

Abstract
Rationale
We previously reported that type VI collagen deposition increases in the infarcted myocardium in vivo. To date, a specific role for this non-fibrillar collagen has not been explored in the setting of myocardial infarction (MI).
Objective
To determine whether deletion of type VI collagen in an in vivo model of post-MI wound healing would alter cardiac function and remodeling in the days to weeks after injury.
Methods and Results
Wild type (WT) and Col6a1-/- mice were subjected to MI followed by serial echocardiographic and histological assessments. At 8 weeks post-MI, infarct size was significantly reduced, ejection fraction was significantly preserved (43.9 ± 3.3% vs. 29.1 ± 4.3% for WT) and left ventricular (LV) chamber dilation was attenuated in the Col6a1-/- MI group (25.8 ± 7.9% increase vs. 62.6 ± 16.5% for WT). The improvement in cardiac remodeling was evident as early as 10 days post-MI in the Col6a1-/- mice. Myocyte apoptosis within the infarcted zones was initially greater in the Col6a1-/- group 3 days post-MI but by day 14 this was significantly reduced. Collagen deposition was also reduced in the infarcted and remote areas of the Col6a1-/- hearts. The reductions in chronic myocyte apoptosis and fibrosis are critical events leading to improved long-term remodeling and functional outcomes.
Conclusions
These unexpected results demonstrate for the first time that deletion of type VI collagen in this knockout model plays a critical protective role following MI by limiting infarct size, chronic apoptosis, aberrant remodeling and fibrosis leading to preservation of cardiac function.
Keywords: post-MI remodeling, cardioprotection, myofibroblast, non-fibrillar collagen, cell-matrix interactions
Introduction
The extracellular matrix (ECM) plays a key role in cardiac remodeling and wound repair following a myocardial infarction (MI). Patients that survive a MI normally develop cardiac fibrosis which contributes to the decline in cardiac function and eventual failure. Matrix turnover is critical in the days and weeks following MI, however, the functions of specific ECM components in this process remain poorly defined. 1-4
It has been accepted that type I and type III collagen are major constituents of the cardiac ECM that provides structural and mechanical support to the heart, and act as signaling conduits between myocardial cells.2-4 While the ECM field has focused upon these fibrillar collagens, our recent studies have demonstrated that type VI collagen induces myofibroblast differentiation in vitro and that its deposition is enhanced in vivo following MI.5,6
Type VI collagen mutations can cause Bethlem Myopathy (BM), an age-related disease characterized by skeletal muscle weakness and limited life span.7 Type VI collagen is a non-fibrillar collagen that assembles end-to-end in a beaded filament arrangement.8 Typically interspersed with types I and III collagen, collagen VI forms a microfilament network that organizes the fibrillar collagens and anchors these to the basement membrane.9 A model for BM was generated by targeted deletion of the Col6a1 gene which causes early-onset myopathy10-11. Although skeletal muscle defects caused by collagen type VI mutations have been described, none have determined the consequences of its absence using MI injury models. The Col6a1 null mutant mouse provides an in vivo model to determine the impact of collagen VI deficiency in skeletal muscle12,13 as well as in other tissues such as adipose, cartilage, brain and tendon14-16. Here, we demonstrate that the absence of type VI collagen provides a profound beneficial effect on post-MI cardiac function and remodeling.
Materials and Methods
Animal model
Seventy-five male Col6a1-/- and wild-type (WT) mice in the CD-1 background and at 12-16 weeks of age were used in this study.
Surgical induction of MI
Mice were anesthetized by i.p. injection of sodium pentobarbital (70 mg/kg) and the heart accessed via left thoracotomy and MI induced by permanent occlusion of the left anterior descending artery (LAD).
Histological assessment of apoptosis, fibrosis and infarct size
Animals were euthanized and cryosections prepared for apoptosis assessment using TUNEL staining method as previously described. 6,7 Additional hearts were embedded in paraffin and entire hearts sectioned serially for assessment of collagen deposition using Masson’s trichrome, picrosirius red (PSR), and PSR under polarized light. Infarct sizes were quantitated by 2,3,5-triphenyltetrazolium chloride (TTC) staining.
Echocardiographic assessment
Two-dimensional echocardiography was performed and calculations carried out offline by double-blinded reviewers using the Vevo 770/3.0 system and software (VisualSonics, Toronto, Canada).
Statistical analysis
Data analysis was performed using Graphpad Prism 4.0 software (Graphpad Software, La Jolla, CA). Significance was determined by ANOVA with Bonferroni’s post-test (p<0.05 considered significant).
Results
Physiological measurements of Col6a1-/- vs. WT mice pre- and post-MI
The physiological features of WT and Col6a1-/- mice are outlined in Table 1. Col6a1-/- mice were consistently smaller than WT mice, and heart weights were lower in Col6a1-/- mice in sham and MI groups when compared to WT. After MI the ejection fraction, fractional shortening and cardiac index were higher in Col6a1-/- hearts whereas LV mass and wall thinning were reduced.
Table 1.
Echocardiographic assessment of WT and Col6a1-/- mice 8 weeks post-MI
| +/+ Sham | -/- Sham | +/+ MI | -/- MI | |
|---|---|---|---|---|
| Body Weight (g) | 44.0 ± 2.8 | 31.0 ± 1.2* | 42.4 ± 1.3 | 32.5 ± 1.0§ |
| Heart Weight (mg) | 206 ± 4 | 161 ± 6* | 242 ± 4 | 172 ± 4§ |
| HW/BW Ratio (g ×100) | .46 ± .02 | .48 ± .01 | .52 ± .01 | .52 ± .01 |
| LV Mass (mg) | 151 ± 7 | 110 ± 5* | 186 ± 21 | 126 ± 7§ |
| HR (bpm) | 506 ± 6 | 510 ± 17 | 554 ± 17 | 536 ± 24 |
| RWT | 0.45 ± 0.02 | 0.46 ± 0.03 | 0.35 ± 0.03* | 0.43 ± 0.05 |
| % FAC | 45.8 ± 4.4 | 45.1 ± 1.6 | 18.4 ± 3.2* | 31.3 ± 2.6§† |
| LVID;d (mm) | 4.1 ± 0.1 | 3.7 ± 0.2 | 4.8 ± 0.3 | 4.0 ± 0.2 |
| LVID;s (mm) | 2.8 ± 0.1 | 2.3 ± 0.3 | 4.1 ± 0.4* | 3.0 ± 0.2§ |
| MV E/A Ratio | 1.38 ± 0.08 | 1.47 ± 0.08 | 1.36 ± 0.16 | 1.26 ± 0.06 |
| MV DT (mm/s2) | 12.56 ± 2.03 | 10.97 ± 1.43 | 7.78 ± 2.89 | 10.63 ± 2.19 |
| CI (ml/min/g) | 0.65 ± 0.01 | 0.63 ± 0.01 | 0.48 ± 0.04* | 0.55 ± 0.02§† |
BW = body weight, HW = heart weight, HR = heart rate, RWT = relative wall thickness, FAC = fractional area change, LVID;d = left ventricular internal diameter; diastole, LVID;s = left ventricular internal diameter; systole, MV = mitral valve, DT = deceleration time, CI = cardiac index.
= p<0.05 vs. WT sham;
= p<0.05 vs. WT MI;
= p<0.05 vs. Col6a1-/- sham. (n=8/group).
Myocardial integrity and cardiac function are significantly preserved in Col6a1 -/- mice
Whole heart images taken 8 weeks post-MI demonstrate preserved LV wall integrity in the Col6a1-/- hearts (Figure 1A). Gross observation and measurement of 2D guided M-mode tracings show improved wall thickness, chamber dimension, and anterior wall kinesis in Col6a1-/- MI mice (Figure 1B), illustrating the preserved myocardial function in the knockout hearts. Area at risk % (AAR) (Online Figure I) and infarct/LV area ratios were not significantly different in the Col6a1-/- mice versus WT 3 days following MI, however these ratios were diminished in the knockouts compared to WT at 8 weeks post-MI (0.25 ± 0.01 vs. 0.37 ± 0.05 respectively, Figure 1C, D).
Figure 1. Cardiac structure and function are preserved in Col6-/- mice following MI.
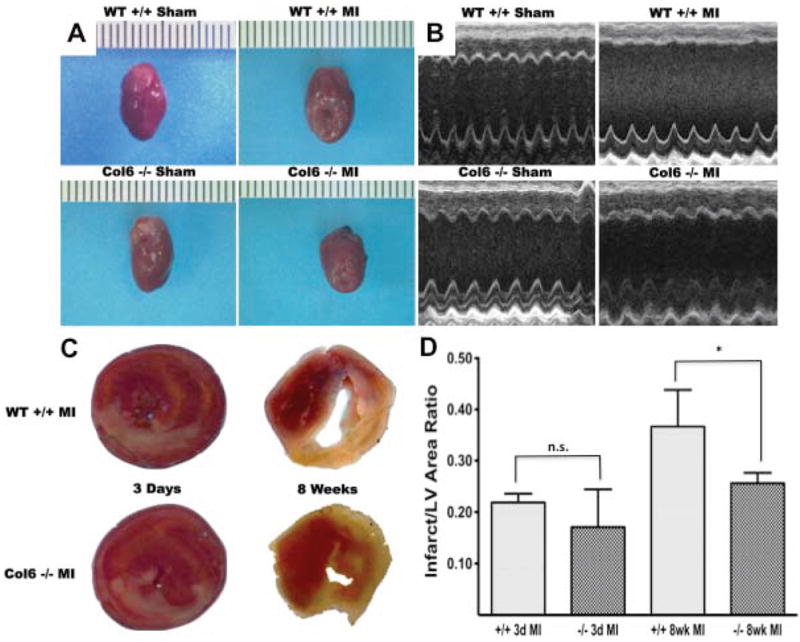
(A and B) Representative images taken at 8 weeks post-MI demonstrate preservation of LV wall integrity, greater wall kinesis, and reduced chamber size in Col6-/- hearts (n=8). (C and D) TTC staining performed on 3 day post-MI hearts revealed no significant differences in Inf/LV area ratio in Col6-/- versus WT mice, however infarct size at 8 weeks is significantly greater in WT (n=4/group). *p<0.05.
Reduced collagen volume and decreased long-term myocyte/non-myocyte apoptosis in the Col6a1-/- hearts
Quantitative analysis of total collagen content in the LV and infarcted zones was visualized by Masson’s trichrome and PSR staining (Figure 2A). Collagen volume (percent collagen/LV area) was significantly reduced in Col6a1-/- MI mice compared to WT (15.89 ± 0.84 vs. 29.42 ± 4.05, Figure 2B; p<0.05). Collagen levels of WT and Col6a1-/- sham mice were not significantly different (data not shown).
Figure 2. Decreased collagen content in Col6-/- mice and temporal apoptotic responses post-MI.
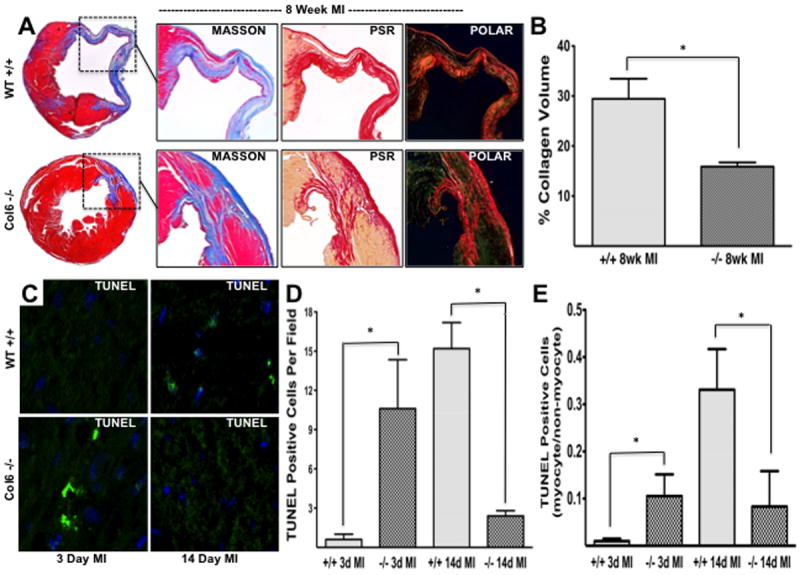
(A) Representative images (2x) of 8 week post-MI WT and Col6-/- hearts stained with Masson’s trichrome illustrates a reduction in infarct size, chamber dilation, and remote fibrosis in Col6-/- mice. Magnified images (4x) of serial sections stained with Masson’s trichrome, PSR, and PSR using polarized microscopy reveal increased collagen deposition and more densely packed collagen in infarct scars of WT mice 8 weeks post-MI. (B) Quantification of collagen volume from stained sections of WT and Col6-/- 8 week post-MI hearts (% stained/LV area). (C) TUNEL positive cells (per field) at 3 days and 14 days post-MI revealed an initial increase in apoptosis in Col6-/- mice at 3 days which was reversed by 14 days. (C) The ratio of myocyte/non-myocyte apoptosis increased at 14 days post-MI in WT mice (n=3/group, 5 fields per heart). *p<0.05.
TUNEL staining revealed an initial increase in apoptosis within the infarcted area of the Col6a1-/- hearts at 3 days (acute phase) followed by a reduction by 14 days (chronic phase) relative to WT hearts (Figure 2C, D; p<0.05). Importantly, the ratio of myocyte/non-myocyte apoptosis by day 14 was significantly decreased in the knockout hearts compared to WT (Figure 2E).
Improved cardiac function and LV dimensions in Col6a1-/- mice 10 days - 8 weeks post-MI
Cardiac function was assessed using 2D and Doppler echocardiography on mice ranging from 3 days to 8 weeks following MI. Echocardiography at 3 days (Figure 3A) revealed that cardiac function is not significantly different between the null and WT MI mice. Differences in LV diastolic volume were also not apparent at this early time (Figure 3B). However, serial measurements of function and remodeling revealed that in Col6a1-/- mice, ejection fraction was preserved as early as 3 weeks and persisted out to 8 weeks post-MI (Figure 3C, E; 43.9 ± 3.3% vs. 29.1 ± 4.3%; p<0.05) and LV chamber volume was reduced beginning at 10 days post-MI (Figure 3D, F; 25.8 ± 7.9% vs. 62.6 ± 16.5% increase in LV volume over shams; p<0.05). Cardiac index increased in Col6a1-/- mice post-MI versus WT (0.55 ± 0.02 vs. 0.48 ± 0.04; p<0.05, Table 1).
Figure 3. Function and geometry of post-MI hearts are improved in Col6-/- mice from 3 days to 8 weeks post-MI.
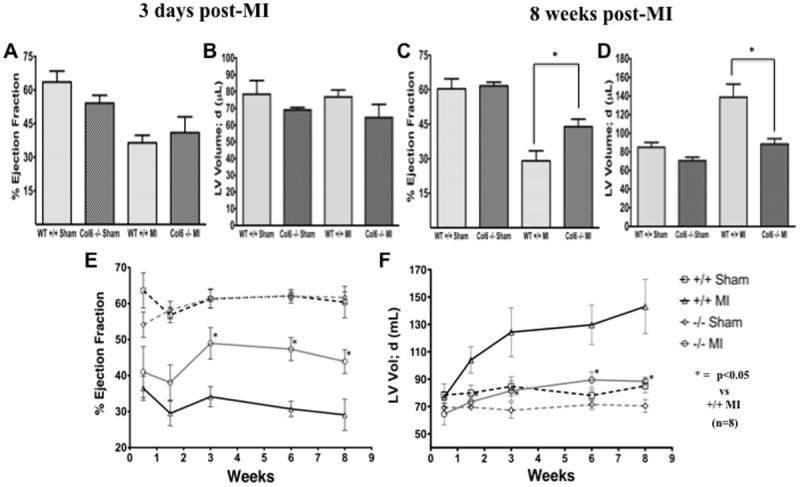
(A and B) Echocardiographic assessment of sham and MI groups at 3 days post-MI show similar function and geometry between MI groups (n=8). (C and D) Echocardiography at 8 weeks post-MI revealed preserved systolic function in Col6-/- MI mice compared to WT MI. Diastolic LV chamber dilation was significantly attenuated in the post-MI Col6-/- MI mice compared WT MI mice (n=8). (E and F) Improved remodeling and cardiac function were evident at 10 days and 3 weeks post-MI, respectively, as measured by serial echocardiography at the indicated post-MI time points. *p<0.05.
Discussion
These data are the first to demonstrate that the lack of collagen VI significantly and paradoxically improves post-MI remodeling in response to permanent LAD occlusion. We originally predicted that the collagen VI deficient mice would suffer deficits in remodeling, since this collagen has been proposed to play a critical role in organizing and anchoring the fibrillar type I and III collagen network.9,17 This along with the information taken from other tissues (skin, tendon, skeletal muscle) and collagen knockouts (XV) have all shaped our original hypothesis that deletion of collagen VI would result in a loss of function.
Elegant EM studies uncovered the function of collagen VI to organize the fibrillar collagen (mainly type I) network in skin.9 Izu et al. (2011) recently reported significantly decreased collagen diameter and compromised mechanical properties in tendons of the Col6a1-/- mice. Another intriguing study demonstrated that collagen XV null mice with experimentally-induced hypertension suffered from a poorly organized fibrillar collagen network, impaired microvascular hemodynamics, and irregularly organized cardiac myocytes.18 Our results are particularly “paradoxical” due to the known skeletal muscle phenotype of this knockout model originally reported as a model of Bethlem Myopathy by Bonaldo and colleagues (1998). This seminal study was followed by several subsequent reports indicating that the skeletal myocytes suffered from premature mitochondrial transition pore opening and apoptosis leading to skeletal muscle dystrophy as the mice aged beyond 32 weeks.12 Prevention of mitochondrial transitional pore opening has been shown to be cardioprotective,19 indicating that this may be a common target for prevention of skeletal and cardiac myocyte dysfunction and death. It is important to note in our study that we utilized young mice for our MI studies (12-16 weeks), an age at which there were no differences in baseline cardiac function and no outward signs of skeletal muscle weakness. It is of interest to perform aging studies on the Col6a1-/- mice, to determine if the cardioprotection seen in the current study persists after the onset of Bethlam Myopathy.
The improvement in post-MI remodeling in the Col6a1-/- hearts is, at least in part, likely due to accelerated remodeling and apoptosis followed by a reduction in chronic apoptosis. The increased apoptosis at 3 days post-MI in the knockouts suggests that injury responses began earlier in these animals, which appears to be beneficial to the long term outcomes. Our data also indicate that less myocyte apoptosis occurs in the knockouts after 14 days which supports the notion that wound healing occurs, and is completed, earlier in the Col6a1-/- mice. Furthermore, the more chronic apoptosis evident at 14 days post-MI in the WT mice may contribute to the increased scarring and fibrosis followed by long-term loss of cardiac function. Other cardioprotective mechanisms may exist as well, since the structure-function relationships relating to collagen VI and the fibrillar collagens have not been established in the myocardium. The previous studies from skin9 and tendon16 (discussed above) suggest that the absence of collagen VI affects fibrillar collagen network organization which, in our case, could be creating a more biomechanically advantageous environment for post-MI wound healing. Alternatively, the possibility exists that cardioprotection may involve changes in mitochondrial function,19 however mitochondrial function is altered and induces apoptosis in the skeletal muscle of Col6a1-/- mice12. The mechanisms responsible for these contrasting phenotypes in cardiac and skeletal muscle are not known and require further investigation.
In conclusion, the confluence of previous reports and our current findings demonstrate a critical and novel role for type VI collagen in myocardial injury and remodeling. Importantly, our study is the first to describe the cardioprotective effects of collagen VI deletion to enhance post-MI cardiac function and limit aberrant remodeling, and identifies a potentially novel target to treat post-ischemic injury in the myocardium.
Supplementary Material
Novelty and Significance.
What is known?
Collagen VI organizes and anchors the fibrillar collagen network in many tissues; its deficiency causes age-related defects in skeletal muscle function.
Post-infarction remodeling is critically dependent upon collagen and extracellular matrix (ECM) synthesis to stabilize the scar and improve long-term remodeling.
The prevailing idea is that deposition of fibrillar types I and III collagen increase following myocardial infarction (MI) and are the key mediators of infarct scar assembly.
Type VI collagen deposition increases after MI, however, the importance of this non-fibrillar collagen in post-MI remodeling is unknown.
What New Information Does This Article Contribute?
Knockout of type VI collagen improves post-MI remodeling by limiting infarct size, collagen deposition, and chronic myocyte apoptosis.
Absence of collagen VI preserves long-term cardiac performance following MI, prevents left ventricular (LV) wall thinning and limits LV chamber dilation.
The beneficial effects of collagen VI deletion suggest that this non-fibrillar collagen, in addition to types I and III, plays a key role in post-MI wound healing and remodeling.
Type VI collagen is a non-fibrillar, filamentous collagen produced by activated fibroblasts that plays roles in fibrillogenesis, organization and anchoring of fibrillar collagens. Our goal was to determine whether collagen VI contributes significantly to post-MI wound healing by employing a relevant in vivo MI model (collagen VI deficient mice). This study is the first to report an unexpected and novel cardioprotective effect of collagen VI deletion to preserve cardiac structure and prevent pathological remodeling following MI. These findings may provide the basis for the development of collagen-based therapies to limit adverse post-MI remodeling.
Acknowledgments
We express our appreciation to the NEOMED Department of Integrative Medical Sciences Cardiovascular Focus Group for their expert critiques and suggestions, and to Dr. Vahagn Ohanyan and the Echocardiography Core for their expert assistance.
Sources of funding
NIH HL-079969; Ohio Board of Regents Grant (JGM)
Non-Standard Abbreviations and Acronyms
- AMI
acute myocardial infarct
- ECM
extracellular matrix
- EF
ejection fraction
- LAD
left anterior descending artery
- MI
myocardial infarction
- PSR
picrosirius red
- TTC
2,3,5-triphenyltetrazolium chloride
Footnotes
Disclosures
None
References
- Lindsey ML, Mann DL, Entman ML, Spinale FG. Extracellular matrix remodeling following myocardial injury. Ann Med. 2003;35:316–26. doi: 10.1080/07853890310001285. [DOI] [PubMed] [Google Scholar]
- Bowers SL, Banerjee I, Baudino TA. The extracellular matrix: at the center of it all. J Mol Cell Cardiol. 2010 Mar;48(3):474–82. doi: 10.1016/j.yjmcc.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–6. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- Jourdan-Lesaux C, Zhang J, Lindsey ML. Extracellular matrix roles during cardiac repair. Life Sci. 2010;87:391–400. doi: 10.1016/j.lfs.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, Folkesson HG, Horne WI, Doane KJ, Meszaros JG. Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling. Am J Physiol Heart Circ Physiol. 2006;290:H323–30. doi: 10.1152/ajpheart.00321.2005. [DOI] [PubMed] [Google Scholar]
- Bryant JE, Shamhart PE, Luther DJ, Olson ER, Koshy JC, Costic DJ, Mohile MV, Dockry M, Doane KJ, Meszaros JG. Cardiac myofibroblast differentiation is attenuated by alpha(3) integrin blockade: potential role in post-MI remodeling. J Mol Cell Cardiol. 2009;46:186–92. doi: 10.1016/j.yjmcc.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42:673–85. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbatti A, Bonaldo P, Doliana R. Type A modules: interacting domains found in several non-fibrillar collagens and in other extracellular matrix proteins. Matrix. 1993;13:297–306. doi: 10.1016/s0934-8832(11)80025-9. [DOI] [PubMed] [Google Scholar]
- Keene DR, Engvall E, Glanville RW. Ultrastructure of type VI collagen in human skin and cartilage suggests an anchoring function for this filamentous network. J Cell Biol. 1988;107:1995–2006. doi: 10.1083/jcb.107.5.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–40. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- Maraldi NM, Sabatelli P, Columbaro M, Zamparelli A, Manzoli FA, Bernardi P, Bonaldo P, Merlini L. Collagen VI myopathies: from the animal model to the clinical trial. Adv Enzyme Regul. 2009;49:197–211. doi: 10.1016/j.advenzreg.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–71. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–91. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos LG, Youn I, Bonaldo P, Guilak F. Developmental and osteoarthritic changes in Col6a1-knockout mice: biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum. 2009;60:771–9. doi: 10.1002/art.24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JS, Dubal DB, Kim DH, Legleiter J, Cheng IH, Yu GQ, Tesseur I, Wyss-Coray T, Bonaldo P, Mucke L. Collagen VI protects neurons against Aβ toxicity. Nat Neurosci. 2009;12:119–21. doi: 10.1038/nn.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izu Y, Ansorge HL, Zhang G, Soslowsky LJ, Bonaldo P, Chu ML, Birk DE. Dysfunctional tendon collagen fibrillogenesis in collagen VI null mice. Matrix Biol. 2011;30:53–61. doi: 10.1016/j.matbio.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamhart PE, Meszaros JG. Non-fibrillar collagens: key mediators of post-infarction cardiac remodeling? J Mol Cell Cardiol. 2010;48:530–7. doi: 10.1016/j.yjmcc.2009.06.017. [DOI] [PubMed] [Google Scholar]
- Rasi K, Piuhola J, Czabanka M, Sormunen R, Ilves M, Leskinen H, Rysä J, Kerkelä R, Janmey P, Heljasvaara R, Peuhkurinen K, Vuolteenaho O, Ruskoaho H, Vajkoczy P, Pihlajaniemi T, Eklund L. Collagen XV is necessary for modeling of the extracellular matrix and its deficiency predisposes to cardiomyopathy. Circ Res. 2010;107(10):1241–52. doi: 10.1161/CIRCRESAHA.110.222133. [DOI] [PubMed] [Google Scholar]
- Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.