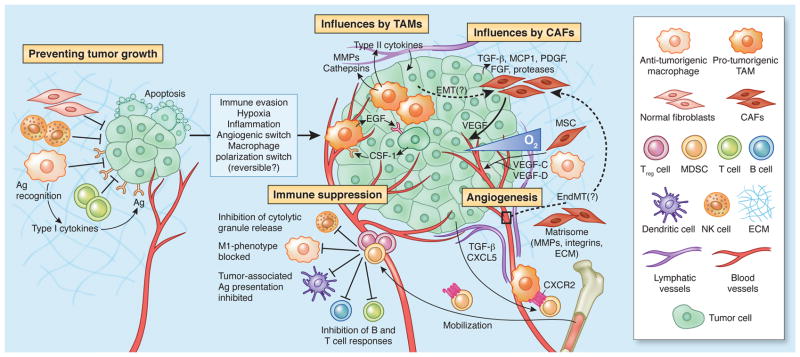
After circumventing cell-intrinsic mechanisms of apoptosis, tumor cells are subject to elimination pressures by the immune system. Tumor cell-specific antigens play a role during this process, which are recognized by cytotoxic immune cells, leading to their destruction. Fibroblasts and macrophages within the TME also contribute to a growth-suppressive state; however, these cells may later become educated by the tumor to acquire pro-tumorigenic functions. For instance, tumor-associated macrophages (TAMs) support diverse phenotypes within the primary tumor including growth, angiogenesis and invasion by secreting a plethora of pro-tumorigenic proteases, cytokines and growth factors (e.g. EGF, which participates in a paracrine signaling loop via tumor-secreted CSF-1). As tumors grow, immune-suppressor cells, including myeloid-derived suppressor cells (MDSCs) and Treg cells are mobilized into circulation in response to activated cytokine axes induced by tumorigenesis (e.g. TGF-β, CXCL5-CXCR2). MDSCs and Treg cells infiltrate the growing tumor to disrupt immune surveillance via multiple mechanisms, including, but not limited to, disruption of antigen presentation by DCs, inhibition of T and B cell proliferation and activation, or inhibition of NK cytotoxicity. Cancer-associated fibroblasts (CAFs), which become activated by tumor-derived factors (e.g. TGF-β, FGF, PDGF, etc), secrete ECM proteins and basement membrane components, regulate differentiation, modulate immune responses, and contribute to deregulated homeostasis. CAFs are also a key source of VEGF, which supports angiogenesis during tumor growth. In addition to cellular contributions, several extracellular properties contribute to tumor progression, including low oxygen tension, high interstitial fluid pressure, and changes in specific components of the ECM.
Macrophage plasticity contributes to tumor growth
Tumor-associated macrophages (TAMs) are important regulators of tumorigenesis that are either tissue-resident, or derived from peripheral reservoirs such as the bone marrow (BM) and spleen. While macrophages are classically regarded as critical effector cells during immune defense, numerous studies have demonstrated a clear role for TAMs in supporting multiple aspects of tumor progression29 (Fig. 1). Perhaps most notable is their role at the leading edge of tumors, where they drive invasive cellular phenotypes30. Indeed, studies in breast cancer and glioma have demonstrated that TAMs facilitate tumor cell invasion via a paracrine signaling loop involving tumor-derived CSF-1 and macrophage-derived EGF30–32. Beyond the leading edge, TAMs are a major source of proteases, such as cysteine cathepsins, which support tumor progression and therapeutic resistance of multiple cancer types33–35.
One explanation for the disparate roles of macrophages during normal tissue homeostasis and tumorigenesis lies in their phenotype. Macrophages are functionally plastic, and can alter their polarization state to accommodate different physiological conditions (Table 1). At the extremes of their phenotypic continuum36, macrophages range from M1 to M2 polarization states: “classically-activated” M1 macrophages produce type I pro-inflammatory cytokines, participate in antigen presentation, and play an anti-tumorigenic role21. Conversely, “alternatively-activated” M2 macrophages produce type II cytokines, promote anti-inflammatory responses, and have pro-tumorigenic functions21. However, it should be noted that while this classification is useful, it is somewhat over-simplified, as it does not fully represent the complexity of macrophage activation, which is often fine-tuned in response to different tissue microenvironments36. Currently, we do not fully understand how macrophages initially switch from tumor-suppressing to tumor-promoting at the onset of disease. It has been suggested that environmental conditions such as tumor hypoxia may mediate this transition. Indeed, TAMs accumulate in regions of hypoxia in growing tumors37, and their recruitment is mediated by an upregulation of macrophage chemoattractants including endothelin-2 and VEGF. Of note, TAM accumulation in these regions correlates with angiogenesis, and the subsequent acquisition of an invasive phenotype37, suggesting that the initial hypoxic response in growing tumors may include a switch in macrophage polarization38.
Table 1.
Immune cell populations in the tumor microenvironment have distinct functions during tumorigenesis.
| Cell type | Markers (Human) | Markers (Mouse) | Function | Refs. |
|---|---|---|---|---|
| Myeloid Lineage | ||||
| TAM | CD11b+ CD68+ CSF1R+ CD163+ EMR1+ |
CD11b+ GR1− CD68+ CSF1R+ F4/80+ |
Classically-activated M1 macrophages are pro-inflammatory and anti-tumorigenic, and secrete TH1 cytokines. Alternatively- activated M2 macrophages are anti-inflammatory and pro- tumorigenic, and secrete TH2 cytokines. TAMs frequently exhibit an M2 phenotype; their presence in tumors supports angiogenesis and invasion. | 1, 29, 36, 44, 57 |
| DC | CD11c+ CD83+ CD123+ |
CD11c+ CD83+ CD123+ |
Dendritic cells (DCs) are monocytic antigen-presenting cells that are derived from the bone marrow. DCs presenting tumor- specific antigens are being developed as vaccines to induce both innate and adaptive immune responses to regress tumors and prevent relapse. | 11, 44, 57, 178 |
| TEM | CD11b+ SCA1+ TIE2+ CD14+ CD16+ |
CD11b+ GR1− SCA1+ TIE2+ |
TIE2 is a receptor for the angiogenic growth factor, angiopoietin. TIE2-expressing monocytes (TEMs) play a role during tumor angiogenesis through a paracrine signaling loop with angiopoietin-expressing endothelial cells. | 1, 10 |
| Neutrophil | CD11b+ CD66b+ CD63+ |
CD11b+ GR1+ 7/4+ |
Neutrophils are the most abundant circulating leukocyte in humans, and are phenotypically plastic. N1 neutrophils are pro-inflammatory and anti-tumorigenic, and secrete TH1 cytokines. N2 neutrophils are anti-inflammatory and pro- tumorigenic, and secrete TH2 cytokines. | 1, 8, 178, 179 |
| Mast cell | CD11b− CD49d+ CD117+ CD203c+ |
CD11b− CD49d+ CD117+ CD203c+ |
Mast cells are best known for their role during allergies and auto-immunity. Mast cells are recruited to tumors where they release factors that enhance proliferation of endothelial cells, to promote tumor angiogenesis. | 1, 9, 178 |
| MDSC | CD11b+ CD33+ HLA-DR- CD14+ (monocytic) CD14− CD15+ (granulocytic) |
CD11b+ GR1+ Ly6G− Ly6C+ (monocytic) Ly6G+ Ly6C− (granulocytic) |
MDSCs are immunosuppressive precursors of dendritic cells, macrophages, and granulocytes. In cancer, their main function is to disrupt tumor immunosurveillance by interfering with T cell activation, cytotoxic activity, antigen presentation, and cell polarization. | 1, 6, 44, 57 |
| Lymphoid Lineage | ||||
| NK cell | CD56+ CD16+ |
CD335+ NK1.1+ |
NK cells are cytotoxic lymphocytes that can kill stressed cells in the absence of antigen presentation. NK cells detect and kill tumor cells through “missing-self” activation (loss of healthy cell markers), or “stress-induced” activation (gain of stressed cell markers). | 1, 57, 180 |
| TH cell | CD3+ CD4+ |
CD3+ CD4+ |
CD4+ helper T (TH) cells can be divided into TH1 and TH2 lineages. TH1 cells secrete pro-inflammatory cytokines and are anti-tumorigenic. TH2 cells secrete anti-inflammatory cytokines and are pro-tumorigenic. The ratio of TH1:TH2 cells in cancer correlates with tumor stage and grade. | 1, 57, 175 |
| Treg cell | CD4+ CD25+ FOXP3+ CTLA-4+ CD45RA+ |
CD4+ CD25+ FOXP3+ CTLA-4+ CD103+ |
Treg cells primarily play pro-tumorigenic roles by suppressing immunosurveillance; however, their presence in tumors is positively correlated with overall survival in certain cancer types. These divergent roles may be attributed to context-dependent functions, or distinct subpopulations that are challenging to identify at present using conventional markers. | 6, 44, 56, 181 |
| TC cell | CD3+ CD8+ |
CD3+ CD8+ |
CD8+ cytotoxic T (TC) cells are effector cells of the adaptive immune system. They specifically recognize and destroy cancer cells through perforin and granzyme-mediated apoptosis. | 1, 57, 182 |
| B cell | CD19+ CD20+ |
B220+ CD19+ CD22+ |
B lymphocytes are important mediators of humoral immunity. In cancer, they can promote disease progression by secreting pro-tumorigenic cytokines and altering TH1:TH2 ratios. Their importance in supporting tumor growth is evident in B cell- deficient mice, which exhibit resistance to engraftment of certain syngeneic tumors. | 1, 44, 183 |
Abbreviations: TAM, Tumor-associated macrophage; TEM, TIE2-expressing monocyte; MDSC, Myeloid-derived suppressor cell; NK cell, Natural killer cell; TC cell, Cytotoxic T cell; TH cell, Helper T cell; Treg cell.
Reversion of TAMs back to an M1 phenotype has also been reported. For example, TAM-specific inactivation of IKKβ, which disrupts NFκB signaling, resulted in an M2-to-M1 switch, recruitment of natural killer (NK) cells and subsequent tumor regression in an ovarian cancer model24. Other studies, using Lewis lung carcinoma (LLC) cells, have implicated TNF-α in regulating this conversion, downstream of Toll-like receptor 3/Toll-IL-1 receptor domain-containing adaptor molecule 1 (TICAM-1)39. Another report on LLC found that miR155 overexpression in TAMs induced re-polarization towards an anti-tumoral M1 state40. We recently showed that macrophage depolarization from an M2 phenotype via CSF-1R inhibition was associated with a robust regression of established high-grade gliomas25. Together these studies highlight a potential therapeutic opportunity, whereby re-educating TME-resident macrophages might have beneficial anti-tumorigenic effects on disease.
Immune suppression by MDSCs and Treg cells
A critical step in the malignant progression of incipient tumors is evasion and suppression of the host immune system6,41 (Fig. 1). This can be achieved through inhibition of various effector immune cells, or via stimulation of immunosuppressive cells (Table 1). One of the most prevalent mechanisms of immune evasion in patients is via the suppressive activity of myeloid-derived suppressor cells (MDSCs)42, which arises as a consequence of the aberrant myelopoiesis that occurs in cancer. MDSCs are functionally defined as immunosuppressive, immature myeloid cells43 that maintain normal tissue homeostasis in response to various systemic insults including infection and traumatic stress. MDSCs are mobilized during tumorigenesis and infiltrate developing tumors where they promote tumor vascularization43 and disrupt major mechanisms of immunosurveillance including antigen presentation by dendritic cells (DCs)44, T cell activation44–46, M1 macrophage polarization47, and inhibition of NK cell cytotoxicity48. The notion that MDSCs promote tumor progression has been demonstrated in several animal models, whereby depletion with various neutralizing antibodies significantly reduces metastasis (reviewed in43), and is supported by the observation that cancer patients exhibit elevated peripheral MDSCs, which positively correlates with advanced disease and therapeutic inefficacy42,49.
Designing therapies that aim to re-educate the immunosuppressive activity of MDSCs is an attractive approach, given that they are composed of mixed subpopulations of cells with varying maturity and plasticity, and can differentiate into multiple cell types. It has been shown in animal models that monocytic MDSCs can be reprogrammed to adopt an anti-tumorigenic phenotype in response to mimicking bacterial stimulation of the immune system50. This transition is accompanied by an increase in pro-inflammatory TH1 cytokines, a reduction in T cell-suppressive factors (e.g. arginase-1, nitric oxide), and differentiation of MDSCs into M1-like macrophages50. These findings suggest that immunomodulatory therapies developed for subverting responses to infection may also be relevant in the context of cancer.
Regulatory T (Treg) cells are another TME cell type that have diverse immune modulatory functions in cancer22,51. Under normal physiological conditions, Treg cells regulate the expansion and activation of T and B cells, and play a critical role in maintaining homeostasis of innate cytotoxic lymphocytes52. Given their complex regulatory roles in response to different environmental stimuli, it is not surprising that Treg cells have diverse effects on tumorigenesis. In some tumor types, including breast cancer and hepatocellular carcinoma, increased Treg cells correlate with reduced overall survival53,54, while in others, such as colorectal cancer, Treg cells are associated with improved survival55. Similar to MDSCs, Treg cells suppress tumor-associated antigen presentation, and also interfere with cytotoxic T cell function by inhibiting cytolytic granule release56.
The mechanisms underlying divergent Treg cell functions in cancer remain elusive; it is not clear if Treg cells exhibit context-dependent functionality, or whether they encompass multiple subpopulations, with distinct functions, that are not differentiated using conventional markers57. Indeed, tumor-associated Treg cell phenotypes are heterogeneous58, suggesting they likely accumulate by various mechanisms such as peripheral recruitment, proliferation of cells in the TME, or differentiation of progenitors in response to tumor-secreted factors. As such, targeting Treg cells via anti-CD25 antibodies or other pharmacological approaches56,59 will likely be most beneficial in the context of improving immunotherapy responses in cancer, similar to MDSCs44.
Cancer-associated fibroblasts elicit pro-tumorigenic functions
Fibroblasts are a predominant, multi-functional cell type in connective tissue, depositing extracellular matrix (ECM) and basement membrane components, regulating differentiation events in associated epithelial cells, modulating immune responses, and mediating homeostasis60,61. In the TME, cancer-associated fibroblasts (CAFs) are present in aberrantly high numbers and are distinct from normal fibroblasts. For example, normal prostate epithelial cells give rise to intraepithelial neoplasia in mice when co-injected with CAFs, but not when co-injected with normal fibroblasts62. Similarly in breast cancer, CAFs confer a mesenchymal-like phenotype and enhance metastasis of both premalignant and malignant mammary epithelial cells, whereas normal fibroblasts promote an epithelial-like phenotype and suppress metastasis63. This highlights the complexity of fibroblast functionality in cancer (Fig. 1), and indicates that CAFs ought to be considered as an entirely different cell type from normal fibroblasts, with potent effects on tumorigenesis.
It is unclear where CAFs arise from during disease progression64. Some studies have suggested that they are generated from endothelial-to-mesenchymal transition (EndMT), whereby tumor-associated endothelial cells delaminate from blood vessels to generate mesenchymal cells with multipotent differentiation potential. Lineage-tracing experiments in mouse melanomas and pancreatic neuroendocrine tumors showed that CAFs in these tumors were of endothelial cell origin65. Epithelial-to-mesenchymal transition (EMT) also promotes the generation of CAFs, whereby tumor cells of epithelial origin (e.g. breast and prostate cancers) dedifferentiate to generate a mesenchymal-like cell population that expresses CAF markers66,67. EMT has also been linked to the generation of fibroblasts that participate in normal tissue homeostasis, for example, in response to chronic injury of renal epithelial cells68.
Once CAFs accumulate in the TME, they are activated by growth factors and cytokines that are present in the surrounding milieu. TGF-β, monocyte chemotactic protein (MCP1), PDGF, FGF, and secreted proteases have all been implicated in CAF activation61,64. In a recent study, induction of the YAP transcription factor was required for the ability of CAFs to remodel ECM to support tumorigenesis69. YAP induction in turn regulates multiple factors that modulate the cytoskeleton and matrix stiffness, which feedback to further enhance YAP production. Following activation, CAFs provide a major source of secreted growth factors that support tumorigenesis, including VEGF, which induces vascular permeability and angiogenesis70. CAFs additionally produce pro-inflammatory factors that activate NFκB signaling to promote tumorigenesis, and a ‘pro-inflammatory’ CAF signature is already evident in pre-neoplastic lesions71. Interestingly, CAFs in the breast TME can select for bone-specific metastatic traits in primary tumor cells, due in part to a selective interaction between breast cancer cells with high Src activity, and primary CAFs that secrete CXCL12 and IGF172. This raises the intriguing possibility that heterotypic signaling in the primary TME enriches for metastatic cells primed to flourish in specific foreign microenvironments, providing further evidence for the interdependency of multiple cell types within the TME.
Extracellular cues influence tumor progression
Beyond the contributions of specific cell types to tumorigenesis, the ECM has a capacity to limit cancer initiation at early stages, and drive disease progression towards malignancy. Indeed, the composition of the extracellular TME is a significant predictor of clinical prognosis. In breast cancer, tumors can be stratified into four subclasses based strictly on ECM composition, which are predictive of patient outcome73. Tumors with high expression of protease inhibitors (e.g. serpin family members) in their ECM are associated with good prognosis, while tumors with high expression of integrins and MMPs correlate with poor prognosis and risk of recurrence73. Different cell types in the TME supply distinct ECM proteins, which has been termed the ‘matrisome’, as identified via proteomics strategies74. Interestingly, primary tumors of diverse metastatic potential differ in composition of both tumor- and stroma-derived ECM components74. Together, these results suggest that disrupting the extracellular environment surrounding and infiltrating tumors may provide an additional level of therapeutic intervention.
The tumor vasculature is supported by the TME and maintains tumor growth
In 1971, Judah Folkman published a revolutionary article proposing that all tumors are angiogenesis-dependent75, initiating a paradigm shift throughout the cancer research community despite initial resistance. Angiogenesis is now accepted as a hallmark of cancer4 in response to a growing need for oxygen and nutrients from the bloodstream, without which tumors would succumb to dormancy. Tumor vascularization requires co-operation of multiple TME cell types, including vascular endothelial cells (which form tight adhesions to ensure vessel integrity), pericytes (which provide vessel coverage and dictate vessel maturity), and BM-derived precursor cells, whose orchestration is often regulated by hypoxia (Fig. 1)5,76,77.
In addition to the cell types comprising the actual vessels, accessory cells including TAMs, mesenchymal stem cells (MSCs), and CAFs, also contribute to tumor vascularization by releasing a plethora of pro-angiogenic signals into the TME. The dichotomous role of MSCs in modulating angiogenesis is particularly intriguing; while co-injection of MSCs and colon cancer cells into mice induced a significant increase in tumor volume and microvascular density78, when MSCs were co-injected with glioma cells this actually prevented vascularization, compared to glioma cells injected with normal astrocytes79. These inhibitory effects were mediated by a reduction of endothelial progenitor cell (EPC) recruitment to gliomas, and a decrease in key signaling molecules involved in gliomagenesis, such as PDGF-BB79. Conversely, in patients with advanced highly-vascularized breast cancer, there is substantial mobilization of MSCs into circulation, which is associated with chemoresistance80. These results, among many others81, demonstrate that MSC functions in tumor progression are also highly contextual, and likely dependent on the stromal composition or impending need for oxygenation.
Lymphangiogenesis is another mode of vascularization in tumors, and lymphatic vessels represent an alternate route for cancer cell dissemination82. Activated macrophages produce VEGF-C and VEGF-D, which correlates with peritumoral inflammation and lymphangiogenesis in human cervical cancer83. Moreover, myeloid cell populations can have critical influences on lymphatic endothelial cells (LECs), not only by modulating their signal transduction, but also by transdifferentiating into functional LEC-like cells. In one retrospective study, tissues were analyzed from patients who received sex-mismatched renal transplants exhibiting transplant rejection, lymphatic activation, and inflammation, and showed incorporation of recipient LEC progenitors into lymphatic vasculature in transplanted tissue84. Similarly, in mouse cancer models, myeloid cell incorporation and transdifferentiation into LECs were reported, mediated via VEGF-C and heparanase, among other factors85,86. Interestingly, the ability of peripheral LEC progenitors to incorporate into newly formed lymphatic vessels is reminiscent of de novo vascularization of tumors by EPC incorporation into vessel walls, and of early embryonic vasculogenesis, highlighting the parallels between these physiological and pathological processes.
Breaking away: cancer cell dissemination and survival in the periphery
Once the primary tumor acquires a capacity to evade host immune defenses and cancer cells enter the circulation, metastatic dissemination is underway. Prior to this event, the primary tumor may have already primed premetastatic sites to be receptive to incoming tumor cells87. Furthermore, recruited cell types that once were destined to destroy the primary tumor, have now been hijacked to facilitate its journey through the body (Fig. 2). In this section we will discuss how the TME supports cancer cells in leaving the primary tumor site and seeding successfully in secondary organs.
Figure 2. The microenvironment supports metastatic dissemination and colonization at secondary sites.

Macrophages, platelets, and mesenchymal stem cells (MSCs) contribute to epithelial-to-mesenchymal transition (EMT) at primary sites, allowing for tumor cells to separate from neighboring epithelial cell-cell contacts and acquire a mobile/invasive phenotype. One major mediator of this event is TGF-β, which is secreted by the tumor stroma and participates in a paracrine signaling loop with tumor cells. TAMs, CAFs and myeloid progenitor cells also tend to cluster at the invasive/leading edge of the primary tumor, where they play an immunosuppressive role by interfering with dendritic cell differentiation. During intravasation of tumor cells into circulation, intravital imaging studies have shown that macrophages are localized to perivascular areas within tumors, where they help tumor cells traverse vessel barriers. In the circulation, platelets and components of the coagulation system support tumor cell survival by protecting them from cytotoxic immune cell recognition. Platelets escort tumor cells in circulation to the site of extravasation, where they bind to areas of vascular retraction and help tumor cells exit circulation into secondary organs. At secondary sites such as the lung, fibroblasts upregulate fibronectin, which serves as a docking site for hematopoietic progenitor cells (HPCs) and the subsequent arrival of tumor cells. Immunosuppressive cell types, such as MDSCs and NK cells, also populate premetastatic niches where they help to direct metastatic dissemination by creating a niche permissive to tumor colonization. Recent studies have demonstrated that primary and secondary sites can communicate through exosomes, shed not only by primary tumor cells but also by immune and stromal cells such as NK cells, CAFs and dendritic cells. Factors contained in exosomes have the capacity to direct organ tropism, modulate immune evasion, support mesenchymal-to-epithelial transition (MET), and are predictive of metastasis and patient outcome.
Stromal influences on phenotypic switching
One of the initiating steps of primary tumor invasion is the EMT, during which tumor cells lose epithelial markers and gain mesenchymal traits that confer stem-like properties and a migratory phenotype88 (Fig. 2). This program recapitulates many processes involved in mammalian development and adult tissue remodeling89, suggesting that tumor-associated EMT is similarly an attempt to reorganize tissue and maintain homeostasis. At later stages of metastasis, however, secondary lesions often display an epithelial-like phenotype, suggesting that this mesenchymal-epithelial transition (MET) is important for metastatic outgrowth90–92. This underscores the importance of phenotypic switching for successful metastasis, rather than EMT per se, and suggests that tumor cells may fluctuate along an epithelial-mesenchymal continuum in response to cues from different environments.
Indeed, several studies have demonstrated the importance of the stroma during phenotypic transition events in cancer, generally by supplying or inhibiting TGF-β89. For example, macrophage accumulation in teratocarcinomas causes EMT due to elevated TAM-derived TGF-β93. EMT can additionally be induced by platelet-tumor cell interactions, via platelet-secreted TGF-β94,95. In gastric carcinoma, the proportion of CD133+ tumor cells is regulated by paracrine TGF-β and Wnt signaling with MSCs96. In breast cancer, BM-derived myeloid progenitors are recruited to the pre-metastatic lung87, where they induce MET of tumor cells through downregulation of SMAD2 signaling (the canonical TGF-β pathway), and a switch to macrometastatic growth90.
Given that many patients rebound after chemotherapy, supposedly due to an inability of chemotherapeutic agents to target “stem-like” cell populations, an understanding of how stromal cell compartments contribute to the acquisition of a mesenchymal phenotype could inspire innovative combination therapies targeting both rapidly-dividing and tumor-initiating cell populations. Alternatively, taking advantage of stromal-mediated epithelialization may be advantageous in combination with chemotherapy, whereby re-programming the premetastatic “soil” may have multiple beneficial consequences for subverting metastasis.
Stromal cells lead the way at the invasive edge
The tumor margin is an important meeting place in the TME where recruited immune and stromal cells are highly active and interactive with the tumor (Fig. 2). Immature myeloid cells accumulate in this region, and prevent differentiation of antigen-presenting DCs, thus supporting tumor immune evasion44. Macrophages are another major cell type at the invasive edge of tumors, and are recruited by tumor-derived chemoattractants30. Upon their arrival, TAMs promote invasion of tumor cells by supplying pro-migratory factors such as EGF, by regulating the production of fibrillar collagen to accelerate tumor motility, and by promoting ECM proteolytic remodeling34,97,98. CAFs are similarly abundant at the tumor margin where they release pro-invasive factors for tumor cells; in hepatocellular carcinoma, CAFs participate in a TGF-β/PDGF signaling crosstalk with tumor cells to support EMT and the acquisition of an invasive phenotype99.
The microenvironment at the invasive edge of tumors is quite different than that of the tumor core. Hypoxia tends to be associated with the center of a tumor, while oxygen is largely available at the periphery. Given that low oxygen is a major driving force for stromal cell behavior and recruitment to tumors100–102, it is intriguing that many stromal cells do not necessarily favor hypoxic regions once they arrive at the tumor site. For instance, in colorectal cancers, while subsets of lymphocytes are present at both the leading edge and tumor core in primary tumors, these cells have been shown to be predominant at the tumor periphery in liver metastases where they correlate with response to therapy57,103. In addition, while macrophages play important roles during invasion at the tumor margin, as previously discussed, they have also been shown to thrive in hypoxic conditions in tumors and ischemic tissues, and thereby facilitate angiogenesis37,38,104. The relationship between oxygen and immune cells may inadvertently promote metastatic dissemination in multiple respects: first, while hypoxia mediates immune cell recruitment, these cells concentrate at the tumor periphery to support cancer invasion at the leading edge. Meanwhile, in the tumor core, hypoxia contributes to cancer cell escape by providing an aggressive selection pressure for resilient stem-like tumor cells that subsequently migrate away to the tumor margin. In light of this complexity, the spatial contexture of immune cells at the tumor edge versus the tumor center may be critical for fully understanding tumor-stroma dynamics.
Dynamic interplay between macrophages and tumor cells during intravasation
Beyond the initial acquisition of invasiveness in primary tumors, the next major rate-limiting step in the metastatic cascade is intravasation into circulation105 (Fig. 2). Multiphoton intravital imaging techniques have been used to observe macrophage-tumor interactions during metastatic dissemination in live animals106,107. These approaches have been instrumental in showing that macrophages are primarily localized in the peripheral tumor stroma and decrease in number towards the center. In deep regions of the tumor, not visible by conventional confocal microscopy, macrophages localize to blood vessels, where they help tumor cells intravasate into circulation107. Moreover, clusters of these three different cell types, termed the tumor microenvironment of metastasis (TMEM), are also found in human breast cancer, where their increased density correlates with distant metastasis108.
Recent studies indicate that entry into circulation may not be a late event in tumor progression, as previously believed109–111. If intravasation indeed requires macrophage-tumor cell interactions with endothelial barriers, it follows that macrophages also play an early role in tumorigenesis. Indeed, in a mouse model of chemically-induced lung cancer, resident pulmonary macrophages became activated in response to signals from incipient tumors within days of their onset. However, macrophage number in these lesions did not change until months later, indicating that recruitment of BM-derived macrophages is a late event during tumorigenesis112. This suggests that if circulating tumor cells are present in early disease, it is possible that this event is mediated by resident, rather than recruited, macrophage populations that are locally activated within the tumor. The kinetics of tumor cell entrance into the bloodstream, and the mechanisms that mediate this putative early event, remain elusive.
Survival in the blood and arrival at secondary organs
Metastasis is a highly inefficient process; only 0.01% of cells that intravasate into circulation are capable of forming detectable metastases113. During dissemination, platelets play an important role in the hostile microenvironment of the bloodstream, where they directly interact with tumor cells and enhance survival94 (Fig. 2). Platelets in circulation form protective aggregates with tumor cells, which interferes with NK cell-mediated cytotoxicity by enhancing fibrin deposition and impeding immune cell recognition94,114. These platelet-tumor cell clusters thereby provide an additional layer of immune evasion, which may contribute to disease progression.
During the physiological response to vascular injury, platelets become activated by thrombin, allowing them to attach to the endothelium, and aggregate to form a fibrin clot. Reminiscent of their role during this process, platelets mediate attachment to the endothelium when tumor cells arrive at secondary organs. Integrins expressed on platelets interact with collagen that becomes exposed at stripped regions of the endothelium, causing platelet activation. Thus, endothelial cell retraction at the secondary site, induced by circulating tumor cells or tumor cell-associated leukocytes, may be an important component of the premetastatic niche that dictates where metastatic tumor cells will exit circulation94,115. Indeed, a recent study found that platelets promote tumor cell extravasation via ATP-dependent activation of the endothelial P2Y2 receptor, which opens the vessel barrier to enable metastatic seeding116. In light of the various contributions of platelets to cancer progression, it is not surprising that in patients, thrombocytosis (high platelet count) is associated with poor prognosis across multiple cancers, including breast cancer, GBM, and pancreatic cancer117–119.
Extravasation of tumor cells and secondary seeding is requisite for metastatic outgrowth. Primary tumors upregulate fibronectin expression by resident fibroblasts in secondary organs, which serves as a docking site for VEGFR1+ hematopoietic progenitor cell (HPC) clusters and the subsequent arrival of tumor cells120. Additional studies have demonstrated that during metastasis of breast cancer to lung, VCAM-1-positive cancer cells associate with VLA-4-expressing macrophages121. This interaction activates PI3K/Akt signaling in tumor cells, protecting them from caspase-induced apoptosis. Indeed, interruption of this interaction renders metastatic cells susceptible to apoptotic insult121. Interestingly, VCAM-1 also interacts with a different integrin partner, α4β1, in osteoclasts, contributing to bone metastasis122. Together, these findings suggest that disruption of adhesion signaling axes between stromal cells and tumor cells may serve to prevent metastatic colonization of multiple organs.
Metastatic colonization and organ tropism: establishing a secondary niche
Organ tropism, more classically known as the seed-and-soil hypothesis, was first proposed by Stephen Paget in 1889, when he concluded that the distribution of metastases was not random, but rather displayed clear organ-preference. Paget’s hypothesis later spawned the idea that prior to metastatic dissemination, primary tumors secrete factors that contribute to the development of a premetastatic niche, characterized by an abundance of BM-derived cell types, increased fibroblasts, and secreted oncoproteins and cytokines that render the secondary environment receptive to tumor growth (Fig. 2). In alignment with this concept, it has been shown that BM-derived VEGFR1+ cells colonized premetastatic sites prior to tumor cell arrival120, suggesting that communication between primary and secondary sites likely occurs. The site of VEGFR1+ HPC colonization was consistent with the typical metastatic location for each experimental cancer type (LLC and B16 melanoma in this study), and remarkably, conditioned media from melanoma induced LLC metastasis to atypical sites that recapitulated melanoma metastasis patterns. Depleting VEGFR1+ cells interfered with formation of these premetastatic clusters and blocked metastasis120. In a subsequent study in breast cancer, expression of lysyl oxidase (LOX), a major target of HIF signaling, facilitated myeloid cell recruitment and subsequent tumor cell colonization in the lung123 LOX inhibition in tumor cells blocked myeloid cell recruitment to premetastatic niches, and reduced lung metastasis.
Additional studies have examined whether primary tumor hypoxia influences the premetastatic niche124. Specifically, simply injecting mice with conditioned media from breast cancer cells cultured under hypoxic conditions induced infiltration of bone marrow-derived cells (BMDCs) into the lung, despite the absence of an actual primary tumor. Major BMDC contributors to this hypoxia-induced pseudo-premetastatic niche included MDSCs and NK cells with impaired cytotoxicity. Collectively, these and other studies87 demonstrate a critical role for BMDCs in predicting and directing sites of future metastasis, and illustrate the importance of systemic communication between primary and secondary organs. It follows that in early stages of disease, patients may benefit from therapeutic intervention that aims to disrupt the premetastatic niche before tumor cells have a chance to arrive.
Another hypothesis regarding organ tropism stems from the observation that primary TMEs can share commonalities with secondary microenvironments. For example, it was recently shown that breast cancer stem cells attempting to colonize secondary sites induce periostin in the resident fibroblasts to recreate the environment of the primary niche125. Periostin induction is necessary for colonization and subsequent outgrowth, as it facilitates Wnt signaling in tumor cells. Notably, periostin knockout mice exhibited ~90% reduction in lung metastasis, whereas metastasis was unchanged when periostin was specifically inhibited in tumor cells in a wild-type (WT) mouse. Similarly, bone metastasis ‘seeds’ can be selected via mesenchymal-derived factors in the primary breast TME72, as discussed above. Whether tropic selection in the primary tumor occurs for other sites of metastasis via this intriguing type of ‘microenvironment mimicry’ is an exciting avenue of ongoing research.
Modulating tumorigenesis through exosomes
Many of the examples of heterotypic signaling within the TME discussed here involve classical paracrine signaling loops of cytokines or growth factors and their receptors. While these signaling mechanisms undoubtedly operate as a key means of intercellular communication within the TME, more recently, exosome shedding has emerged as another mode of cell-cell signaling. In cancer, tumor-derived exosomes from the primary tumor educate their environment to form a pro-tumorigenic niche, and direct BM-derived progenitors to enhance and direct metastatic dissemination15 (Fig. 2). Exosomes derived from aggressive melanoma cells increased growth and metastasis of primary tumors, and programmed BMDCs at the premetastatic site to assume a pro-angiogenic phenotype126. This was dependent on the receptor tyrosine kinase MET, as its inhibition in exosomes impaired pro-metastatic effects. Remarkably, exosomes also altered organ tropism; when exosomes from one melanoma cell line were injected into mice intravenously, they dictated the site of spontaneous metastasis of different subcutaneously-implanted melanoma cell lines. Critically, this study identified a prognostic exosome signature, detectable in patient blood, which accurately predicts stage and metastatic outcome126.
Various stromal cells are also capable of exosome release; for example, fibroblast-secreted exosomes promote breast cancer cell migration via Wnt-PCP signaling127. NK cell-derived exosomes from human blood contain proteins that induce cytotoxicity of tumor and activated-immune cell sex vivo128. Notably, NK cell-derived exosomes are not cytotoxic to resting-immune cells, suggesting their cytolytic effects are specifically directed to activated cells. Exosomes released from DCs, termed dexosomes, are being investigated in clinical trials for their potential as a cancer vaccine129,130. This concept is supported by preclinical melanoma models, whereby dexosome immunization induced CD8+ cytotoxic T cells and delayed tumor growth131.
Overcoming tumor dormancy and metastatic outgrowth
Even if tumor cells successfully seed secondary organs, this does not ensure their survival or expansion. The microenvironment at the secondary site can actively suppress metastatic cell survival and outgrowth, for example by neutrophil-mediated killing of tumor cells132, or thrombospondin-1 secretion by BM-derived Gr1+ cells133. Alternatively, as we will discuss in the following section, tumor cells can survive initial cell-clearing defense mechanisms in secondary organs, and subsequently exist as asymptomatic dormant micrometastases that can persist in the body for years without detection. Tumor dormancy is mediated by several processes that are driven in part by the microenvironment (Fig. 3) including tumor mass dormancy (proliferation is balanced by apoptosis), cellular dormancy (cells are arrested in G0), or immune dormancy (immunoediting leads to a state of equilibrium)134–136.
Figure 3. Overcoming tumor dormancy, and initiation of secondary outgrowth in metastatic niches.
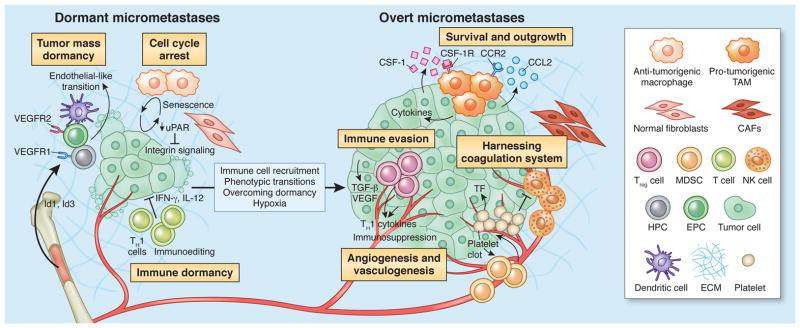
Dormant micrometastases are held in check by several mechanisms. Tumor mass dormancy, or angiogenic dormancy, is when proliferation is balanced by apoptosis, owing to a lack of vasculature and limited supply of nutrients and oxygen. Multiple cell types contribute to the re-establishment of vascularity at the secondary site, including hematopoietic and endothelial progenitor cells (HPCs; EPCs) expressing VEGF receptors, and dendritic cell precursors which can differentiate into an endothelial-like state. Tumor cells can also exist in a state of cellular dormancy, whereby proliferation is arrested in G0. This can be overcome via several mechanisms, for example, fibronectin-integrin interactions and activation of EGFR signaling, or re-polarization of macrophages from an anti- to a pro-tumorigenic state within the TME. Last, tumor cells can enter immune-induced dormancy whereby immunogenic cells are cleared, and cells that are able to survive enter a state of equilibrium. Immune suppressor cells are recruited to tumors in response to this process, and contribute to the establishment of an immunosuppressive state within secondary tissues. Treg cells and MDSCs are depicted here, which produce anti-inflammatory cytokines and suppress the anti-tumorigenic capacities of other immune cell types. Once micrometastases overcome dormancy, they become receptive to signals and cell types within their microenvironment to further support their expansion. For instance, TAMs are abundant in metastases of multiple cancer types, and support different tumorigenic processes to allow for outgrowth, including vascularization, impaired immunogenicity, and enhanced survival in overt metastases. Platelets, and components of the coagulation system, such as tissue factor (TF), are also important mediators of metastatic outgrowth, as they interfere with the ability of NK cells to destroy micrometastases, and support clot formation, which in turn causes the recruitment of MDSCs.
Flipping the angiogenic switch at the secondary site
One major limiting step of metastasis is outgrowth in secondary locations, which is in part dependent on establishing a blood supply, as in the primary TME. An inability of tumors to grow beyond a certain size due to insufficient vascularization is termed angiogenic/tumor mass dormancy134. At secondary sites, angiogenic dormancy is characterized by avascular micrometastatic lesions that do not grow beyond 1–2 mm in diameter (in accordance with the diffusion limit of oxygen137) due to a balance between proliferation and apoptosis. The angiogenic switch marks the transition out of this dormant state, at which point metastases are said to be “macro” (i.e. larger than 1–2 mm due to vascular infiltration), and exhibit elevated proliferation rates compared to apoptosis134,137. That tumor growth is dependent on the ability to recruit a vasculature has been reported extensively in multiple tumor types, and is closely linked to the composition of the tumor-associated stroma5. Supporting this concept, a recent study elegantly demonstrated that a dormant niche was associated with mature vessels and endothelial-derived thrombospondin-1, whereas metastatic outgrowth was associated with sprouting neovessels, and production of periostin and TGF-β138.
Several studies have demonstrated critical roles for recruited BMDCs in overcoming angiogenic dormancy (Fig. 3)139–141. For instance, it has been suggested that VEGFR1+ HPCs and VEGFR2+ EPCs are dually-required for mediating neovascularization at metastatic sites141. This requires Id1 and Id3, as Id1+/−/Id3−/− mice exhibited angiogenic defects concomitant with significantly impaired tumor growth, and BM transplantation from WT donors restored metastatic outgrowth141. More recently, when MMTV-PyMT mice were reconstituted with GFP+ BM and pulmonary metastases analyzed, this demonstrated that micrometastases were avascular, while macrometastases exhibited angiogenesis with GFP+ EPC incorporation140. Notably, Id1 inhibition prevented the transition from micro- to macrometastatic growth, by blocking EPC recruitment and angiogenic switching140. Another study reported that recruited CD11c+ DC precursors assembled into tumor-associated neovessels in an ovarian carcinoma model139. Interestingly, these cells contributed to vascularization via a phenotypic transition towards a more endothelial cell-like state139. While these and many other studies document roles for EPCs and BMDCs in promoting tumor-associated angiogenesis and establishing the premetastatic niche, it should be noted that other reports have come to different conclusions142,143, and therefore this remains an active area of discussion in the TME field144.
Mechanisms of cellular dormancy by cell cycle arrest
Studies in breast cancer found that dormant cells exist in a non-proliferative state in the BM of ~36–56% of patients145,146, irrespective of lymph node status, and detection of these cells is predictive of metastasis and worse survival. Analyses of micrometastases in mouse models demonstrated that these lesions are often completely negative for proliferation markers, suggesting they are in a state of G0-G1 arrest147. This arrest is mediated by microenvironment-mediated signals (Fig. 3), such as integrin interactions through the ECM or deregulated mitogenic/stress signals, and not necessarily dictated by insufficient vascularization. For example, uPAR can induce EGFR in a ligand-independent manner to promote tumor cell proliferation via fibronectin/αvβI integrin and ERK activation. Accordingly, uPAR downregulation results in tumor dormancy134,148,149. Recently, it was shown that senescent p53-positive stellate cells in the liver secrete a cocktail of cytokines that cause macrophages to become M2 polarized. This subsequently promotes proliferation of premalignant hepatocellular carcinoma cells, whereas disruption of p53-induced senescence blocks this progression150. Additional insight into the role of secreted factors in helping maintain, and subsequently overcome, dormancy came from a recent screen in metastatic breast cancer151. BMP signaling suppressed tumor stem cell traits, whereas Coco, a secreted BMP antagonist, re-activated tumor cells leading to metastatic outgrowth. Interestingly, these signals were only operational in the lung, and not the brain or bone151, indicating that metastasis-initiating cells may override microenvironment-mediated suppression in an organ-specific manner.
Immune-induced tumor dormancy
The notion that tumor cells can be recognized and destroyed by the immune system was first proposed by Burnet and Thomas in the 1950s. This theory was met with great controversy in the field, as early experiments were not successful in supporting their hypothesis. However, we now know that tumor cells are indeed recognized and destroyed through immunosurveillance, which molds the tumor and selects for less-immunogenic cells135. This process, termed immunoediting, parallels Darwinian selection, whereby tumor cells that are vulnerable to attack by the immune system are cleared, while cells that have or acquire a capacity to circumvent surveillance can survive and propagate a new tumor that is primed to evade the immune system. The concept of immunoediting is supported by the finding that tumors derived from immunodeficient mice are more immunogenic than tumors derived from immunocompetent mice, owing largely to the differences in immune selection pressures within each type of host152.
Tumor cell variants that are able to survive immunosurveillance enter a state of dynamic equilibrium, which is a form of immune-mediated tumor dormancy that prevents outgrowth of remaining tumor cells135,153. This tumor-immune equilibrium is driven by a strong selective pressure from the adaptive arm of the immune system, including T cells and TH1 cytokines (e.g. IFNγ or IL-12), and does not require recognition of effector cells (Fig. 3)153. Evidence for this mechanism came from observations that carcinogen-induced tumors frequently remain dormant and asymptomatic in immunocompetent mice; however, when mice are treated with antibodies against T cells and IFNγ, tumors grow at the site of induction153. Clinically, it is known that tumors can remain dormant and asymptomaticin patients for years or even decades before relapsing, and that patients who appear to be in full remission still have tumor cells circulating in their blood. Although it is unlikely that these phenomena are dictated solely by immune pressures, these observations are provoking; perhaps directing the immunoediting process to remain in the equilibrium phase could synergize with standard-of-care therapy to manage incurable disease and maximize the remission period for patients.
Tumor awakening and metastatic outgrowth
Immunoedited tumors are at risk of exiting their equilibrium (Fig. 3). Given that tumor cells are inherently genetically unstable, the strong immune pressure placed on tumor cells in equilibrium makes them susceptible to acquiring mutations that may allow for immune evasion and outgrowth. These adapted tumors frequently exhibit defects in antigen presentation and/or processing, for example, through loss of MHC class I or latent membrane protein (LMP)-family molecules154, rendering them undetectable by the adaptive immune system. They are also capable of establishing a global immunosuppressive state in the TME, by secreting a plethora of anti-inflammatory cytokines including TH1 cytokines, TGF-β, and VEGF, or by recruiting immunosuppressive cell types including Treg cells and MDSCs which further contribute to the anti-inflammatory cytokine milieu, as described earlier. Indeed, immune therapies have been utilized extensively in cancer patients, aiming to curtail aberrant immune responses to growing tumors155.
Besides escaping immunosurveillance, tumors are similarly prone to exiting angiogenic- or cell cycle-mediated mechanisms of dormancy, enabling development of lethal macrometastases. Several factors contribute to this process in addition to immune suppression, including sustained vascularization and enhanced survival. One cell type that is important for all of these processes are recruited TAMs. In pancreatic cancer patients, peripheral M2-polarized macrophages are associated with establishment and growth of liver metastases156. Similarly, in breast cancer models, TAMs mediate extravasation and metastatic outgrowth of tumor cells in the lung, and blocking TAMs with clodronate liposomes or through genetic ablation of CSF-1 significantly interfered with both processes157. More recently, a novel population of metastasis-associated macrophages (MAMs) was identified, which promoted the extravasation, seeding and outgrowth of breast cancer cells in the lung158. Interestingly, inhibition of CCL2-CCR2 signaling specifically prevented MAM accumulation and reduced metastasis in mice158. In a comparison of tumor associated lympho-monocytes (TALMs) in cancer patients versus autologous peripheral blood mononuclear cells, it was found that TALMs were associated with impaired immunogenic function and secreted elevated levels of cytokines reported to enhance tumor growth159. Together these studies illustrate the multifaceted functions of immune cells in advanced disease stages.
Interestingly, a role for the coagulation system has been demonstrated not only in circulation, but also during metastatic outgrowth. One coagulation protein in particular, tissue factor (TF), correlates with poor prognosis in patients, as it interferes with NK cell-mediated lysis of micrometastases160,161. TF inhibition with recombinant Tissue Factor Pathway Inhibitor or TF-targeted shRNAs in murine melanomas blocked lung metastasis162. Furthermore, TF induced platelet clots leading to BM-derived macrophage recruitment to support melanoma survival in the lung160. These clots also recruited MDSCs to secondary lesions, thereby suppressing immune rejection of the tumor160. That tumors use the coagulation system to support disease progression is yet another example of normal tissue homeostasis being hijacked in cancer.
Therapeutic strategies for re-educating the TME
Most therapeutic strategies against cancer have focused on targeting various aspects of tumor cells directly; however, stromal cells within the TME are genetically stable compared to tumor cells, and are thus likely to be less susceptible to classical mechanisms of therapeutic resistance. Moreover, given the accumulating evidence of overwhelming heterogeneity at every level in cancer cells163,164, targeting the TME becomes an even more compelling option (Fig. 4)165. Therapies aiming to deplete stromal cells, including various angiogenesis inhibitors166, have had limited benefits, possibly because they generally block the pro-tumorigenic effects of the TME. Given the paradoxical capacity of the TME to both promote and impair tumor growth, an avenue of therapeutic intervention worth exploring may be to harness this inherent plasticity, by developing strategies to manipulate and re-educate the TME, rather than to simply target TME components for depletion or destruction. Immunotherapies that are currently generating much excitement in the clinic11,155,167,168 are a classic example of such a reprogramming approach.
Figure 4. Therapeutic strategies to re-educate the tumor microenvironment.

Multiple strategies to target the TME are either currently in clinical use, or at different stages of clinical development, as indicated here, and referenced throughout the review. The tumor vasculature can be targeted with multiple drugs, such as bevacizumab (targets VEGF-A), CXCR2 antagonists, Sunitinib (a multi-target RTK inhibitor), and VEGF-Trap (soluble decoy receptor for VEGF). Immune activation, marked by an induction of T cells, is also a promising avenue of therapeutic intervention. This can be achieved through blockade of CTLA-4 (ipilimumab), PD1 receptor (nivolumab), or PD1 Ligand (lambrolizumab). Repolarization or re-education of cells within the TME, in particular macrophages or other myeloid cells, can be achieved by CSF-1R inhibition (e.g. BLZ945), or monoclonal antibodies that activate CD40. Alternatively, immune cell recruitment and expansion can be blocked through inhibition of critical cytokine axes, such as CXCR4 (AMD3100), CXCR2 (S-265610), CSF-1R and/or KIT (PLX3397), and the chemotherapeutic agent trabectedin, whose anti-tumor activity is proposed to be a result of selective depletion of monocytes/macrophages177. Likewise, metastatic seeding and outgrowth can be blocked by inhibition of key cytokine axes, such as CCR2 (MLN1202).
Various therapies attempt to block mechanisms of immune evasion by tumors, many of which are currently focused on advanced melanoma patients given their high numbers of lymphocytes167. Ipilimumab is an FDA-approved antibody that targets cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), which activates T cells and promotes anti-tumor immunity167. In the first clinical trial for ipilimumab in patients with inoperable metastatic melanoma, overall survival increased to ~10 months, compared to 6.4 months for those patients not on ipilimumab therapy169,170. Several drugs have been combined with ipilimumab to further improve patient outcome. For example, nivolumab, an antibody targeting Programmed Death 1 (PD1) receptor, was used in combination or in sequence with ipilimumab to treat advanced melanoma patients. PD1 represses T cell effector function and limits the immune response to tumors, thus enabling immune evasion. Results from this phase I trial revealed that patients receiving a maximum dose combination regimen exhibited >80% tumor volume reduction171. Similarly, lambrolizumab, a blocking antibody against PD1 ligand (PD-L1), was used in metastatic melanoma patients, who had either been previously treated with ipilimumab or not172. In the optimal dosing group, the response rate was 52%, and overall progression-free survival was >7 months172. Another immunotherapy success story involvesa different approach, using agonistic antibodies to activate CD40, a TNF receptor superfamily member broadly expressed on immune cells. The CD40 mAb reverses immune suppression by collectively activating antigen-presenting cells, promoting antitumor T-cell responses and re-educating cytotoxic myeloid cells173. Interestingly, preclinical pancreatic cancer studies demonstrated that the combined efficacy of CD40 mAb and gemcitabine required macrophage activation174. Taken together, these studies suggest that combining therapies targeting tumor mechanisms of immune evasion (e.g. removing suppressive cell types via PD1/PD-L1 neutralization) with activation of normal immune cell functionality (i.e. T cell activation) may provide optimal benefits for patients; however, these trials are still in early stages, and our mechanistic understanding of how these drugs contribute to efficacy and overall survival remains incomplete. Moreover, the long-term impact of these therapies on patient safety and survival will take several years to fully evaluate.
Further emerging examples of TME-directed therapies that do not focus on target cell depletion include the concept of neutralizing tumor-associated chronic inflammation175. Among the cell types and signaling molecules we have discussed here, strategies that block the NFκB pathway, or inhibition of key cytokine pathways that dictate recruitment and/or immune cell function, e.g. via CSF-1R, CCR2, CXCR2 etc. are being investigated in clinical trials (Fig. 4), in part based on preclinical successes25,158,176. For example, in a recent study, a CSF-1R inhibitor was used to target macrophages/microglia in the TME of gliomas25. This resulted in a robust decrease in tumor volume concomitant with a significant prolongation in survival in preclinical trials, and reprogramming rather than depletion of macrophages25. In contrast, CSF-1R inhibitors deplete TAMs in preclinical models of breast cancer, yet this has no effect on primary tumor growth, unless used in combination with other therapies176. Given that breast and brain cancers respond very differently to CSF-1R inhibition in their primary environments, whether a similar treatment modality would work for brain metastases from a breast primary tumor would be interesting to investigate, since these treatments target the microenvironment specifically. It is plausible that TME-targeted therapies should be administered to patients depending on where their tumor is located, in addition to what type of tumor they have, and that dual-targeting of tumor cells and their local environment may have robust consequences for mitigating metastasis.
Conclusions
This is an exciting time for the TME field, as illustrated by the examples discussed here, which have revealed new biological concepts and identified novel therapeutic strategies to target the TME. Nonetheless, with these advances come new challenges, the most obvious being how to identify and target susceptible nodes in the increasingly complex and interconnected TME (Fig. 4). Indeed, given that TMEs and key signaling pathways are broadly diverse between different tumor types and tissues, insights into how to manage this diversity, and how different TMEs may alter response to current standard-of-care therapies will be important areas to investigate going forward. Another largely unexplored question in the field is how the TME may be educated and sculpted by specific oncogenic driver(s) in the tumor cells, and how this could result in stromal cell diversity- even within the same tissue. Additional points to consider include determining which patients to target, which anti-cancer therapies to combine with TME-targeted agents, and how to overcome intrinsic or acquired resistance in the TME. However, from the studies highlighted in this Review, we now have the roadmap to convert these challenges into opportunities. For example, patient selection will require analysis of the entire TME57,176, not simply individual cell types in isolation, to determine specific therapies to use. Tumor cell-directed agents will also need to be combined with TME-therapies in a manner that considers how stromal cells modulate efficacy of a broad range of standard chemotherapies and targeted agents. Looking forward, perhaps the greatest promise may come from the notion that re-educating a dysfunctional TME could yield striking results in cancer control and remission as evidenced by the accumulating success stories in the cancer immunotherapy field.
BOX 1. Emerging stromal cell types that influence tumorigenesis.
There are several stromal cell types that are emerging as having robust influences on cancer. For instance, adipocytes and their progenitors, and the apoptotic crown-like structures that they form with phagocytic macrophages in obese individuals, contribute to tumor progression across a variety of obesity-associated cancers184–187. Moreover, adipose stromal cells can be recruited to growing tumors where they differentiate into pericytes and incorporate into vessel walls188, demonstrating that like tumor cells, stromal cells in the TME can exhibit cellular plasticity that contributes to tumorigenesis. Non-classical stem-promoting functions of nerves in both the BM and the local environment189,190 may also mediate cancer malignancy, given the association between perineural invasion and neurogenesis in several tumor types including GI, pancreatic and prostate cancers191–194. Therapies that mitigate nerve impulses, such as Botulinum toxin (Botox), may therefore have beneficial effects in some patients. Finally, the gut microbiome and associated inflammation are now accepted as major influences in the outcome of colorectal cancers. Indeed, the US National Institutes of Health has initiated an effort to fully characterize the human microbiome in various anatomical sites, including the gut, in health and disease. Treatment with anti-inflammatory (such as aspirin) or antimicrobial agents mitigates colorectal cancer tumorigenesis and extends patient survival195. Rather than completely disrupting the gut microbiome, it is possible that these agents are capable of augmenting the microbiome composition, thus creating an unfavorable environment for tumor growth. Re-education of the TME, in this case through alterations in cellular proportions, may again have favorable consequences for cancer. Interestingly, whether reintroduction of pro-inflammatory microbiota (i.e. those that existed before treatment) allows tumorigenesis to resume has not been established, and might suggest that long-term management of the gut microbiota could be critical to patient outcome. Given the emergence of non-classical stromal cell types in cancer, more creative combination tumor therapies may have an untapped benefit in managing disease progression. Furthermore, the TME should not be viewed simply as a local result of tumor burden; the systemic microenvironment is also capable of modulating disease progression and ought to be considered for therapeutic intervention.
BOX 2. Mechanisms of resistance in response to TME-targeted therapies.
While some successful TME-targeted therapies currently exist, specific depletion of one stromal cell type within tumors can lead to a state of imbalance within the TME, thereby leading to alterations in other stromal cell populations that contribute to intrinsic or acquired resistance. Moreover, some TME-targeted therapies, such as ipililumab, only work in a subset of patients, and uncovering the mechanisms of therapeutic resistance in non-responders is currently a high priority. Ipililumab depends strongly on T cell infiltration in responding patients, whereas patients that do not respond to therapy lack this infiltration response, and present with an abundance of tumor-associated immunosuppressive cell types. As one example, indoleamine 2,3-dioxygenase (IDO) derived from tumor cells, macrophages and DCs, was identified as a major contributor to an immunosuppressed state, defined by reduced CD8+ T cells and high Treg cells196. In mouse models, dual targeting of CTLA-4 and IDO caused rejection of established melanoma and breast tumors, mediated by reduced Treg cells and increased CD8+ T cells. This effect extends to other T cell-targeted strategies, including anti-PD1 or anti-GITR (glucocorticoid-induced tumor necrosis factor receptor family–related protein; a T cell inducer that mediates tumor immunity). Dual targeting of PD1/GITR and IDO causes a greater reduction in tumor growth compared to either therapy alone, suggesting that IDO mediates resistance to multiple immune checkpoint therapies, and that its effects are not limited to ipililumab. In another example of how the TME can be subverted to interfere with T-cell therapy, it was shown that melanomas acquire resistance via inflammation-induced loss of melanocytic antigens, leading to dedifferentiation through a TNF-α-mediated mechanism197. Finally, several groups have demonstrated TME alterations following traditional anti-cancer therapies, e.g. TAM accumulation35,176,198, which severely impair therapeutic efficacy. Together, these and other recent studies underscore how the microenvironment can subvert therapeutic efficacy and ultimately abrogate patient outcome, emphasizing the necessity of investigating the global TME response in all aspects of therapeutic targeting in future studies.
Acknowledgments
We apologize to the many authors whose work we could not cite owing to space constraints. D.F.Q. is supported by a Canadian Institutes of Health Research fellowship. Research in J.A.J’s lab is supported by the National Cancer Institute, American Cancer Society, Breast Cancer Research Foundation, and Cycle for Survival.
References
- 1.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature reviews. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nature medicine. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 6.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105–115. doi: 10.1111/imm.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes & development. 2011;25:2559–2572. doi: 10.1101/gad.169029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nature reviews. Immunology. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 9.Khazaie K, et al. The significant role of mast cells in cancer. Cancer metastasis reviews. 2011;30:45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 10.De Palma M, Naldini L. Tie2-expressing monocytes (TEMs): novel targets and vehicles of anticancer therapy? Biochimica et biophysica acta. 2009;1796:5–10. doi: 10.1016/j.bbcan.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews. Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sangiovanni A, et al. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology. 2004;126:1005–1014. doi: 10.1053/j.gastro.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 14.Beaugerie L, et al. Risk of Colorectal High-Grade Dysplasia and Cancer in a Prospective Observational Cohort of Patients With Inflammatory Bowel Disease. Gastroenterology. 2013;145:166–175. doi: 10.1053/j.gastro.2013.03.044. [DOI] [PubMed] [Google Scholar]
- 15.Barcellos-Hoff MH, Lyden D, Wang TC. The evolution of the cancer niche during multistage carcinogenesis. Nature reviews. Cancer. 2013;13:511–518. doi: 10.1038/nrc3536. [DOI] [PubMed] [Google Scholar]
- 16.de Martel C, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. The lancet oncology. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 17.Stewart T, Tsai SC, Grayson H, Henderson R, Opelz G. Incidence of de-novo breast cancer in women chronically immunosuppressed after organ transplantation. Lancet. 1995;346:796–798. doi: 10.1016/s0140-6736(95)91618-0. [DOI] [PubMed] [Google Scholar]
- 18.Gallagher B, Wang Z, Schymura MJ, Kahn A, Fordyce EJ. Cancer incidence in New York State acquired immunodeficiency syndrome patients. Am J Epidemiol. 2001;154:544–556. doi: 10.1093/aje/154.6.544. [DOI] [PubMed] [Google Scholar]
- 19.Schulz TF. Cancer and viral infections in immunocompromised individuals. International journal of cancer. Journal international du cancer. 2009;125:1755–1763. doi: 10.1002/ijc.24741. [DOI] [PubMed] [Google Scholar]
- 20.Vajdic CM, van Leeuwen MT. Cancer incidence and risk factors after solid organ transplantation. International journal of cancer. Journal international du cancer. 2009;125:1747–1754. doi: 10.1002/ijc.24439. [DOI] [PubMed] [Google Scholar]
- 21.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 22.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nature reviews. Immunology. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang HW, Joyce JA. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell cycle. 2010;9:4824–4835. doi: 10.4161/cc.9.24.14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagemann T, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature medicine. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cook J, Hagemann T. Tumour-associated macrophages and cancer. Current opinion in pharmacology. 2013;13:595–601. doi: 10.1016/j.coph.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 27.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature medicine. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Developmental cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Goswami S, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer research. 2005;65:5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 32.Coniglio SJ, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Molecular medicine. 2012;18:519–527. doi: 10.2119/molmed.2011.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joyce JA, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 34.Gocheva V, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes & development. 2010;24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shree T, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes & development. 2011;25:2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews. Immunology. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. The American journal of pathology. 2005;167:627–635. doi: 10.1016/S0002-9440(10)62038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Escribese MM, Casas M, Corbi AL. Influence of low oxygen tensions on macrophage polarization. Immunobiology. 2012;217:1233–1240. doi: 10.1016/j.imbio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Shime H, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2066–2071. doi: 10.1073/pnas.1113099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai X, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;4:341–343. doi: 10.1093/jmcb/mjs044. [DOI] [PubMed] [Google Scholar]
- 41.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. Journal of immunology. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 43.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nature reviews. Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews. Immunology. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazzoni A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. Journal of immunology. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 46.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. Journal of immunology. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 47.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. Journal of immunology. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 48.Liu C, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–4342. doi: 10.1182/blood-2006-09-046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz-Montero CM, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. Journal of immunology. 2012;188:1592–1599. doi: 10.4049/jimmunol.1101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whiteside TL, Schuler P, Schilling B. Induced and natural regulatory T cells in human cancer. Expert opinion on biological therapy. 2012;12:1383–1397. doi: 10.1517/14712598.2012.707184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gasteiger G, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. 2013;210:1179–1187. doi: 10.1084/jem.20122462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bates GJ, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24:5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 54.Fu J, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 55.Frey DM, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. International journal of cancer. Journal international du cancer. 2010;126:2635–2643. doi: 10.1002/ijc.24989. [DOI] [PubMed] [Google Scholar]
- 56.von Boehmer H, Daniel C. Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nature reviews. Drug discovery. 2013;12:51–63. doi: 10.1038/nrd3683. [DOI] [PubMed] [Google Scholar]
- 57.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nature reviews. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 58.Blatner NR, et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Science translational medicine. 2012;4:164ra159. doi: 10.1126/scitranslmed.3004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rech AJ, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Science translational medicine. 2012;4:134ra162. doi: 10.1126/scitranslmed.3003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 61.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 62.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer research. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dumont N, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia. 2013;15:249–262. doi: 10.1593/neo.121950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochimica et biophysica acta. 2013;1832:1070–1078. doi: 10.1016/j.bbadis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer research. 2007;67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 66.Petersen OW, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. The American journal of pathology. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orr B, et al. Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene. 2012;31:1130–1142. doi: 10.1038/onc.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeisberg M, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature medicine. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 69.Calvo F, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature cell biology. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fukumura D, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 71.Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 72.Zhang XH, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–1073. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bergamaschi A, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol. 2008;214:357–367. doi: 10.1002/path.2278. [DOI] [PubMed] [Google Scholar]
- 74.Naba A, et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Molecular & cellular proteomics: MCP. 2012;11:M111 014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Folkman J. Tumor angiogenesis: therapeutic implications. The New England journal of medicine. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 76.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. 2012 doi: 10.1038/onc.2012.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu W, et al. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006;80:267–274. doi: 10.1016/j.yexmp.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 79.Ho IA, et al. Human Bone Marrow-Derived Mesenchymal Stem Cells Suppress Human Glioma Growth Through Inhibition of Angiogenesis. Stem Cells. 2012;31:146–155. doi: 10.1002/stem.1247. [DOI] [PubMed] [Google Scholar]
- 80.Roodhart JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer cell. 2011;20:370–383. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 81.Cuiffo BG, Karnoub AE. Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell adhesion & migration. 2012;6:220–230. doi: 10.4161/cam.20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alitalo A, Detmar M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene. 2012;31:4499–4508. doi: 10.1038/onc.2011.602. [DOI] [PubMed] [Google Scholar]
- 83.Schoppmann SF, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. The American journal of pathology. 2002;161:947–956. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kerjaschki D, et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nature medicine. 2006;12:230–234. doi: 10.1038/nm1340. [DOI] [PubMed] [Google Scholar]
- 85.Zumsteg A, et al. Myeloid cells contribute to tumor lymphangiogenesis. PloS one. 2009;4:e7067. doi: 10.1371/journal.pone.0007067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hunter KE, et al. Heparanase promotes lymphangiogenesis and tumor invasion in pancreatic neuroendocrine tumors. Oncogene. 2013 doi: 10.1038/onc.2013.142. [DOI] [PubMed] [Google Scholar]
- 87.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature reviews. Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 90.Gao D, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer research. 2012;72:1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chao Y, Wu Q, Acquafondata M, Dhir R, Wells A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012;5:19–28. doi: 10.1007/s12307-011-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chaffer CL, Thompson EW, Williams ED. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs. 2007;185:7–19. doi: 10.1159/000101298. [DOI] [PubMed] [Google Scholar]
- 93.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nature reviews. Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nishimura K, Semba S, Aoyagi K, Sasaki H, Yokozaki H. Mesenchymal stem cells provide an advantageous tumor microenvironment for the restoration of cancer stem cells. Pathobiology. 2012;79:290–306. doi: 10.1159/000337296. [DOI] [PubMed] [Google Scholar]
- 97.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nature reviews. Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 98.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer research. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 99.van Zijl F, et al. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene. 2009;28:4022–4033. doi: 10.1038/onc.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chouaib S, et al. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol. 2012;3:21. doi: 10.3389/fimmu.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Facciabene A, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 102.Corzo CA, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Halama N, et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer research. 2011;71:5670–5677. doi: 10.1158/0008-5472.CAN-11-0268. [DOI] [PubMed] [Google Scholar]
- 104.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 105.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nature reviews. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 106.Condeelis J, Weissleder R. In vivo imaging in cancer. Cold Spring Harbor perspectives in biology. 2010;2:a003848. doi: 10.1101/cshperspect.a003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia. 2006;11:151–163. doi: 10.1007/s10911-006-9021-5. [DOI] [PubMed] [Google Scholar]
- 108.Robinson BD, et al. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:2433–2441. doi: 10.1158/1078-0432.CCR-08-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lucci A, et al. Circulating tumour cells in non-metastatic breast cancer: a prospective study. The lancet oncology. 2012;13:688–695. doi: 10.1016/S1470-2045(12)70209-7. [DOI] [PubMed] [Google Scholar]
- 110.Krishnamurthy S, et al. Detection of minimal residual disease in blood and bone marrow in early stage breast cancer. Cancer. 2010;116:3330–3337. doi: 10.1002/cncr.25145. [DOI] [PubMed] [Google Scholar]
- 111.Stoecklein NH, et al. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer cell. 2008;13:441–453. doi: 10.1016/j.ccr.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 112.Redente EF, et al. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. The American journal of pathology. 2010;176:2972–2985. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chambers AF, et al. Critical steps in hematogenous metastasis: an overview. Surg Oncol Clin N Am. 2001;10:243–255. vii. [PubMed] [Google Scholar]
- 114.Palumbo JS, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–185. doi: 10.1182/blood-2004-06-2272. [DOI] [PubMed] [Google Scholar]
- 115.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 116.Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-Derived Nucleotides Promote Tumor-Cell Transendothelial Migration and Metastasis via P2Y2 Receptor. Cancer cell. 2013;24:130–137. doi: 10.1016/j.ccr.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 117.Taucher S, et al. Impact of pretreatment thrombocytosis on survival in primary breast cancer. Thromb Haemost. 2003;89:1098–1106. [PubMed] [Google Scholar]
- 118.Brown KM, Domin C, Aranha GV, Yong S, Shoup M. Increased preoperative platelet count is associated with decreased survival after resection for adenocarcinoma of the pancreas. Am J Surg. 2005;189:278–282. doi: 10.1016/j.amjsurg.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 119.Brockmann MA, et al. Preoperative thrombocytosis predicts poor survival in patients with glioblastoma. Neuro Oncol. 2007;9:335–342. doi: 10.1215/15228517-2007-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer cell. 2011;20:538–549. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lu X, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer cell. 2011;20:701–714. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 124.Sceneay J, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer research. 2012;72:3906–3911. doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- 125.Malanchi I, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 126.Peinado H, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nature medicine. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luga V, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151:1542–1556. doi: 10.1016/j.cell.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 128.Lugini L, et al. Immune surveillance properties of human NK cell-derived exosomes. Journal of immunology. 2012;189:2833–2842. doi: 10.4049/jimmunol.1101988. [DOI] [PubMed] [Google Scholar]
- 129.Morse MA, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9. doi: 10.1186/1479-5876-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Escudier B, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Naslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. Journal of immunology. 2013;190:2712–2719. doi: 10.4049/jimmunol.1203082. [DOI] [PubMed] [Google Scholar]
- 132.Granot Z, et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer cell. 2011;20:300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Catena R, et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer discovery. 2013;3:578–589. doi: 10.1158/2159-8290.CD-12-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nature reviews. Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 136.Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nature reviews. Clinical oncology. 2013;10:41–51. doi: 10.1038/nrclinonc.2012.207. [DOI] [PubMed] [Google Scholar]
- 137.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell cycle. 2006;5:1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 138.Ghajar CM, et al. The perivascular niche regulates breast tumour dormancy. Nature cell biology. 2013;15:807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Conejo-Garcia JR, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nature medicine. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 140.Gao D, et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 141.Lyden D, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nature medicine. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 142.Purhonen S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6620–6625. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dawson MR, Duda DG, Fukumura D, Jain RK. VEGFR1-activity-independent metastasis formation. Nature. 2009;461:E4. doi: 10.1038/nature08254. discussion E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kerbel RS, et al. Endothelial progenitor cells are cellular hubs essential for neoangiogenesis of certain aggressive adenocarcinomas and metastatic transition but not adenomas. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:E54. doi: 10.1073/pnas.0804876105. author reply E55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pierga JY, et al. Clinical significance of proliferative potential of occult metastatic cells in bone marrow of patients with breast cancer. Br J Cancer. 2003;89:539–545. doi: 10.1038/sj.bjc.6601121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Braun S, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. The New England journal of medicine. 2000;342:525–533. doi: 10.1056/NEJM200002243420801. [DOI] [PubMed] [Google Scholar]
- 147.Naumov GN, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer research. 2002;62:2162–2168. [PubMed] [Google Scholar]
- 148.Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer cell. 2002;1:445–457. doi: 10.1016/s1535-6108(02)00072-7. [DOI] [PubMed] [Google Scholar]
- 149.Ranganathan AC, Adam AP, Aguirre-Ghiso JA. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell cycle. 2006;5:1799–1807. doi: 10.4161/cc.5.16.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lujambio A, et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gao H, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 2012;150:764–779. doi: 10.1016/j.cell.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shankaran V, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 153.Koebel CM, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 154.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nature reviews. Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yoshikawa K, et al. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012;103:2012–2020. doi: 10.1111/j.1349-7006.2012.02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Qian B, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PloS one. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mantovani G, et al. Tumor-associated lympho-monocytes from neoplastic effusions are immunologically defective in comparison with patient autologous PBMCs but are capable of releasing high amounts of various cytokines. International journal of cancer. Journal international du cancer. 1997;71:724–731. doi: 10.1002/(sici)1097-0215(19970529)71:5<724::aid-ijc6>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 160.Gil-Bernabe AM, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119:3164–3175. doi: 10.1182/blood-2011-08-376426. [DOI] [PubMed] [Google Scholar]
- 161.Palumbo JS. Mechanisms linking tumor cell-associated procoagulant function to tumor dissemination. Semin Thromb Hemost. 2008;34:154–160. doi: 10.1055/s-2008-1079255. [DOI] [PubMed] [Google Scholar]
- 162.Amirkhosravi A, et al. Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb Haemost. 2002;87:930–936. [PubMed] [Google Scholar]
- 163.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nature reviews. Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 164.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 165.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer research. 2013;73:4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nature reviews. Cancer. 2011;11:805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews. Immunology. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hwu P. Treating cancer by targeting the immune system. The New England journal of medicine. 2010;363:779–781. doi: 10.1056/NEJMe1006416. [DOI] [PubMed] [Google Scholar]
- 171.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:1035–1043. doi: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.DeNardo DG, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Germano G, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 178.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nature reviews. Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 179.Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33:949–955. doi: 10.1093/carcin/bgs123. [DOI] [PubMed] [Google Scholar]
- 180.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 181.Bos PD, Rudensky AY. Treg cells in cancer: a case of multiple personality disorder. Science translational medicine. 2012;4:164fs144. doi: 10.1126/scitranslmed.3005283. [DOI] [PubMed] [Google Scholar]
- 182.Mahmoud SM, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011;29:1949–1955. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- 183.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 184.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 185.Behan JW, et al. Adipocytes impair leukemia treatment in mice. Cancer research. 2009;69:7867–7874. doi: 10.1158/0008-5472.CAN-09-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Morris PG, et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer prevention research. 2011;4:1021–1029. doi: 10.1158/1940-6207.CAPR-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang Y, et al. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer research. 2012;72:5198–5208. doi: 10.1158/0008-5472.CAN-12-0294. [DOI] [PubMed] [Google Scholar]
- 189.Katayama Y, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 190.Yamazaki S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147:1146–1158. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 191.Liebig C, et al. Perineural invasion is an independent predictor of outcome in colorectal cancer. J Clin Oncol. 2009;27:5131–5137. doi: 10.1200/JCO.2009.22.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ayala GE, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:7593–7603. doi: 10.1158/1078-0432.CCR-08-1164. [DOI] [PubMed] [Google Scholar]
- 193.Demir IE, Friess H, Ceyhan GO. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front Physiol. 2012;3:97. doi: 10.3389/fphys.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Magnon C, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 195.Liao X, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. The New England journal of medicine. 2012;367:1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–1402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Landsberg J, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–416. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 198.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]