

Abstract
To determine if patients with myasthenia gravis (MG) have antibodies to agrin, a proteoglycan released by motor neurons and is critical for neuromuscular junction (NMJ) formation, we collected serum samples from 93 patients with MG with known status of antibodies to acetylcholine receptor (AChR), muscle specific kinase (MuSK) and lipoprotein-related 4 (LRP4) and samples from control subjects (healthy individuals and individuals with other diseases). Sera were assayed for antibodies to agrin. We found antibodies to agrin in 7 serum samples of MG patients. None of the 25 healthy controls and none of the 55 control neurological patients had agrin antibodies. Two of the four triple negative MG patients (i.e., no detectable AChR, MuSK or LRP4 antibodies, AChR-/MuSK-/LRP4-) had antibodies against agrin. In addition, agrin antibodies were detected in 5 out of 83 AChR+/MuSK-/LRP4- patients but were not found in the 6 patients with MuSK antibodies (AChR-/MuSK+/LRP4-). Sera from MG patients with agrin antibodies were able to recognize recombinant agrin in conditioned media and in transfected HEK293 cells. These sera also inhibited the agrin-induced MuSK phosphorylation and AChR clustering in muscle cells. Together, these observations indicate that agrin is another autoantigen in patients with MG and agrin autoantibodies may be pathogenic through inhibition of agrin/LRP4/MuSK signaling at the NMJ.
Introduction
Autoimmune MG is the most common disorder of NMJ, affecting nearly 20 per 100,000 people in various populations [1]–[5]. MG patients show characteristic fatiguing weakness of voluntary ocular, bulbar and limb muscles, dysarthria, dysphagia and in severe cases death from difficulty with breathing. The symptoms and pathology of MG are known to be due to an antibody-mediated, autoimmune attack directed against molecules at the NMJ. Autoantibodies against AChR can be detected in the circulation of ∼80–90% of MG patients [6], [7]. Evidence from classic experiments indicates the anti-AChR antibodies are pathogenic [8]–[11].
However, AChR antibodies cannot be detected in ∼10–20% of generalized MG patients. Recent studies shed light on understanding the pathology in these “seronegative” MG. Approximately 40–70% of the seronegative patients have antibodies against MuSK [4], [5], [12]–[15]. Our group and others also reported that 2–50% of AChR and MuSK double seronegative patients have anti-LRP4 antibodies [16]–[19].
However, in at least 2–5% of MG patients identifiable antibodies to a known autoantigen have not been detected. The NMJ is a cholinergic synapse that rapidly conveys signals from motoneurons to muscle cells [20]–[26]. Previous studies suggest a critical role of the agrin/LRP4/MuSK pathway in formation of the NMJ. Neuronal agrin is a large extracellular matrix protein utilized by motoneurons to induce AChR clustering and postjunctional differentiation [27]–[32]. Agrin binds to LRP4 to form a tetrameric complex, which interacts with and activates MuSK to initiate downstream signaling cascades mediating AChR clustering [33], [34]. Ablation of the genes encoding for agrin, MuSK or LRP4 prevents NMJ formation [35]–[41]. We posit that agrin may be a potential autoantigen for its function at the NMJ and spatial proximity with AChR, MuSK and LRP4.
Here we show that approximately 50% of known triple seronegative MG patients (i.e., no detectable AChR, MuSK or LRP4 antibodies, AChR-/MuSK-/LRP4-) have serum antibodies against agrin, representing approximately 2–3% of all MG patients in our study. The agrin autoantibodies recognized agrin protein expressed in transfected HEK293 cells and inhibited agrin-induced AChR clustering in cultured myotubes. Our results indicate the potential involvement of agrin antibody in the pathogenesis of AChR/MuSK/LRP4-seronegative MG, thus defining one novel immunological form of the disease. Measurement of agrin antibodies would also substantially aid diagnosis and clinical management. In addition, agrin antibodies are also found in the serum of patients with antibodies to other components of the NMJ such as AChR, although not to date in our studies in those with MuSK antibodies. Studies of those patients might contribute to understanding the pathogenic mechanisms of the disease.
Materials and Methods
Ethics statement
Serum samples from Wayne State University were all archival and had been previously collected as part of prior Wayne State University IRB approved research studies or as additional serum obtained at the time of diagnostic studies, with informed consent for all samples. All samples were anonymized.
Patient sera
Serum of 93 patients with MG had previously been tested for anti-AChR and anti-MuSK antibodies or tested for these antibodies for this study. Additionally we tested serum of 6 patients with MG in whom we had no data on antibody status to AChR, MuSK but were known to be negative for LRP4 antibodies. All of these were negative for agrin but since we have no data on the antibody status of these sera, they have not been included in the statistical analysis. Patients and healthy volunteers gave their written informed consent. Serum samples were assayed for AChR binding antibody at ARUP Laboratories (Salt Lake City, UT; positive ≥0.5 nM/L), at the Mayo Clinic (Rochester, MN; positive >0.02 nM/L) or at Athena laboratories (≥0.5 nM/L). Anti-MuSK was either assayed by Dr. Angela Vincent as part of a multi-institutional study of serum from MG patients (positives as defined previously [12]) or by a commercial laboratory (Athena). LRP4 antibodies were examined in our previous report [16]. Seropositive MG was defined as AChR, MuSK and/or LRP4 antibodies positive. Normal control sera were obtained from age and gender-matched volunteers serving as controls of other studies of MG. In addition, sera from patients with the following diseases were examined: amyotrophic lateral sclerosis (ALS) (n = 9); chronic inflammatory demyelinating polyneuropathy (CIDP) (n = 4); primary CNS Sjogrens syndrome (n = 2); Guillain-Barre syndrome (GBS)/acute inflammatory demyelinating polyneuropathy/(AIDP) (n = 6); acute motor axonal neuropathy (AMAN) (n = 1); GBS with concomitant Isaac's syndrome (n = 1); CNS Lyme disease (n = 1); multiple sclerosis (MS) (n = 20); paraneoplastic neuropathies (n = 2); polymyositis in a patient with primary Sjogrens syndrome (n = 1); polychondritis with CNS vasculitis (n = 1); neuromyelitis optica spectrum disorder (NMOSD) (n = 1); inflammatory myelopathies (not transverse myelitis or NMO)(n = 3); peripheral neuropathy of unknown etiology (n = 1); and neuroscarcoisis (n = 1). Overall, we tested sera from 93 immunologically-characterized MG patients, including AChR+/MuSK-/LRP4- (n = 83), AChR-/MuSK+/LRP4- (n = 6) and AChR-/MuSK-/LRP4- (n = 4). There were also 4 who were previously shown to be LRP4- but of unknown status re: AChR and MuSK antibody (negative for agrin as noted in Results, these were not among the 93 in the final data analysis) and sera from normals (n = 25) and other disease controls (n = 55) as indicated above were assayed for antibodies to agrin.
Recombinant protein production and purification
pFlag-agrin construct was described previously [42]. Of note, this construct contains 6XHis tag between Flag-tag and agrin coding sequence, enabling metal affinity chromatography for recombinant agrin protein. HEK293 cells were transfected with pFlag-agrin and 24 hr later, cells were switched to Dulbecco's Modified Eagle Medium supplemented with reduced concentration (0.5%) of fetal bovine serum. Conditioned media containing secreted agrin proteins were harvested 24 hr later and were purified by affinity chromatography using TALON Resins (BD Biosciences). Expression and purification of agrin proteins were verified by Western blot with anti-Flag antibody (Sigma).
ELISA detection of antibodies to agrin
Maxi-Sorp Immuno 96-well Plates (Nunc) were coated with 50 μl of 1 μg/ml agrin in the coating buffer containing 50 mM carbonate (pH 9.6) at 40C overnight, washed six times with TBST (0.1% Tween 20 in 50 mM Tris, 150 mM NaCl, pH 7.6) and incubated with the blocking buffer containing 5% nonfat milk in TBST to block non-specific binding. Sera were diluted 1∶10 in the blocking buffer (100 μl per well) and incubated for 1 hr at 370C. After six washes with TBST, the wells were incubated with alkaline phosphatase (AP)-goat anti-human IgG+IgM+IgA as the secondary antibody (Abcam), diluted 1∶30,000 in TBST, at 37°C for 1 hr. Activity of immobilized AP was measured by optical density (OD) assay (at 405 nm) following incubation in the substrate buffer containing 0.5 mM MgCl2, 3 mg/ml p-nitrophenyl phosphate (pNPP) and 1 M diethanolamine (DEA), at room temperature for 30 min. Each sample was assayed in duplicate and repeated more than three times. Nonspecific signal was determined by OD reading of wells coated with the coating buffer alone followed by incubation of secondary antibody and substrate. Cut-off value was set as mean +3 standard deviation (SD) of control normal human serum, representing confidence of 99.7%.
Immunoprecipitation of agrin by serum samples with agrin autoantibodies
Conditioned media containing Flag-agrin were incubated with 10 μl of sera (sera 1–21, 1–106, 2–17, 2–27 and normal human serum control) at 4°C overnight with agitation, followed by 2 hr incubation with 50 μl Protein-G beads at 4°C. Bead-immobilized proteins were subjected to SDS-PAGE and western blotting with anti-Flag antibody.
Immunostaining of agrin-transfected HEK293 cells by serum samples
HEK293 cells were transfected with pFlag-agrin and 72 hr later, cells were washed briefly with PBS and fixed in 4% paraformaldehyde. After permeabilization with 0.5% Triton X-100 in PBS for 5 min, cells were blocked with blocking buffer containing 10% normal goat serum and 1% BSA in PBS. MG patient and normal human control serum samples were diluted 1∶10 in blocking buffer containing rabbit anti-Flag antibody (1∶500 dilution) and incubated with cells at 4°C overnight. After wash, FITC labeled goat anti-human IgG secondary antibody (Southern Biotech) and Alexa 594 labeled donkey anti-rabbit secondary antibody (Invitrogen) were added and incubated for 1 hr. After wash, cells were mounted and viewed under a Zeiss epifluoresence microscope. At least 5 views per dish and at least 2 dishes were scored in two independent experiments. All samples were examined blindly without previous information of the diagnosis.
Effects of agrin positive sera on agrin-induced MuSK phosphorylation and AChR clustering
Agrin-induced MuSK phosphorylation and AChR clustering were assayed as previously described [33], [43], [44]. Briefly, C2C12 myotubes were treated with neural agrin (10 ng/ml) [33] together with agrin positive sera (1∶100 dilution) (sera 1–21, 1–106, 2–17, 2–27) or normal human control serum for 30 min. After brief wash, cells were lyzed in RIPA buffer and incubated with anti-MuSK antibody at 4°C overnight with agitation, followed by 2 hr incubation with 50 μl Protein-G beads at 4°C. Bead-immobilized proteins were subjected to SDS-PAGE and western blotting with anti-phospho-tyrosine antibody 4G10 (Millipore). For AChR clustering assay, myotubes were treated with neural agrin (10 ng/ml) together with agrin positive sera (1∶100 dilution) (sera 1–21, 1–106, 2–17, 2–27) or normal human control serum for 16 hr, then fixed in 4% paraformaldehyde, and incubated with 50 nM rhodamine-conjugated-bungarotoxin (R-BTX) (Invitrogen) to label AChR clusters. Myotubes were viewed under a Zeiss epifluoresence microscope and AChR clusters with diameters or a longer axis ≥4 μm were scored. At least 10 views per dish and at least 2 dishes were scored in each of three independent experiments.
Statistical Analysis
For ELISA examination of control and MG patient sera, all samples were tested in triplicate in three independent experiments. The cut-off value was set as mean +3 SD of normal human serum samples to represent 99.7% confidence. For AChR clustering assay, data of multiple groups was analyzed by ANOVA, followed by a student-New-man-Keuls test. Differences were considered significant at p<0.05.
Results
Detection of agrin autoantibodies in sera of MG patients
To determine whether sera of MG patients have agrin autoantibodies as well as to characterize those sera with regard to antibodies to other known autoantigens at the NMJ, we generated Flag/His-tagged rat agrin (His1137 to Pro1940). The purified protein resolved around 120 kDa on SDS-PAGE. Moreover, it could be detected by a commercial antibody against the Flag epitope (data not shown). The agrin protein was used in ELISA assays for autoantibodies in sera from MG patients as well as various groups of control individuals. With the mean plus 3 SD of normal sera as cut-off, none of the serum samples from normal individuals were positive for agrin antibodies. No positive sera were detected from patients with non-MG neurological disorders as defined in methods (Fig. 1). Of 93 MG patients, 7 were positive for agrin autoantibodies: 5 were AChR+, but MuSK- and LRP4- patients and 2 were from patients who were ‘triple seronegative’ (no AChR, MuSK or LRP4 antibodies) (Fig. 2). As noted earlier, there were sera from 6 patients with MG who were known to be negative for LRP4 antibodies but had not been tested for AChR and MuSK antibodies and they were all negative for agrin antibodies (data not shown in Figs. 1 or 2).
Figure 1. Detection of agrin autoantibodies in MG patient samples.
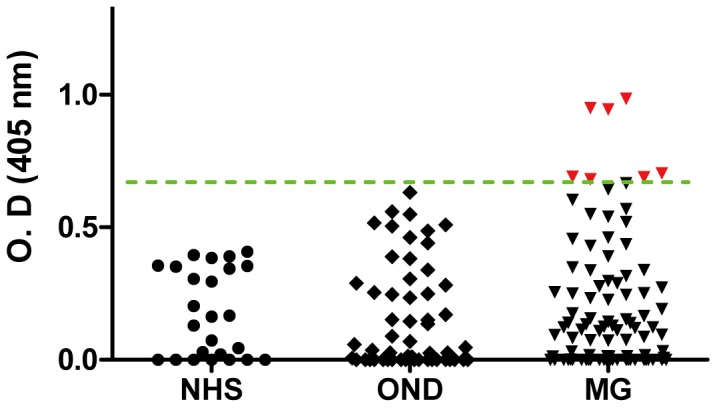
Optical density readings of normal human serum were 0.18 ± 0.16 (mean ± SD, n = 25). The green dotted line was set as mean + 3 SD to indicate the cut-off. The red dots indicate positive for agrin antibodies. NHS, normal human serum; OND, other neurological diseases, n = 55; MG, myasthenia gravis, n = 93.
Figure 2. Distribution of agrin autoantibodies among MG patients.
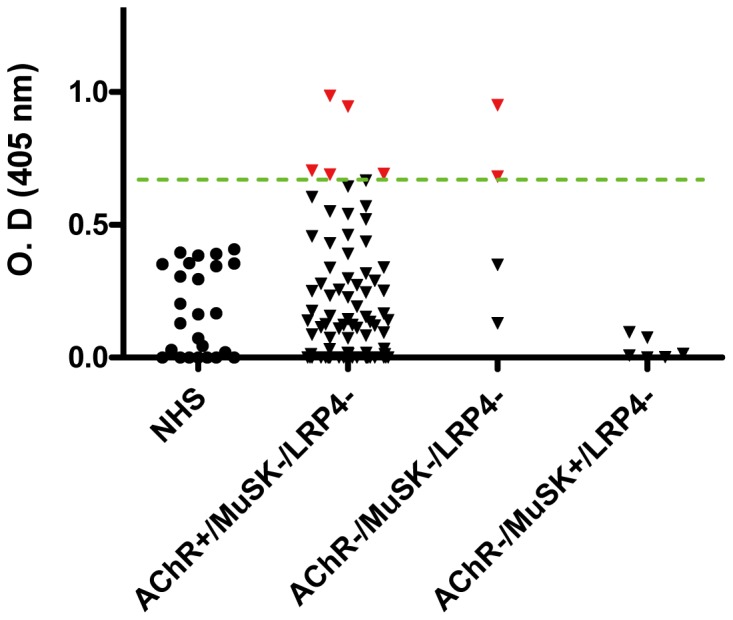
Of 93 MG samples previously analyzed for antibody to AChR, MuSK and LRP4, 83 were AChR+/MuSK-/LRP4-; 4 were triple seronegative (AChR-/MuSK-/LRP4-) and 6 were AChR-/MuSK+/LRP4-. The cut-off, indicated by the green line, was set as mean + 3 SD. The red dots indicate positive for agrin antibodies.
To confirm that the target antigen of these sera was agrin, rather than any contaminant in the agrin preparation, we examined whether the agrin positive sera could recognize agrin in soluble form. Four agrin+ sera were incubated with Flag-agrin conditioned media. The immunocomplex was purified by protein G immobilized on beads, resolved by SDS-PAGE and subjected to western blot analysis with anti-Flag antibody. As expected, agrin was not detectable in the immunocomplex by normal human serum. However, Flag-tagged agrin was detected in the precipitates by 4 agrin positive sera, indicating that agrin autoantibodies were able to recognize agrin protein in solution from transfected cells (Fig. 3).
Figure 3. Recognition of agrin protein by serum samples with agrin autoantibodies.

Conditioned media from Flag-agrin-transfected HEK293 cells were incubated with serum samples with agrin antibodies or normal human serum samples. Immunocomplex and conditioned media (to indicate equal amounts of input) were subjected to Western blotting with anti-Flag antibody.
To further confirm that the sera are able to recognize agrin in natural condition, we examined whether the agrin positive sera could detect agrin in transfected cells. One agrin positive serum (2–17) was used to stain HEK293 cells transfected with Flag-agrin construct. As expected, agrin positive serum was able to detect transfected agrin in HEK293 cells, co-staining with anti-Flag antibody. However, Flag-tagged agrin was not detected by normal human serum, indicating that agrin autoantibodies were able to recognize agrin protein in transfected cells (Fig. 4).
Figure 4. Recognition of agrin from transfected HEK293 cells by agrin+ serum.

Agrin positive serum 2–17 stained positively with HEK293 cells transfected with Flag-agrin construct, co-staining with anti-Flag antibody. Normal human serum cannot recognize agrin-transfected cells.
Alteration of agrin-induced MuSK phosphorylation and AChR clustering by agrin autoantibodies
Agrin induces Tyrosine phosphorylation of MuSK, which is critical for downstream cascades activation and agrin-induced AChR clustering [45]. We speculated the autoantibodies may change agrin-induced MuSK phosphorylation and thus AChR clustering. To test this hypothesis, C2C12 myotubes were treated with neural agrin alone or together with control or agrin+ sera, and examined for MuSK phosphorylation and AChR clusters. As shown in Fig. 5A, neural agrin induced MuSK phosphorylation in myotubes without serum treatment or treated with normal human serum. However, the phosphorylation was decreased in anti-agrin sera treated myotubes, especially 1–21 and 2–17 samples, indicating the blocking effect of the autoantibodies on agrin signaling. In AChR clustering assay (Figs 5B and 5C), agrin-induced AChR clusters in myotubes were not altered by normal human sera, but were inhibited by all agrin positive sera. These results suggest that agrin autoantibodies may have pathogenic role through its inhibition on AChR clustering induced by agrin.
Figure 5. Serum samples with agrin antibodies inhibit agrin-induced MuSK phosphorylation and AChR clustering in myotubes.

A, Anti-agrin autoantibodies inhibit agrin-induced MuSK phosphorylation. C2C12 myotubes were incubated without or with agrin and serum samples. Endogenous MuSK was precipitated by MuSK antibody and its phosphorylation was examined by 4G10 antibody. NS, no serum. B, Anti-agrin autoantibodies inhibit agrin-induced AChR clustering. Representative images. C, Quantitative data of basal (W/O agrin, green) and induced (W/agrin, red) AChR clusters. Data shown were mean ± SEM. *, p < 0.05, compared with control.
Discussion
About 80–90% of MG patients have detectable serum antibodies against AChRs with 40–70% of the remaining patients being positive for anti-MuSK antibodies and 2–50% for anti-LRP4 antibodies [12], [16]–[18], [46]. This would leave approximately 2–5% of the MG patients triple seronegative, i.e., without detectable antibodies against any known autoantigen (AChR, MuSK or LRP4) at the NMJ. This study presents evidence that anti-agrin autoantibodies exist in sera of the triple seronegative MG patients, as well as in patients with AChR antibodies. In our cohort of 93 serologicallly characterized patients, 7 were found positive for anti-agrin antibodies, accounting for about 7–8% of all MG patients. The presence of agrin antibodies in 2 out of 4 ‘triple seronegative’ patients with MG suggests that agrin may be a novel antigen in some triple seronegative MG patients. It is worth noting that we found no agrin antibody in any of our patients who had MuSK antibodies. Since none of the 93 patients tested for agrin antibodies in this study were positive for LRP4 antibody as tested in our previous paper [16], we do not know if some LRP4+ patients will be found to have agrin antibodies in future studies.
During the preparation of this study, a group from the United Kingdom reported the detection of agrin autoantibody in seronegative MG patients [47] using a cell-based assay. They found that in triple seronegative MG patients, 15% were anti-agrin positive. Also they showed high percentage of overlapping between AChR+ and agrin+ patients (13 AChR+ in total 24 agrin+ patients). Although detailed methodology was not included, the results from the report support what we observed in current study. Due to the time consuming nature of cell-based methods, our ELISA-based assay reported here would provide a convenient yet reliable clinical diagnostic test with quantitative value.
Pathogenic mechanisms of AChR antibodies have been well studied. In rabbit, mouse, and rat models of experimental autoimmune myasthenia gravis (EAMG), anti-AChR antibodies block the activity of the AChR [48], [49] and may accelerate the internalization and degradation of AChRs [50]. In addition, the autoantibodies may fix complement, leading to complement activation causing damage and simplification of the postsynaptic membrane of the NMJ [10], [51]–[54]. The AChR deficiency decreases the amplitude of miniature end-plate potentials (mEPPs) and hence that of end-plate potentials (EPPs), which consequently reduces the safety margin of neuromuscular transmission [10], [11]. On the other hand, MuSK antibodies seem to inhibit the activity of MuSK, leading to attenuation of agrin-induced AChR clustering thus reducing AChR levels at the junctional folds [55]–[59]. In addition, NMJs and AChR scaffolds are disrupted in MuSK antibody induced EAMG. However MuSK antibodies in MG patients are predominantly of the IgG4 subclass [60], [61] which does not bind and activate complement. Thus, it seems that MuSK antibody-associated MG may have different etiological and pathological mechanisms from those of the AChR antibody associated MG. This concept is also supported by the observation that MG patients with MuSK antibodies patients do not appear to have thymic hyperplasia or thymoma [62]–[66]. The pathogenic role of LRP4 autoantibodies has been presented in the EAMG recently by our lab [67].
Whether and how agrin autoantibodies are pathogenic requires further study. We have demonstrated that some agrin+ sera were able to inhibit agrin-induced AChR clustering which provides one possible pathologic role of these antibodies in vivo. It is of note that agrin protein has multiple isoforms and can be secreted by muscle and motor neuron (muscle and neural agrin, respectively) [68]. Neural agrin has up to 1000-fold greater AChR clustering activity compared to other isoforms and was used throughout this study. However the primary sequences between neural and muscle agrin mainly differ at the Z insert, only 8 amino acids [69]. Considering the large size of agrin, it is likely that the agrin autoantibodies also recognize muscle agrin. Whether the antibodies against muscle agrin are pathogenic would also be interesting to explore.
Funding Statement
This work was supported in part by grants from National Institutes of Health (WCX, LM), and by Parker Webber Chair in Neurology Endowment and the Mary Parker Neuroscience Fund (SR, RPL). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vincent A, Palace J, Hilton-Jones D (2001) Myasthenia gravis. Lancet 357: 2122–2128. [DOI] [PubMed] [Google Scholar]
- 2. Carr AS, Cardwell CR, McCarron PO, McConville J (2010) A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol 10: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phillips LH Jr (2003) The epidemiology of myasthenia gravis. Ann N Y Acad Sci 998: 407–412. [DOI] [PubMed] [Google Scholar]
- 4. Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ (2009) Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 72: 1548–1554. [DOI] [PubMed] [Google Scholar]
- 5. Richman DP, Agius MA (2003) Treatment of autoimmune myasthenia gravis. Neurology 61: 1652–1661. [DOI] [PubMed] [Google Scholar]
- 6. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD (1976) Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology 26: 1054–1059. [DOI] [PubMed] [Google Scholar]
- 7. Vincent A, Newsom-Davis J (1985) Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry 48: 1246–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patrick J, Lindstrom J (1973) Autoimmune response to acetylcholine receptor. Science 180: 871–872. [DOI] [PubMed] [Google Scholar]
- 9. Christadoss P, Krco CJ, Lennon VA, David CS (1981) Genetic control of experimental autoimmune myasthenia gravis in mice. II. Lymphocyte proliferative response to acetylcholine receptor is dependent on Lyt-1+23- cells. J Immunol 126: 1646–1647. [PubMed] [Google Scholar]
- 10. Lindstrom JM, Lennon VA, Seybold ME, Whittingham S (1976) Experimental autoimmune myasthenia gravis and myasthenia gravis: biochemical and immunochemical aspects. Ann N Y Acad Sci 274: 254–274. [DOI] [PubMed] [Google Scholar]
- 11. Kao I, Drachman DB (1977) Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Science 196: 527–529. [DOI] [PubMed] [Google Scholar]
- 12. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, et al. (2001) Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7: 365–368. [DOI] [PubMed] [Google Scholar]
- 13. Sanders DB, El-Salem K, Massey JM, McConville J, Vincent A (2003) Clinical aspects of MuSK antibody positive seronegative MG. Neurology 60: 1978–1980. [DOI] [PubMed] [Google Scholar]
- 14. Kumar V, Kaminski HJ (2011) Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 11: 89–96. [DOI] [PubMed] [Google Scholar]
- 15. Kim JY, Park KD, Richman DP (2011) Treatment of myasthenia gravis based on its immunopathogenesis. J Clin Neurol 7: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, et al. (2012) Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 69: 445–451. [DOI] [PubMed] [Google Scholar]
- 17. Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, et al. (2012) Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 259: 427–435. [DOI] [PubMed] [Google Scholar]
- 18. Higuchi O, Hamuro J, Motomura M, Yamanashi Y (2011) Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69: 418–422. [DOI] [PubMed] [Google Scholar]
- 19.Zisimopoulou P, Evangelakou P, Tzartos J, Lazaridis K, Zouvelou V, et al.. (2013) A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. Journal of autoimmunity. [DOI] [PubMed]
- 20.Hall ZW, Sanes JR (1993) Synaptic structure and development: the neuromuscular junction. Cells 72 (Suppl): : 99–121. [DOI] [PubMed] [Google Scholar]
- 21. Sanes JR, Lichtman JW (1999) Development of the vetebrate neuromuscular junction. Annual Review of Neuroscience 22: 389–442. [DOI] [PubMed] [Google Scholar]
- 22. Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791–805. [DOI] [PubMed] [Google Scholar]
- 23. Fallon JR, Hall ZW (1994) Building synapses: agrin and dystroglycan stick together. [Review]. Trends in Neurosciences 17: 469–473. [DOI] [PubMed] [Google Scholar]
- 24. Rotundo RL (2003) Expression and localization of acetylcholinesterase at the neuromuscular junction. J Neurocytol 32: 743–766. [DOI] [PubMed] [Google Scholar]
- 25. Wu H, Xiong WC, Mei L (2010) To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137: 1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Froehner SC (1993) Regulation of ion channel distribution at synapses. Annual Review of Neuroscience 16: 347–368. [DOI] [PubMed] [Google Scholar]
- 27. McMahan UJ, Horton SE, Werle MJ, Honig LS, Kroger S, et al. (1992) Agrin isoforms and their role in synaptogenesis. [Review]. Curr Opin Cell Biol 4: 869–874. [DOI] [PubMed] [Google Scholar]
- 28. McMahan UJ (1990) The agrin hypothesis. [Review]. Cold Spring Harb Symp Quant Biol 55: 407–418. [DOI] [PubMed] [Google Scholar]
- 29. Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ (1984) Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. Journal of Cell Biology 99: 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wallace BG (1988) Regulation of agrin-induced acetylcholine receptor aggregation by Ca++ and phorbol ester. J Cell Biol 107: 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Campanelli JT, Hoch W, Rupp F, Kreiner T, Scheller RH (1991) Agrin mediates cell contact-induced acetylcholine receptor clustering. Cells 67: 909–916. [DOI] [PubMed] [Google Scholar]
- 32. Fallon JR, Gelfman CE (1989) Agrin-related molecules are concentrated at acetylcholine receptor clusters in normal and aneural developing muscle. J Cell Biol 108: 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, et al. (2008) LRP4 serves as a coreceptor of agrin. Neuron 60: 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, et al. (2008) Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135: 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, et al. (1996) The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85: 501–512. [DOI] [PubMed] [Google Scholar]
- 36. Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, et al. (2001) Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410: 1057–1064. [DOI] [PubMed] [Google Scholar]
- 37. Yang X, Arber S, William C, Li L, Tanabe Y, et al. (2001) Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30: 399–410. [DOI] [PubMed] [Google Scholar]
- 38. Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, et al. (1996) Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85: 525–535. [DOI] [PubMed] [Google Scholar]
- 39. Ruegg MA, Bixby JL (1998) Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci 21: 22–27. [DOI] [PubMed] [Google Scholar]
- 40. Weatherbee SD, Anderson KV, Niswander LA (2006) LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 133: 4993–5000. [DOI] [PubMed] [Google Scholar]
- 41. Wu H, Lu Y, Shen C, Patel N, Gan L, et al. (2012) Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 75: 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luo Z, Wang Q, Zhou J, Wang J, Liu M, et al. (2002) Regulation of AChR Clustering by Dishevelled Interacting with MuSK and PAK1. Neuron 35: 489–505. [DOI] [PubMed] [Google Scholar]
- 43. Luo S, Zhang B, Dong XP, Tao Y, Ting A, et al. (2008) HSP90 beta regulates rapsyn turnover and subsequent AChR cluster formation and maintenance. Neuron 60: 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang B, Luo S, Dong XP, Zhang X, Liu C, et al. (2007) Beta-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J Neurosci 27: 3968–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, et al. (1996) Agrin acts via a MuSK receptor complex. Cell 85: 513–523. [DOI] [PubMed] [Google Scholar]
- 46. Meriggioli MN, Sanders DB (2009) Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 8: 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cossins J, Belaya K, Zoltowska K, Koneczny I, Maxwell S, et al. (2012) The search for new antigenic targets in myasthenia gravis. Ann N Y Acad Sci 1275: 123–128. [DOI] [PubMed] [Google Scholar]
- 48. Gomez CM, Richman DP (1983) Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci U S A 80: 4089–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lindstrom JM (2000) Acetylcholine receptors and myasthenia. Muscle Nerve 23: 453–477. [DOI] [PubMed] [Google Scholar]
- 50. Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ (1978) Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 298: 1116–1122. [DOI] [PubMed] [Google Scholar]
- 51. Aharonov A, Tarrab-Hazdai R, Abramsky O, Fuchs S (1975) Immunological relationship between acetylcholine receptor and thymus: a possible significance in myasthenia gravis. Proc Natl Acad Sci U S A 72: 1456–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Engel AG, Tsujihata M, Lambert EH, Lindstrom JM, Lennon VA (1976) Experimental autoimmune myasthenia gravis: a sequential and quantitative study of the neuromuscular junction ultrastructure and electrophysiologic correlations. J Neuropathol Exp Neurol 35: 569–587. [DOI] [PubMed] [Google Scholar]
- 53. Engel AG, Lambert EH, Howard FM (1977) Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc 52: 267–280. [PubMed] [Google Scholar]
- 54. Engel AG, Arahata K (1987) The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci 505: 326–332. [DOI] [PubMed] [Google Scholar]
- 55. ter Beek WP, Martinez-Martinez P, Losen M, de Baets MH, Wintzen AR, et al. (2009) The effect of plasma from muscle-specific tyrosine kinase myasthenia patients on regenerating endplates. Am J Pathol 175: 1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Punga AR, Lin S, Oliveri F, Meinen S, Ruegg MA (2011) Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol 230: 207–217. [DOI] [PubMed] [Google Scholar]
- 57. Jha S, Xu K, Maruta T, Oshima M, Mosier DR, et al. (2006) Myasthenia gravis induced in mice by immunization with the recombinant extracellular domain of rat muscle-specific kinase (MuSK). J Neuroimmunol 175: 107–117. [DOI] [PubMed] [Google Scholar]
- 58. Shigemoto K, Kubo S, Jie C, Hato N, Abe Y, et al. (2008) Myasthenia gravis experimentally induced with muscle-specific kinase. Ann N Y Acad Sci 1132: 93–98. [DOI] [PubMed] [Google Scholar]
- 59. Cole RN, Reddel SW, Gervasio OL, Phillips WD (2008) Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol 63: 782–789. [DOI] [PubMed] [Google Scholar]
- 60. McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, et al. (2004) Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 55: 580–584. [DOI] [PubMed] [Google Scholar]
- 61. Tsiamalos P, Kordas G, Kokla A, Poulas K, Tzartos SJ (2009) Epidemiological and immunological profile of muscle-specific kinase myasthenia gravis in Greece. Eur J Neurol 16: 925–930. [DOI] [PubMed] [Google Scholar]
- 62. Zhou L, McConville J, Chaudhry V, Adams RN, Skolasky RL, et al. (2004) Clinical comparison of muscle-specific tyrosine kinase (MuSK) antibody-positive and -negative myasthenic patients. Muscle Nerve 30: 55–60. [DOI] [PubMed] [Google Scholar]
- 63. Lavrnic D, Losen M, Vujic A, De Baets M, Hajdukovic LJ, et al. (2005) The features of myasthenia gravis with autoantibodies to MuSK. J Neurol Neurosurg Psychiatry 76: 1099–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leite MI, Strobel P, Jones M, Micklem K, Moritz R, et al. (2005) Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol 57: 444–448. [DOI] [PubMed] [Google Scholar]
- 65.Saka E, Topcuoglu MA, Akkaya B, Galati A, Onal MZ, et al. (2005) Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology 65: : 782–783; author reply 782–783. [DOI] [PubMed] [Google Scholar]
- 66. Suhail H, Subbiah V, Singh S, Behari M (2010) Serological and clinical features of patients with myasthenia gravis in north Indian population. Int J Neurosci 120: 115–119. [DOI] [PubMed] [Google Scholar]
- 67. Shen C, Lu Y, Zhang B, Figueiredo D, Bean J, et al. (2013) Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest 123: 5190–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gesemann M, Denzer AJ, Ruegg MA (1995) Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. Journal of Cell Biology 128: 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z (1993) The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron (USA) 11: 491–502. [DOI] [PubMed] [Google Scholar]