

Abstract
Complexes [MnMST(NH3)]n-3 (Mn = FeII, FeIII, GaIII) were prepared and each contains a intramolecular hydrogen bonding network involving the ammonia ligand. Deprotonation of the FeIII–NH3 complex afforded a putative [FeIIIMST(NH2)]− species whose reactivity has been explored.
Monomeric Fe–NHn (n = 2,3) complexes have been targeted as key intermediates in a variety of chemical transformations. They have potential use as precursors for N-atom transfer reactions, including those that activate C–H bonds. Terminal Fe–NH2 or Fe–NH3 species are also proposed to be significant in biological nitrogen fixation, whereby release of NH3 represents the final step in the reduction of N2.1–5 Such amido and ammine complexes have been proposed as intermediates in this process, and have been studied in various synthetic small molecule systems.6–14 However, few of these have been structurally characterized, with only a single example of a complex containing a FeIII–NH3 center. In this report we describe the preparation and properties of a redox-pair of FeII/III–NH3 complexes and a related GaIII–NH3 species. We demonstrate that these complexes contain an intramolecular hydrogen bonding (H-bonding) network surrounding the M–NH3 unit that persists in both solution and the solid state. Preliminary evidence has provided that deprotonation of the FeIII–NH3 complex produces a putative amido analog, which has moderate activity to cleave N–H bonds from an external substrate.
Our group investigates the influences of the secondary coordination sphere on metal-mediated processes. We have developed several multidentate ligands that incorporate intramolecular H-bonds within the secondary coordination sphere. One example is the sulfonamide-based tripodal ligand N,N′N″ -[2,2′,2″ nitrilotris(ethane-2,1-diyl)]tris(2,4,6-trimethyl-benzenesulfonamido) ([MST]3−) that upon binding a metal ion forms a C3-symmetric cavity proximal to the metal center. The [MST]3− ligand can also form up to three intramolecular H-bonds with an external ligand (Fig. 1).15–19 Because of these structural features, we reasoned that complexes of [MST]3− should be ideally suited to stabilize species with a terminal ammonia ligand.
Fig. 1.

General diagram of the [MnMST(NH3)]n–3 complexes.
The iron complex, [FeIIMST]−, was prepared by treating a solution of H3MST in N, N-dimethylacetamide (DMA) with three equivalents of NaH followed by metallation with Fe(OAc)2. After work up and removal of two equivalents NaOAc, Na[FeIIMST] was obtained as a white powder. Treating a suspension of Na[FeIIMST] in THF with one equiv of NH3 in THF resulted in a clear, colorless solution of [FeIIMST(NH3)]− (Scheme 1). Parallel mode EPR spectroscopy of the complex at 4K displayed a sharp signal at g= 9.4, indicative of a new high-spin FeII species with an S = 2 spin state (Fig. S1). FTIR studies showed two distinct ν(NH) vibrations at 3382 and 3408 cm−1 in the solid state, suggesting the possibility of an unsymmetrically H-bonding network involving the ammine ligand.
Scheme 1.
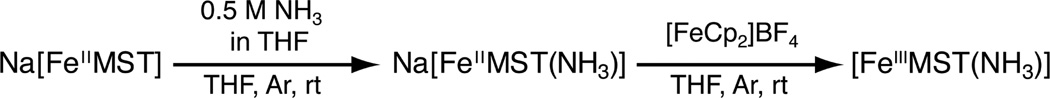
Synthesis of Fe-NH3 Complexes
The redox properties of [FeIIMST(NH3)]− were investigated using cyclic voltammetry. A reversible one-electron redox event at −0.645 V versus [FeCp2]+/0 was observed, which was assigned to the FeII/FeIII couple. (Fig. 2A). This analysis suggested that the analogous FeIII–NH3 complex could be prepared in bulk. Thus, treating the colorless [FeIIMST(NH3)]− complex with either [FeCp2]+ or [C7H7]+ in THF resulted in an immediate color change to afford a red-orange species having a λmax (εM) = 398 nm (8000) (Fig. 2B). FTIR analysis of the isolated solid showed a single ν(NH) peak at 3348 cm−1. According to perpendicular mode EPR spectroscopy performed at 77 K, the new species is a high-spin FeIII species having axial symmetry with g-values at 5.59 and 1.99 (Fig. S2).
Fig. 2.

(A) Cyclic voltammogram of [FeIIIMST(NH3)] recorded in THF at a scan velocity of 0.01 V s−1. (B) Electronic absorption spectra of [FeIIMST(NH3)]− (dashed line) and [FeIIIMST(NH3)] (solid line), collected in THF at 298 K.
The molecular structures of the Fe–NH3 complexes were determined using X-ray diffraction (XRD) methods. According to the solid-state structure of [FeIIMST(NH3)]−, the FeII center possesses an N5 primary coordination sphere with distorted trigonal-bipyramidal geometry (Fig. S3, Table S4). The nitrogen atoms of the [MST]3− ligand coordinate to the FeII ion with an apical Fe1—N1 bond length of 2.224(1)Å and an average Fe1—Neq bond distance of 2.099(3) Å. The primary sphere of the FeII center is completed by an ammonia ligand having a Fe1—NH3 bond distance of 2.145(1)Å and a N5—Fe1—N1 bond angle of 177.58(5)°. The solid-state structure also supports our FTIR findings that the FeII–NH3 unit is involved in an unsymmetrical H-bonding network with the SO2Mes groups of [MST]3−. We observed two relatively long H-bonds having O3⋯N5 and O5⋯N5 distances of 2.914(2) and 2.918(2) Å; the third H-bond was statistically shorter with an O1⋯N5 distance of 2.810(2) Å. Note that Peters has also prepared a trigonal bipyramidal FeII–NH3 complex, [FeIISi(PR)3(NH3)]+ that contains a significantly shorter Fe–NH3 bond of 2.063(2) Å.12, ‡ The 0.082 Å difference between the Fe–NH3 bond distances in the two complexes is caused, in part, by the intramolecular H-bonding network surrounding the Fe–NH3 unit in [FeIIMST(NH3)]−.
[FeIIIMST(NH3)] crystallized in the space group hexagonal P, which requires the complex to have a C3 center of rotation about the N1–Fe1–N3 axis (Fig. 3). The ferric complex also has a trigonal bipyramidal coordination geometry, yet there are significant differences in the metrical parameters between the two Fe–NH3 complexes. In [FeIIIMST(NH3)], an Fe1–N1 bond distance of 2.295(3) Å was observed, a lengthening of 0.071 Å compared to that found in [FeIIMST(NH3)]−. In contrast, the Fe1–Neq and Fe–NH3 bond lengths of 1.979(2) and 2.080(3) Å are shorter compared to those observed in [FeIIMST(NH3)]−. These observations are consistent with oxidation of [FeIIMST(NH3)]− to [FeIIIMST(NH3)]. The intramolecular H-bonds involving the FeIII–NH3 and SO2R groups remain intact upon oxidation, and a C3-symmetric H-bonding network is present in [FeIIIMST(NH3)] with an O1⋯NH3 distance of 2.881(15) Å.
Fig. 3.

Thermal ellipsoid diagram depicting the molecular structure of [FeIIIMST(NH3)]. Ellipsoids are drawn at the 50% probability level, and for clarity only the ammine hydrogen atoms are shown. Selected bond lengths (Å) and angles (°): Fe1—N3, 2.080(3); Fe1—N2, 1.979(2); Fe1—N1, 2.295(3); N3⋯O1, 2.881(15); N2—Fe1—N2, 117.33(3); N2—Fe1—N3, 99.51(5); N2—Fe1—N1, 80.49(5); N3—Fe1—N1, 180.00(1).
The analogous [GaIIIMST(NH3)] complex was prepared by treating a dichloromethane (DCM) solution of [GaIIIMST] complex18 with 0.5 M NH3 in THF. The molecular structure of [GaIIIMST(NH3)] determined by XRD is isostructural to that of [FeIIIMST(NH3)], with nearly identical crystallographic and metrical parameters (Fig. S5, Table S5). For instance, in [GaIIIMST(NH3)] the bond distances for Ga1–N1 (2.261 Å) and Ga1–NH3 (2.022 Å) are slightly shorter than those of the FeIII analog. These findings agree with the 0.03 Å difference in ionic radii between the FeIII and GaIII ions.20 In addition, a symmetric intramolecular H-bonding network is present in [GaIIIMST(NH3)] with an O1⋯N3 distance of 2.837(15) Å, which is supported by a vibrational study showing a single NH vibration at 3339 cm−1.
Our results showed that the solid-state properties of the [MIIIMST(NH3)] complexes (MIII = Fe, Ga) are nearly identical. Since [GaIIIMST(NH3)] is diamagnetic, we were also able to evaluate its molecular structure in solution using nuclear magnetic resonance (NMR) spectroscopy. The complex possesses C3 symmetry in CDCl3 at 298 K, with a singlet at 4.76 ppm attributed to the ammine protons (Fig. S6). Nuclear Overhauser Effect (NOE) spectroscopy was used to assess the spatial relationship of the Ga–NH3 unit to the rest of the complex. The protons at the o-methyl positions of the mesityl units showed a 1% NOE enhancement with the ammine protons (Fig. S7). This result is indicative of a ~4 Å distance between these groups as was observed in the solid-state structure of [GaIIIMST(NH3)], suggesting that the NH3 ligand remains coordinated to the GaIII center in solution on the NMR timescale. Similar structural properties should be operative for the FeIII–NH3 analog in solution.
The relative acidity of the ammine ligand in [FeIIIMST(NH3)] was assessed by treatment of the complex with various organic bases in THF at room temperature. No reaction was observed between the complex and either pyrrolidine (pKaTHF = 13.5) or 2-phenyl-1,1,3,3-tetramethylguanidine (pKaTHF = 14). In contrast, reactions were observed when [FeIIIMST(NH3)] was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKaTHF = 16.8), 1,5,7-triazobicyclo[4.4.0]dec-5-ene (TBD, pKaTHF = 21.0) or sodium hexamethyldisilazide (NaHMDS, pKaTHF = 26) to produce a new ferric species that we assign as [FeIIIMST(NH2)]− (see below). These experiments allowed the pKa(NH3) to be bracketed between 14.0 and 16.8 in THF, representing a decrease in pKa value of greater than 30 orders of magnitude compared to that of free ammonia.□
The properties of the deprotonated product were further examined for the reaction of [FeIIIMST(NH3)] and TBD, which at room temperature in THF resulted in clean generation of a yellow-orange species with a λmax = 368 nm (Fig. 4A). Solution FTIR studies of the new species in 1:1 DCM:THF displayed a new broad ν(NH) peak at 3422 cm−1, representing a major shift from the ν(NH) peaks at 3339 and 3309 cm−1 found for [FeIIIMST(NH3)] in solution (Fig. S10). Monitoring the deprotonation using perpendicular-mode EPR spectroscopy, new features at g = 9.37 and 4.21 were observed at 77 K (Fig. 4B). The rhombic spectrum is consistent with the formation of a new high-spin ferric complex but one that does not have C3-symmetry as was found in the original [FeIIIMST(NH3)] complex. Furthermore, treatment of the deprotonated species with an acid, such as HNEt3BF4 (pKaTHF = 12.5) rapidly regenerated [FeIIIMST(NH3)] (Fig. S11). Taken together, these findings suggest that the deprotonated species is the FeIII–amido complex, [FeIIIMST(NH2)]−.
Fig. 4.

(A) Electronic absorption spectrum of 0.125 mM [FeIIIMST(NH3)] in THF treated with 1 equiv. TBD at 298 K. (B) Titration of TBD into 9.8 mM [FeIIIMST(NH3)] in 1:1 DCM:THF. Perpendicular-mode X-band EPR spectra collected as a frozen glass at 77K.
Preliminary results showed that the putative FeIII–amido complex reacts poorly with substrates containing X–H bonds. The [FeIII–NH2]− species obtained from the deprotonation with TBD did not react with 9,10-dihydroanthracene (DHA, BDEC–H = 78 kcal/mol)21 or 2,6-di-t-butyl-p-cresol (BHT, BDEO–H = 81 kcal/mol).22 In the presence of diphenylhydrazine (DPH, BDEN–H = 69 kcal/mol)23 a small amount of azobenzene was detected but the yield was less than 10%. Using NaHMDS to prepare [FeIIIMST(NH2)]− gave similar results but the reaction with DPH was qualitatively more rapid. It is possible that the presence of the Na(I) ion in this reaction could affect the rate of the reaction. Note that non-redox active metal ions have been shown to affect the rates in other complexes containing the [MST]3− ligand.18
The two [FeII/IIIMST(NH3)]n complexes represent the first example of a pair of Fe–NH3 complexes differing by only one electron. Other reported Fe–NH3 complexes do not display a reversible redox couple; rather, some systems such as [FeIITPB(NH3)]+‡ release NH3 upon reduction.14 The intramolecular H-bonding networks surrounding the Fe–NH3 units in these complexes undoubtedly influence their overall stability, an effect that is comparable to those found in related Fe–O(H) complexes.24–26 Deprotonation of [FeIIIMST(NH3)] to form a putative FeIII–NH2 species, and its subsequent ability to cleave N–H bonds, demonstrates the potential reactivity of these systems toward external substrates. The scope of their reactivity is currently under investigation.
Supplementary Material
Acknowledgments
Acknowledgement is made to the National Institutes of Health USA (GM050781 to ASB) for financial support.
Footnotes
Electronic Supplementary Information (ESI) available: Detailed experimental procedures and spectra for complexes and experiments. CCDC 971643-971645. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/b000000x/
[Si(PR)3]−, tris(2-(diisopropylphosphanyl)phenyl)silanato, R = i-Pr, or tris(2-(diphenylphosphanyl)phenyl)silanato, R = Ph; [TBP]−, tris(2-(diisopropylphosphanyl)phenyl)boranato.
Notes and references
- 1.Peters JC, Mehn MP. In: Activation of Small Molecules. Tolman WB, editor. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2006. pp. 81–120. [Google Scholar]
- 2.Seefeldt LC, Hoffman BM, Dean DR. Annu. Rev. Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crossland JL, Tyler DR. Coord. Chem. Rev. 2010;254:1883–1894. [Google Scholar]
- 4.MacLeod KC, Holland PL. Nat. Chem. 2013;5:559–565. doi: 10.1038/nchem.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84–87. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olmstead MM, Sigel G, Hope H, Xu X, Power PP. J. Am. Chem. Soc. 1985;107:8087–8091. [Google Scholar]
- 7.Chetcuti PA, Liégard A, Rihs G, Rist G, Schweiger A. Helv. Chim. Acta. 1991;74:1591–1599. [Google Scholar]
- 8.Sellmann D, Soglowek W, Knoch F, Ritter G, Dengler J. Inorg. Chem. 1992;31:3711–3717. [Google Scholar]
- 9.Fox DJ, Bergman RG. J. Am. Chem. Soc. 2003;125:8984–8985. doi: 10.1021/ja035707a. [DOI] [PubMed] [Google Scholar]
- 10.Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J. Am. Chem. Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]
- 11.Bowman AC, Bart SC, Heinemann FW, Meyer K, Chirik PJ. Inorg. Chem. 2009;48:5587–5589. doi: 10.1021/ic9003017. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saouma CT, Moore CE, Rheingold AL, Peters JC. Inorg. Chem. 2011;50:11285–11287. doi: 10.1021/ic2016066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson JS, Moret M-E, Peters JC. J. Am. Chem. Soc. 2013;135:534–537. doi: 10.1021/ja307714m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park YJ, Ziller JW, Borovik AS. J. Am. Chem. Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS. J. Am. Chem. Soc. 2012;134:17526–17535. doi: 10.1021/ja304525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sickerman NS, Henry RM, Ziller JW, Borovik AS. Polyhedron. 2013;58:65–70. doi: 10.1016/j.poly.2012.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park YJ, Cook SA, Sickerman NS, Sano Y, Ziller JW, Borovik AS. Chem. Sci. 2013;4:717–726. doi: 10.1039/C2SC21400H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sano Y, Weitz AC, Ziller JW, Hendrich MP, Borovik AS. Inorg. Chem. 2013;52:10229–10231. doi: 10.1021/ic401561k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon RD. Acta Crystallogr. 1976;A32:751–767. [Google Scholar]
- 21.Bordwell FG, Cheng J, Ji GZ, Satish AV, Zhang X. J. Am. Chem. Soc. 1991;113:9790–9795. [Google Scholar]
- 22.Lucarini M, Pedrielli P, Pedulli GF, Cabiddu S, Fattuoni C. J. Org. Chem. 1996;61:9259–9263. [Google Scholar]
- 23.Zhao Y, Bordwell FG, Cheng J-P, Wang D. J. Am. Chem. Soc. 1997;119:9125–9129. [Google Scholar]
- 24.MacBeth CE, Gupta R, Mitchell-Koch KR, Victor G Young, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 25.Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 26.Dey A, Hocking RK, Larsen P, Borovik AS, Hodgson KO, Hedman B, Solomon EI. J. Am. Chem. Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x. [DOI] [PubMed] [Google Scholar]
- 27.Bordwell FG, Drucker GE, Fried HE. J. Org. Chem. 1981;46:632–635. [Google Scholar]
- 28.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J. Org. Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.