

SUMMARY
Genes mutated in patients with Fanconi anemia (FA) interact with the DNA repair genes BRCA1 and BRCA2/FANCD1 to suppress tumorigenesis, but the molecular functions ascribed to them cannot fully explain all of their cellular roles. Here, we show a repair-independent requirement for FA genes, including FANCD2, and BRCA1 in protecting stalled replication forks from degradation. Fork protection is surprisingly rescued in FANCD2-deficient cells by elevated RAD51 levels or stabilized RAD51 filaments. Moreover, FANCD2-mediated fork protection is epistatic with RAD51 functions, revealing an unanticipated fork protection pathway that connects FA genes to RAD51 and the BRCA1/2 breast cancer suppressors. Collective results imply a unified molecular mechanism for repair-independent functions of FA, RAD51, and BRCA1/2 proteins in preventing genomic instability and suppressing tumorigenesis.
INTRODUCTION
Replication stalling is central to the mechanism of efficacy of many commonly used cancer chemotherapeutics. These include agents that induce DNA lesions, such as camptothecin and cisplatin, as well as those that stall replication progression by perturbing the composition and/or concentration of nucleotide pools, such as gemcitabine and 5-fluorouracil (Stathis and Moore, 2010). Tumor suppressors mutated in Fanconi anemia (FA) are crucial for preventing genomic instability upon replication stalling (Moldovan and D'Andrea, 2009), thus providing a context in which to understand cellular responses to perturbed DNA replication.
The FA pathway involves monoubiquitination of FANCD2-FANCI proteins by the FA core complex in addition to a parallel or downstream function of homologous recombination (HR) proteins, including the breast cancer suppressor BRCA2/FANCD1 (Moldovan and D'Andrea, 2009) (Figure 1A). Together, FA and HR proteins suppress cellular sensitivity to DNA replication poisons that induce DNA interstrand crosslinks (ICLs), such that mechanistic studies have largely focused on the connection between these proteins in the context of ICL repair. In vitro studies have demonstrated that FANCD2 promotes break formation at ICLs and translesion synthesis by an unknown mechanism, while HR proteins act downstream of ICL processing in repairing the collapsed fork caused by strand breakage (Long et al., 2011).
Figure 1. FA/BRCA Gene Network Protects Stalled DNA Replication Forks.

(A) A graphical representation of the FA/BRCA gene network depicts FA core complex proteins (FANCA, B, C, E, F, G, L, M), which promote the monoubiquitination (Ub) of FANCD2 and FANCI. BRCA-related proteins (FANCD1/BRCA2, FANCN/PALB2, and FANCJ/BRIP1) and recently identified FANCO/RAD51C and FANCP/SLX4 are not required for FANCD2–FANCI monoubiquitination and act downstream or in parallel to canonical FA proteins. While BRCA1 is part of the FA/BRCA gene network, BRCA1 mutations have not been found in FA patients. BLM interacts with the gene network, but its loss causes a distinct syndrome.
(B) Graphical sketch of experimental design of fork protection assay. Lengths of nascent replication tracts (labeled with IdU) are measured by DNA spreading after 5 hr of replication stalling with HU. Representative DNA fiber images are given. Scale bars (white) correspond to 4 μm.
(C) Preformed IdU tract lengths measuring replication fork stability by DNA spreading in patient-derived FANCD2-defective PD20 cells, but not cells complemented with the wild-type protein, shorten with HU. Median IdU tract lengths are given in parentheses here and in subsequent figures.
(D) Nascent tract length distribution curve measured by DNA spreading in patient-derived FANCA-defective GM6914 cells show nascent strand shortening with HU, unlike cells complemented with the wild-type protein.
(E) Nascent tract length distribution curve measured by DNA spreading in PD20 cells expressing the FANCD2 K561R mutant defective for monoubiquitination show nascent strand shortening with HU.
See also Figure S1.
Paradoxical to these functions in promoting DNA breakage and subsequent break repair, the FA/BRCA protein network is also highly activated by replication stalling from depletion of nucleotide pools, such as from hydroxyurea (HU), which does not elicit physical DNA lesions that require removal (Howlett et al., 2005; Naim and Rosselli, 2009), as well from other lesions (Langevin et al., 2011; Rosado et al., 2011), including UV damage, which primarily is removed by other repair pathways. Moreover, FANCD2 functionally interacts with the RAD51-mediator protein BRCA2 (Hussain et al., 2004; Wang et al., 2004). However, FA proteins are not canonical HR factors, as cells derived from FA patients are not severely defective in HR repair of double-strand breaks (Nakanishi et al., 2011). Thus, the functional relationship between FA and HR proteins during replication stalling remains enigmatic. The importance of replication stalling to tumor development is underscored by the observation that oncogene activation in general induces replication stress (Bartkova et al., 2006), and specifically by the recent finding that precancerous oncogene expression reduces nucleotide pools (Bester et al., 2011).
To address general roles of FA/BRCA proteins during perturbed replication and to provide a more accurate and complete appreciation of how these proteins function in replication fork fidelity with implications for therapeutic strategies, we examined the functional connection between FANCD2 and HR proteins at replication forks stalled by nucleotide depletion with HU as well as replication stalling chemotherapeutic agents.
RESULTS
Fanconi Anemia Proteins Protect Stalled DNA Replication Forks
BRCA2 and RAD51 can act in replication fork stabilization independent of double-strand break repair (Hashimoto et al., 2010; Lomonosov et al., 2003; Schlacher et al., 2011). Specifically, RAD51 recombinase filament stabilization by the BRCA2 C terminus (C-ter) protects against nucleolytic degradation of stalled replication forks. Stalled replication forks that are not protected by BRCA2 lead to chromosomal instability. We reasoned that if this distinct mechanism is involved in disease suppression, defects may be found associated with disease susceptibility genes other than BRCA2. Because BRCA2 is a suppressor of FA and as such is also known as FANCD1, we tested a possible function for FA genes in fork stability. Specifically, nascent replication tracts were IdU-labeled before replication stalling with hydroxyurea (HU) (Figure 1B); the retention of the label after HU treatment serves as a measure for fork stability using DNA fiber spreading (Schlacher et al., 2011).
To test an involvement of FA proteins in protecting stalled replication forks, we monitored the stability of nascent replication tracts in FA patient-derived cells defective in FANCD2. Replication stalling causes a dramatic shortening of the median IdU tract length in FANCD2-defective PD20 cells compared either to mock treatment (Figure 1C, 4.18 μm and 8.12 μm, p < 0.0001) or to cells complemented with FANCD2 (Figure 1C, 8.08 μm and 8.32 μm, p = 0.2735). These results with FANCD2-defective cells are similar to those obtained with BRCA2 (FANCD1)-defective patient cells (Figure S1 available online) and with BRCA2-defective rodent cells (Schlacher et al., 2011).
FA pathway activation involves monoubiquitination of the FANCD2/FANCI proteins by FA core complex proteins (D'Andrea, 2010). Similar to FANCD2-defective cells, we found that patient-derived GM6914 cells defective in the core complex protein FANCA show degradation of newly synthesized DNA strands when treated with HU, but not with mock treatment (Figure 1D, 4.15 μm and 7.85 μm, p < 0.0001). Yet nascent strands are maintained intact in FANCA-complemented cells (Figure 1D, 8.37 μm and 8.26 μm, p = 0.3702). This suggests a functional requirement for FA proteins upstream of FANCD2 monoubiquitination in maintaining fork stability.
To directly assess if fork protection requires FA pathway activation by monoubiquitination of FANCD2, we analyzed nascent replication tracts in PD20 cells expressing mutant FANCD2-K561R incapable of being ubiquitinated (Ub) (Garcia-Higuera et al., 2001). We found that these cells fail to maintain the integrity of nascent DNA tracts during replication stalling with HU (Figure 1E, 4.89 μm and 9.44 μm, p < 0.0001). Thus, in addition to BRCA2, fork stabilization requires FA pathway activation through FANCD2 monoubiquitination.
FA Pathway Suppresses Genomic Instability When Replication Is Stalled
We tested the cellular consequence of replication stalling by nucleotide depletion on FA-defective cells. Metaphase spreads of FANCA-defective GM6914 cells show significantly elevated levels of spontaneous chromosomal aberrations compared to cells that are complemented with wild-type FANCA (Figure 2A, p = 0.0107). Upon treatment with HU, the load of DNA breaks and radial structures in GM6914 cells considerably increases from an average of 0.6 to 2.5 aberrations per cell (Figure 2A, p < 0.0001), while only a moderate elevation is observed in FANCA-complemented cells from 0.34 to 0.72 aberrations per cell (Figure 2A, p = 0.0836). Thus, replication stalling by nucleotide depletion selectively elevates genomic instability in FA-defective cells.
Figure 2. FA Pathway Suppresses Genomic Instability upon Replication Stalling.

(A) Chromosomal aberrations measured by metaphase chromosome spreads with FANCA-deficient and –complemented GM6914 cells with HU (± SD, n = 40). Representative images of chromosomal aberrations of metaphase chromosomes are given. Scale bars (gray) correspond to 2 μm.
(B) Cell survival analysis of FANCA-defective, patient-derived GM6914 cells and cells complemented with FANCA upon continuous HU treatment (± SEM, n = 4).
(C) Cell survival analysis of FANCA-defective cells and cells complemented with FANCA upon continuous gemcitabine (GEM) treatment (± SEM, n = 3).
(D and E) Nascent tract length distribution curves measured by DNA spreading in patient-derived FANCA-defective GM6914 cells and cells complemented with the wild-type protein with gemcitabine [GEM, (D)] and camptothecin [CPT, (E)].
See also Figure S2.
Genomic instability upon HU, however, is not accompanied by acute cell death. While FANCA-defective cells are exquisitely sensitive to ICL-inducing reagents such as mitomycin-C (MMC, Figure S2A), they show no substantial difference in cellular survival rates compared to FANCA-proficient cells upon treatment with HU (Figure 2B). Interestingly, when we tested BRCA2/FANCD1-defective cells for MMC sensitivity, we found that cells containing the BRCA2 S3291A variant, which is proficient for double-strand break repair but cannot protect stalled forks, shows only moderate sensitivity to high concentrations of MMC compared to cells with BRCA2 truncation (Figure S2B), suggesting two separable functions for BRCA2/FANCD1 during ICL repair, one of which involves replication fork protection.
Other replication stalling agents such as the chemotherapeutics gemcitabine (Figure 2C), which inhibits replication elongation, or camptothecin (Figure S2C), a replication poison that prevents DNA ligation and elicits a roadblock to replication by covalently locking topoisomerase I to DNA, show similar results compared to HU with no to very mild acute cellular death. These results suggest that immediate cell death is not an obligate immediate consequence of replication stalling in FA-defective cells.
We next examined if FA proteins are required for fork protection when replication is stalled by agents other than HU. Replication tracts are maintained intact in FANCA-proficient cells when treated with the chemotherapeutic gemcitabine (Figure 2D, p = 0.612 with and without gemcitabine). In contrast, the nascent strands shorten dramatically in FANCA-defective GM6914 cells with gemcitabine (Figure 2D, p < 0.0001). Likewise, exposure to camptothecin shortens replication tracts in FA-defective cells (Figure 2E, p < 0.0001). Thus, replication stalling caused by various agents elicit fork instability in FANCA-defective cells.
Parallel and Downstream Functions of FA-Associated Proteins
Several proteins have been identified to associate with FA components, although they are not considered to be FA proteins as mutations have not been found as yet in FA patients. The BRCA1 breast cancer suppressor associates in complexes with several FANC proteins, including FANCD2 and BRCA2 (Moldovan and D'Andrea, 2009). We found that BRCA1-defective mouse embryonic stem (ES) cells show shortened nascent tracts with replication stalling (Figure 3A, 5.52 μm and 8.88 μm, p < 0.0001), unlike cells with a functional BRCA1 (Figure 3A, 8.82 μm and 8.73 μm, p = 0.831). Thus, both BRCA1 and BRCA2—the major hereditary breast cancer suppressors—stabilize replication forks, providing a mechanistic link between tumor suppression and the protection of stalled replication forks.
Figure 3. Parallel and Downstream Functions of FA-Associated Proteins.

(A) Replication fork stability analysis by DNA spreading of preformed IdU tracts in BRCA1-deficient mouse ES cells (mES Brca1−/−) and mouse ES cells containing wild-type BRCA1 (mES Brca1+/−) with HU. Median IdU tract lengths are given in parenthesis here and in subsequent graph panels.
(B) Replication fork stability analysis by DNA spreading of preformed IdU tracts in BLM-depleted mouse ES cells with negative doxycycline (DOX) control of BLM expression (mES Blmtet/tet +DOX) and BLM proficient ES cells (mES Blmtet/tet). See inset, western blot for BLM expression.
(C) Replication recovery analysis after fork stalling with HU as measured by DNA spreading of CldU replication tracts in BLM-depleted (mES Blmtet/tet +DOX) and BLM proficient mouse ES cells (mES Blmtet/tet). Median CldU tract lengths are given in parentheses here and in subsequent graph panel.
(D) Replication recovery analysis (CldU tract length) after fork stalling is in FANCD2-defective and in FANCD2-complemented PD20 cells.
See also Figure S3.
BLM helicase interacts with both FA and BRCA networks (Chu and Hickson, 2009; Deans and West, 2009; Moldovan and D'Andrea, 2009). Loss of BLM causes Bloom syndrome, a developmental disorder with high cancer predisposition (Chu and Hickson, 2009), but is phenotypically distinct from FA. On the cellular level, BLM, in partnership with TopIIIα, decatenates fully replicated chromosomes (Chu and Hickson, 2009). We used mouse ES cells expressing BLM under negative doxycycline control (Figure 3B, Blmtet/tet inset) to assess if BLM plays a role in the protection of stalled forks from degradation. BLM-depleted cells maintain IdU tracts intact when exposed to HU (Figure 3B, Blmtet/tet+DOX; 7.93 μm and 7.72 μm, p = 0.338), similar to cells expressing BLM (Figure 3B, Blmtet/tet; 8.09 μm and 8.12 μm, p = 0.831). Thus, unlike BRCA and FANC deficiency, loss of BLM does not result in degradation of stalled replication forks.
BLM helicase, however, is required for efficient replication restart after HU (Davies et al., 2007) (Figure S3A). Related to this, we find that BLM-depleted cells have a defect in replication recovery as measured by substantially shorter CldU tracts after exposure to HU when compared with cells expressing wild-type BLM (Figure 3C, 3.59 μm and 6.28 μm, p < 0.0001 and Figure S3B). Thus, BLM deficiency results in defects in replication recovery that can be observed by measuring either the length of the replication tracts (Figure 3C) or the frequency of forks that restart (Davies et al., 2007; Figure S3A).
Because BLM interacts with both FA and BRCA networks, we further tested FA (Figure 3D and Figure S3C) and BRCA1-defective cells (Figure S3D) for replication recovery after stalling with HU. Similar to BRCA2 deficiency (Schlacher et al., 2011) but in contrast to BLM deficiency (Figure 3C), no defect in replication recovery is observed: replication tracts formed after HU are similarly short in both FANCD2-defective and -complemented PD20 cells after exposure to HU (Figure 3, 4.12 μm and 3.73 μm, p = 0.0029 and Figure S3E). Similar results were obtained for FANCA (Figures S3C and S3F) and BRCA1-defective cells (Figures S3D and S3G). These data suggest that BLM acts downstream of the BRCA and FANC proteins, subsequent to the protection of stalled forks, perhaps in decatenation of structures elicited by positive supercoiling ahead of the fork.
FA Genes Protect Against MRE11-Dependent Fork Degradation
MRE11 nuclease promotes degradation of stalled replication forks when either BRCA2 or RAD51 function is impaired (Hashimoto et al., 2010; Schlacher et al., 2011). We found that degradation in FANCD2-defective cells occurs at ~2.2 kb/hr (Figure 4A, inset), which is reminiscent of the slow kinetics of MRE11-dependent degradation in BRCA2-defective cells (Schlacher et al., 2011). Moreover, double-labeling experiments suggest that both leading and lagging strand degradation occurs, as the most recently incorporated nucleotides are excised first (Figures S4A–S4C). These results suggest that MRE11 nuclease, which promotes both 3′–5′ and 5′–3′ end processing (Williams et al., 2008), mediates the nascent replication tract shortening in FA-defective cells. We chemically inhibited MRE11 nuclease with mirin (Dupré et al., 2008), and found that it blocks nascent tract shortening upon fork stalling with HU (Figure 4B, 16.46 μm and 16.59 μm, p = 0.738). Therefore, FANCD2, like BRCA2 and RAD51, protects nascent strands at stalled replication forks from degradation by MRE11.
Figure 4. FA Genes Protect Against MRE11-Dependent Fork Degradation.
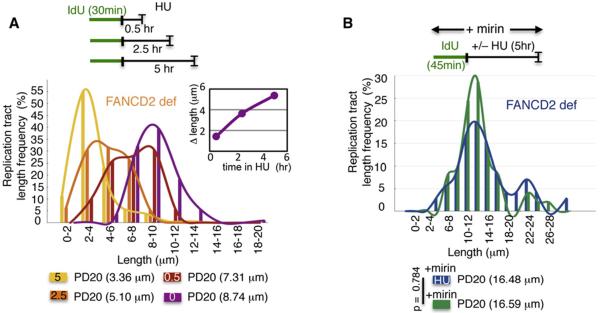
(A) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-deficient PD20 cells during various exposure times to HU. Inset; the rate of IdU tract length change is 0.87 μm/hr, estimated to be ~2.2 kb/hr.
(B) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-deficient PD20 with chemical inhibition of MRE11 nuclease by treatment with mirin, with and without HU.
See also Figure S4.
Functional Interaction of FA and RAD51 Proteins at Stalled Forks
RAD51 was recently shown to act downstream of FANCD2 during ICL repair (Long et al., 2011). Yet, RAD51 is recruited to ICLs prior to FANCD2 such that a second function upstream of ICL incision could not be excluded (Long et al., 2011). We therefore sought to address whether FANCD2 acts in synergy with RAD51 when protecting stalled forks or whether they act epistatically, within a common pathway. To test this, we expressed the BRC4 peptide (Saeki et al., 2006), which suppresses DNA binding of RAD51 and thus disrupts RAD51 filaments.
Replication tracts are dramatically shortened upon HU in FANCD2-complemented PD20 cells expressing the BRC4 peptide (Figure 5A, 3.86 μm and 8.18 μm p < 0.0001), demonstrating efficient RAD51 depletion from filaments leading to fork destabilization. Moreover, RAD51 depletion in PD20 cells complemented with FANCD2 mimics the replication tract shortening seen with deficiency of FANCD2 itself (compare Figure 5A, 3.86 μm, and Figure 5B, 4.17 μm, p = 0.104). Although replication tracts are significantly shorter in FANCD2-defective cells with HU compared to without HU (Figure 5B, 4.17 μm and 8.9 μm, p < 0.0001), the tract shortening is not exacerbated by depletion of RAD51 from filaments (Figure 5B, 4.11 μm, p = 0.324). Taken together, the data suggest that RAD51 and BRCA2 act in epistasis with FANCD2 for replication fork stabilization.
Figure 5. Functional Interaction of FA and RAD51 Proteins at Stalled Forks.

(A) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-complemented PD20 cells expressing flag-tagged BRC4-peptide (see western blot, inset), which disrupts RAD51 binding to DNA.
(B) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-deficient PD20 cells expressing flag-tagged BRC4-peptide (see western blot, inset).
(C) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-deficient PD20 cells expressing mutant RAD51 K133R (see western blot, inset), which forms stable filaments, upon fork stalling with HU.
(D) Replication fork stability analysis by DNA spreading of IdU replication tracts in FANCD2-deficient PD20 cells overexpressing wild-type (WT) RAD51 (see western blot, inset), which promotes filament assembly, upon fork stalling with HU.
Given that perturbing RAD51 in FANCD2-defective cells results in a phenotype comparable to FANCD2 deficiency alone, we hypothesized that FANCD2 may play a role in RAD51 filament stabilization subsequent to RAD51 loading onto DNA. To gain insight into the mechanism of FANCD2-mediated fork stabilization, we expressed the RAD51 K133R mutant. This mutant is devoid of ATPase activity required for dissociation from DNA (Morrison et al., 1999), and thus forms hyperstable filaments. We found that RAD51 K133R renders IdU tracts in PD20 cells resistant to degradation, maintaining replication tract lengths comparable to those observed in PD20 cells expressing wild-type FANCD2 (compare Figure 5C, 7.95 μm, and Figure 5A, 8.18 μm, p = 0.825). Thus, fork instability caused by FANCD2 deficiency can be compensated for by RAD51 filament stabilization.
Elevated RAD51 protein levels are often found in tumor cells (Raderschall et al., 2002b; Brown and Holt, 2009), which perhaps drives more stable filament formation (Raderschall et al., 2002a), suggesting the potential for even wild-type RAD51 to effect fork protection. We therefore examined the effect of higher levels of wild-type RAD51 on replication tract stability in FA-defective cells. Strikingly, we found that overexpression of wild-type RAD51 in FANCD2-defective PD20 cells can partially rescue replication fork instability upon HU and primarily maintain replication tracts intact (Figure 5D, 4.17 and 6.50 without and with increased RAD51 expression, p < 0.0001). Taken together, the data show that FANCD2 and RAD51 support each other at replication forks such that both FANCD2 and RAD51 positively regulate replication tract stability.
DISCUSSION
Because of the exquisite sensitivity of FA genes to ICL lesions, most studies thus far have focused on their role in ICL repair. Yet, the FA machinery prevents genome instability and lethality caused by replication blocks other than ICLs with uncharacterized implications for tumorigenesis (Howlett et al., 2005; Langevin et al., 2011; Naim and Rosselli, 2009; Rosado et al., 2011). Here the discovery of a role for FANCD2-Ub in preventing degradation of nascent DNA strands in vivo independent of ICL processing complements and extends existing results. Notably, FANCD2-Ub functions epistatically with RAD51 at stalled forks within this distinct pathway, as does BRCA2, which provides a more complete understanding of how these proteins maintain replication fork fidelity in the context of ICL and other DNA stresses.
FANCD2 monoubiquitination involves an interaction with the replisome component proliferating cell nuclear antigen (PCNA) (Howlett et al., 2009). Because BRCA2/RAD51 functionally interacts with FANCD2 (Long et al., 2011; Wang et al., 2004) and BRCA2/RAD51 alone is insufficient for fork protection in the absence of FANCD2-Ub, a testable mechanistic model is that as part of a protein supercomplex FANCD2-Ub connects the BRCA and RAD51 proteins to replisome components to stabilize stalled forks and prevent fork collapse (Figure 6). This protection mechanism provides a functional explanation for the observations that upon replication stalling both BRCA1 and BRCA2 rapidly relocalize to replication foci containing PCNA (Chen et al., 1998). Moreover, FANCD2 colocalizes with PCNA and RAD51 to foci in response to HU (Hussain et al., 2004) and FANCD2 is found localized to sister chromatids upon replication stress (Naim and Rosselli, 2009). It recently was reported that an FA component stabilizes a specialized translesion synthesis (TLS) DNA polymerase at nuclear foci upon DNA damage (Kim et al., 2012). Thus, given the data and model presented here, it will be interesting to see if this polymerase stabilizing function could hold true for other DNA polymerases and in particular non-specialized polymerases, because TLS polymerases are unlikely to be involved when replication is stalled without eliciting DNA lesions such as by HU.
Figure 6. Model of FA/BRCA Gene Network Functions in Replication Fork Stability.
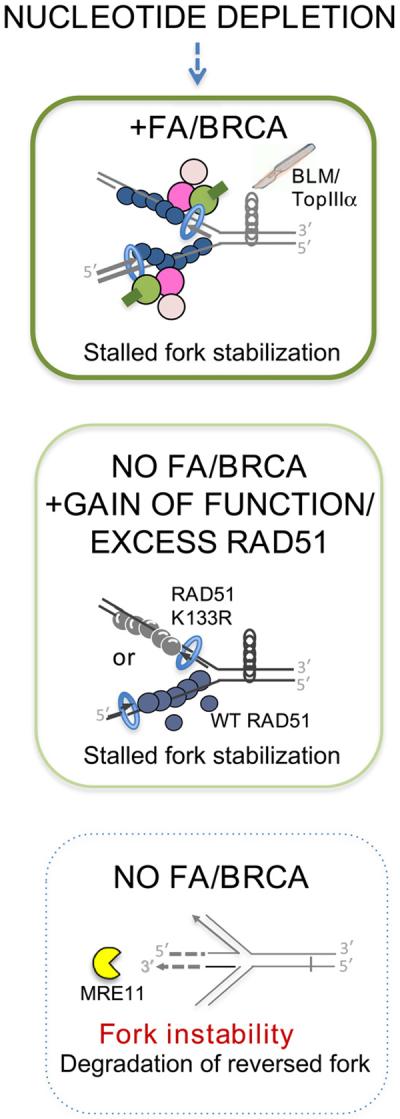
Nucleotide depletion, as caused by oncogene activation or chemotherapeutic agents, stalls replication forks. FA/BRCA proteins stabilize RAD51 at stalled replication forks to protect nascent strands from MRE11-dependent degradation. RAD51 filament stabilization in the absence of FA/BRCA proteins is sufficient for fork protection. This can be achieved by gain of function mutant RAD51 or overexpression of wild-type RAD51, as commonly seen in tumor cells. BLM-TopIIIα acts downstream in the restart of stalled forks. Protein colors: BRCA2, pink; FANCD2, green; ubiquitin, dark green; BRCA1, light pink; wild-type RAD51, dark blue; RAD51 K133R, steel gray; PCNA, blue (doughnut); MRE11 yellow (pacman). BLM-TopIIIα, scalpel.
See also Figure S5.
Other FA proteins may be found to act in this distinct pathway of fork stabilization. FANCJ is important in processing DNA secondary structures at G-rich regions (Hiom, 2010). In the context of replication fork stability, disruption of such G-rich structures may be crucial to create single-strand DNA stretches long enough to support sufficiently stable RAD51 filaments. FANCP (SLX4) binds to several endonucleases implicated in ICL processing (Cybulski and Howlett, 2011). However, it is feasible that FANCP (SLX4) may have additional functions that support the structural maintenance of replication fork structures through Smc5/6 (Ohouo and Smolka, 2011), as how Smc5/6 promotes chromosome stability is largely unknown and it may have a role in the maintenance of undamaged chromosomes. Structural destabilization of replication forks and fork degradation indeed provide feasible mechanisms for both spontaneous fork breakage and deletion mutations, both of which are hallmarks of FA-defective cells (Papadopoulo et al., 1990; Schroeder et al., 1964).
FA is a disease with life-threatening consequences on hematopoiesis, marked by stem cell attrition before the development of tumors. Consistent with a nonlethal phenotype during embryogenesis and the small stature of FA patients, we find that replication stalling does not elicit acute but only marginal cell death in FA-defective cells (Figure 2B). Yet, unprotected replication forks result in DNA damage as indicated by the marked increase in genomic aberrations in FA-defective cells upon treatment with HU (Figure 2A). As DNA damage has recently been shown to promote hematopoietic stem cell aging (Wang et al., 2012), we propose in particular that nonlethal DNA damage may promote hematopoietic stem cell attrition by accumulated differentiation rather than cell death. Nonlethal DNA damage in other tissues on the other hand could eventually promote tumorigensis by prolonged cellular exposure to mutagenesis without cell death and account for the high tumor susceptibility in FA patients. Thus, replication fork protection potentially provides a mechanism to resolve the apparent paradox involving the seemingly opposing phenotypes of stem cell death and mutagenesis promoting tumor predisposition.
Fork stabilization is likely also an important event during ICL repair, consistent with data demonstrating RAD51 recruitment to stalled forks before ICL processing (Long et al., 2011). Our observation that a variant of BRCA2/FANCD1 defective in fork protection sensitizes cells to ICLs, however to a much lesser extent than mutant BRCA2/FANCD1 defective in both DSB repair and fork protection, suggests that both fork protection and DSB repair contribute to the suppression of lethality upon ICL. Intriguingly, a dual role during ICL repair beyond DSB repair was also recently reported for BRCA1 (Bunting et al., 2012). The fork protection role for BRCA1 that we report here could therefore be feasibly related to the DSB repair-independent function of BRCA1 during ICL repair. Likewise, sensitivity of FA-deficient cells to ICLs may involve fork protection. While FA proteins clearly have separate roles during ICL repair (Knipscheer et al., 2009; Kim et al., 2012), fork stabilization may not be essential to ensure the complete removal of this lethal type of lesion. Rather, we suggest that during ICL repair, fork stabilization by BRCA and FA proteins may increase access for nucleases and thus the efficiency for incision and subsequent repair.
However, replisome stalling is far more frequent than ICL processes and has particular significance for rapidly cycling cells such as cells of the hematological system or those responding to mitogenic signals like hormones. Moreover, precancerous oncogene expression by HPV infection can induce replication stress by decreasing nucleotide pools (Bester et al., 2011). This direct connection between tumor initiation and replication stalling together with our data showing that FA proteins suppress genomic instability by protecting stalled forks suggests a mechanistic basis for the observed susceptibility of FA patients to oral cancer upon HPV infection (Park et al., 2010). Protection and stabilization of replication forks has critical implications for the maintenance of genomic integrity and thus likely constitutes an unanticipated mechanism of tumor suppression. In support of this hypothesis, we identified the sporadic breast cancer cell line MCF7 to be defective in protecting stalled forks (Figure S5), which implies that this distinct mechanism is also linked to some sporadic cancers.
Cancer therapeutics target DNA replication and dividing cells, so that DNA damage responses are exploited as therapeutic targets and resistance factors. Our experiments reveal that RAD51 stabilization rescues FANCD2 deficiency in protecting stalled forks, despite the fact that FA proteins are clearly not canonical HR factors. This surprising result underscores the emerging importance of an unappreciated aspect in RAD51 filament mediation, which differs from loading of RAD51 onto DNA to promote strand exchange, to instead utilize RAD51 filaments to stabilize DNA structures. Importantly, wild-type RAD51 is often found overexpressed in tumors that acquired resistance to chemotherapeutic drug treatment (Brown and Holt, 2009), which is sufficient in overcoming genetic defects to restore repair functions in these cells and, as we show here, also replication fork stability to a large extent. While defects in replication fork protection elicit genomic instability that initially contribute to tumorigenesis, restoration of the function by RAD51 overexpression after transformation could benefit the proliferative capacity of the tumor cell. Our results provide a mechanistic link between tumor suppression and the protection of stalled replication forks by showing that both BRCA1 and BRCA2, the major hereditary breast cancer suppressors, stabilize replication forks. Thus, our collective results unite breast cancer and FA susceptibility genes in one common molecular process that protect against genomic instability. This integrated function for the FA/BRCA gene network in stabilizing stalled forks is expected to prompt investigations of this distinct pathway in tumorigenesis, stem cell aging and controlling stalled DNA replication processes to suppress genomic instability that will shape emerging therapeutic strategies.
EXPERIMENTAL PROCEDURES
Cell Lines
SV40-transformed FA fibroblasts (GM6914, GM6914+FANCA, PD20F, and PD20F+FANCD2 (Jakobs et al., 1996; Näf et al., 1998; Timmers et al., 2001) were previously described and are also available through the FA Cell Repository at Oregon Health & Science University. Mouse ES cells (Brca1−/− and Brca1+/− [Gowen et al., 1996; Moynahan et al., 1999]; Blmtet/tet [Yusa et al., 2004]) were previously described.
Drugs
5′iodo-2′deoxyuridine (IdU), 5′chloro-2′deoxyuridine (CldU), and HU were purchased from Sigma-Aldrich. Mirin (Dupré et al., 2008) was provided by the MSKCC Organic Synthesis Core Facility.
Cell Transfection
Using Fugene 6 (Roche Applied Science) and following the manufacturer's instructions, 105 PD20 or complemented cells were transfected with 2 μg of flag-BRC4, RAD51 K133R, wild-type RAD51 or empty (pCaggs) expression plasmids. Expression of the peptides was tested 40 hr post transfection by standard western blotting using Anti-Flag Clone M2 (Sigma-Aldrich) antibody against flag-BRC4 or anti-RAD51 antibody (Santa Cruz) against RAD51 K133R or wild-type RAD51.
DNA Fiber Spreads
DNA fiber spreads were prepared as previously described (Schlacher et al., 2011). Briefly, replication tracts of log-phase cells were pulse labeled with 50 μM IdU and CldU before or after replication stalling with 4 mM HU, 1 μM gemcitabine or 0.5 μM camptothecin respectively, as indicated in the sketches. Cells were harvested, lysed and spread to obtain single DNA molecules on microscope slides before standard immunofluorescence with antibodies against IdU and CldU (Novus Biologicals, BD Biosciences).
Statistical Analysis
Between 100 and 1400 nascent DNA tracts were measured using ImageJ software from 1–3 independent experiments. P-values obtained from the Mann-Whitney test and the 95% confidence intervals were calculated using Prism software.
Cellular Survival Assays
For survival assays, 3000 cells were seeded in a 24-well plate the day before continuous treatment with the indicated drugs. The number of viable cells was determined when confluency reached ~80% for the untreated cells using Cell Titer 96 AQueous One Solution Cell Proliferation Assay (Promega).
Metaphase Spread Analysis
For metaphase spreads, 2 × 105 cells were seeded the day before HU treatment (4 mM) and treated with colcemid (0.1 μg/ml, GIBCO), as indicated. For metaphase spreads, cells were swollen with 0.075 M KCL (12 min, 37° C), fixed with methanol/acetic acid (3:1), dropped onto a microscope slide, stained with 5% Giemsa, and mounted with Cytoseal 60 (Fisher Scientific) before imaging with an Olympus BX60 microscope.
Supplementary Material
Significance.
Replication stalling is at the heart of many chemotherapeutic agents, including those that limit nucleotide incorporation into DNA (e.g., gemcitabine) and those that block replication fork progression (e.g., camptothecin and platinum drugs). Replication stalling agents activate the FA/BRCA tumor suppressor pathway and cause genomic instability in cells lacking pathway components. We report here an unexpected function of the FA/BRCA pathway in protecting stalled replication forks from nucleolytic degradation. This finding provides a cellular understanding of expanded roles of these tumor suppressors with significant implications for ongoing research efforts in understanding tumor susceptibility in patients with FA, therapeutic resistance, and emerging therapeutic strategies.
ACKNOWLEDGMENTS
We thank Koji Nakanishi, Jan LaRocque, and other members of the Jasin and Wu labs for reagents and discussions. K.S. is the Berger Foundation Fellow of the Damon Runyon Cancer Research Foundation (DRG 1957-07). This work was supported by NIH grants R01CA121110 (H.W.) and R01GM54668 and P01CA94060 (M.J.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at doi:10.1016/j.ccr.2012.05.015.
REFERENCES
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ET, Holt JT. Rad51 overexpression rescues radiation resistance in BRCA2-defective cancer cells. Mol. Carcinog. 2009;48:105–109. doi: 10.1002/mc.20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 Functions Independently of Homologous Recombination in DNA Interstrand Crosslink Repair. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- Cybulski KE, Howlett NG. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011;10:1757–1763. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N. Engl. J. Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007;14:677–679. doi: 10.1038/nsmb1267. [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC. FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol. Cell. 2009;36:943–953. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiom K. FANCJ: solving problems in DNA replication. DNA Repair (Amst.) 2010;9:250–256. doi: 10.1016/j.dnarep.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- Howlett NG, Harney JA, Rego MA, Kolling FW, 4th, Glover TW. Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J. Biol. Chem. 2009;284:28935–28942. doi: 10.1074/jbc.M109.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004;13:1241–1248. doi: 10.1093/hmg/ddh135. [DOI] [PubMed] [Google Scholar]
- Jakobs PM, Sahaayaruban P, Saito H, Reifsteck C, Olson S, Joenje H, Moses RE, Grompe M. Immortalization of four new Fanconi anemia fibroblast cell lines by an improved procedure. Somat. Cell Mol. Genet. 1996;22:151–157. doi: 10.1007/BF02369905. [DOI] [PubMed] [Google Scholar]
- Kim H, Yang K, Dejsuphong D, D'Andrea AD. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Räschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C, Shinohara A, Sonoda E, Yamaguchi-Iwai Y, Takata M, Weichselbaum RR, Takeda S. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol. Cell. Biol. 1999;19:6891–6897. doi: 10.1128/mcb.19.10.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Näf D, Kupfer GM, Suliman A, Lambert K, D'Andrea AD. Functional activity of the fanconi anemia protein FAA requires FAC binding and nuclear localization. Mol. Cell. Biol. 1998;18:5952–5960. doi: 10.1128/mcb.18.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat. Struct. Mol. Biol. 2011;18:500–503. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Smolka MB. A touching moment for Smc5/6: from ssDNA binding to repair. Cell Cycle. 2011;10:1190–1191. doi: 10.4161/cc.10.8.15380. [DOI] [PubMed] [Google Scholar]
- Papadopoulo D, Guillouf C, Mohrenweiser H, Moustacchi E. Hypomutability in Fanconi anemia cells is associated with increased deletion frequency at the HPRT locus. Proc. Natl. Acad. Sci. USA. 1990;87:8383–8387. doi: 10.1073/pnas.87.21.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Pitot HC, Strati K, Spardy N, Duensing S, Grompe M, Lambert PF. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res. 2010;70:9959–9968. doi: 10.1158/0008-5472.CAN-10-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E, Bazarov A, Cao J, Lurz R, Smith A, Mann W, Ropers HH, Sedivy JM, Golub EI, Fritz E, Haaf T. Formation of higher-order nuclear Rad51 structures is functionally linked to p21 expression and protection from DNA damage-induced apoptosis. J. Cell Sci. 2002a;115:153–164. doi: 10.1242/jcs.115.1.153. [DOI] [PubMed] [Google Scholar]
- Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002b;62:219–225. [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011;18:1432–1434. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- Saeki H, Siaud N, Christ N, Wiegant WW, van Buul PP, Han M, Zdzienicka MZ, Stark JM, Jasin M. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc. Natl. Acad. Sci. USA. 2006;103:8768–8773. doi: 10.1073/pnas.0600298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder TM, Anschütz F, Knopp A. [Spontaneous chromo-some aberrations in familial panmyelopathy] Humangenetik. 1964;1:194–196. doi: 10.1007/BF00389636. [DOI] [PubMed] [Google Scholar]
- Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010;7:163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell. 2001;7:241–248. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D'Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell. Biol. 2004;24:5850–5862. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Horie K, Kondoh G, Kouno M, Maeda Y, Kinoshita T, Takeda J. Genome-wide phenotype analysis in ES cells by regulated disruption of Bloom's syndrome gene. Nature. 2004;429:896–899. doi: 10.1038/nature02646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.