

Abstract
Cell penetrating peptides (CPPs) have been well established as potential carriers for intracellular delivery of protein/peptide therapeutics. However, their lack of selectivity impedes their application in vivo. In order to increase their specificity, a highly pH-sensitive histidine-glutamate (HE) co-oligopeptide was fused with a CPP, i.e. model amphipathic peptide (MAP), and was expressed as fusion protein with glutathione S-transferase (GST) acting as a cargo protein. Compared with two other fusion proteins containing either HE or MAP, only the fused peptide (HE-MAP) could effectively deliver the cargo GST protein to cells at pH 6.5 or below, while maintaining low delivery to cells at pH 7.0 and above. Using a xenograft mouse model of human breast cancer, fluorescent imaging showed that only HE-MAP could effectively target GST to the tumor site, while reducing non-specific association of MAP in other organs. The data presented in this report demonstrates the diagnostic and/or therapeutic potential of the fused peptide, HE-MAP, for targeting the acidic tumor microenvironment. The concise design for this pH-sensitive peptide offers a simple way to overcome CPP’s lack of selectivity, which could lead to increased application of CPPs and macromolecular therapeutics.
Keywords: Drug delivery, Image analysis, Peptide, Recombinant protein, pH-sensitive activation
Introduction
With the advancement of research and biotechnology, increasingly more proteins and peptides have been produced and developed as potential therapeutics. Amongst the over 130 approved protein/peptide drugs, a majority of their therapeutic targets, such as hormones receptors or cell surface markers for antibodies, are outside or on the surface of cells [1]. On one hand, it is not surprising considering that these targets are more readily accessible to protein/peptide drugs. On the other hand, it emphasizes the challenge for proteins or peptides to cross barriers in the form of cellular membranes.
Since their discovery, cell penetrating peptides (CPPs) have been considered as promising carriers for overcoming the intracellular drug delivery obstacle. Initially found in natural proteins, these short peptides are required and sufficient for the membrane transport of their respective proteins via a yet unknown mechanism termed “membrane transduction” [2, 3]. Currently, many nature-derived, synthetic, and chimeric peptides have been identified for their cell penetrating ability [4, 5], either alone or conjugated with other cargo molecules [6–10]. Although the internalization mechanism for CPPs is peptide and cargo dependent [11, 12], the ubiquitous uptake of CPPs by all kinds of cells has been found in vitro and in vivo [13]. However, the universal effectiveness of CPPs regardless of cell type also reflects their lack of specificity - a major hurdle for application in vivo. Approaches in active targeting methods of CPPs have often failed since the targeting efficiency of ligands directly attached to CPPs is overridden by the strong interaction between CPPs and negatively charged cell membranes [14]. Therefore, improving selectivity is crucial for the clinical application of CPPs, especially for use through systemic delivery.
Our studies have shown that non-specific binding and internalization of CPPs can be minimized through masking their cationic charges [15]. This masking effect has been utilized in several designs to reduce non-specific uptake [16–19] (reviewed in [14, 20]). For example, an activatable CPP has been designed for tumor imaging in which the cationic charge is reversibly masked by attachment of an anionic sequence through an enzyme-sensitive linkage [21], but the application of this approach may be limited by the presence and cleavage efficiency of the specific enzymes. Another promising method to activate the masked CPPs is to take advantage of the relatively acidic pH at target sites, such as inside endosomes or the microenvironment of tumor cells or sites of inflammation. However, previous efforts in our laboratory have shown that acid-labile chemical modifications may not be removed efficiently at the relevant physiological pH [22].
In this report, we describe a CPP-containing fusion protein that exhibits a highly pH-sensitive cell association, which could be used to target mildly acidic sites inside the body. The CPP used is model amphipathic peptide (MAP, single letter amino acid sequence: KLALKLALKALKAALKLAY), which has been shown in our laboratory to efficiently deliver cargo proteins to the cytosolic and/or nuclear compartment of cells via an endocytic mechanism [23]. In order to reduce the non-specific cell association at neutral pH, a peptide, (HE)10 (single letter amino acid sequence: HEHEHEHEHEHEHEHEHEHE) was attached to MAP through a pentaglycine linker (HE-MAP) as a recombinant glutathione-S-transferase (GST) fusion protein (“GST-HE-MAP”). At the same time, a (HE)10 peptide or MAP was cloned into the same plasmid and expressed as a GST-fusion protein (“GST-HE” or “GST-MAP”, respectively). In this study, GST served both as a tag for protein purification, as well as a cargo protein to study the cell association mediated by different peptides. This GST-HE-MAP fusion protein has previously been shown to exhibit pH-dependent binding and internalization in vitro. In this report, three fusion proteins were compared in cell assays and in a xenograft mouse model of human breast cancer, demonstrating that HE-MAP can deliver cargo proteins to mildly acidic sites in vivo.
Materials and Methods
Materials
HeLa and MDA-MB-231 cell lines were purchased from ATCC (Manassas, VA). RPMI 1640 media, fetal bovine serum (FBS), and ampicillin were from Mediatech (Manassas, VA). Pfx DNA polymerase, Taq DNA polymerase, T4 DNA ligase, RPMI 1640 powder, L-glutamine, penicillin-streptomycin, and trypsin-EDTA were from Invitrogen (Carlsbad, CA). Restriction enzymes were from New England BioLabs (Ipswich, MA). pGEX-4T-1 vector and Sephadex G50 were from GE healthcare life sciences (Piscataway, NJ). PageRuler™ prestained protein ladder, glutathione (GSH) agarose and HisPur™ Ni-NTA resin were from Thermo Fisher Scientific (Waltham, MA). (HE)10 peptide was synthesized by Genemed (San Antonio, TX). IRDye 800CW NHS ester and IRDye 800CW carboxylate were from LI-COR Biosciences (Lincoln, NE). Matrigel™ basement membrane matrix and peptone were from BD Biosciences (Bedford, MA). Radioactive I-125 was from PerkinElmer (Waltham MA). Isopropyl β-D-1-thiogalactopyranoside (IPTG), yeast extract, and sodium lauroyl sarcosine (sarkosyl) were from Amresco (Solon, OH). Phenylmethylsulfonyl fluoride (PMSF) was from EMD Millipore (Billerica, MA). Reduced glutathione was from Alfa Aesar (Ward Hill, MA). Dialysis regenerated cellulose tubing, 12,000–14,000 molecular weight cut off (MWCO), was from Spectrum Laboratories (Rancho Dominguez, CA). Microsep™ centrifugal device, MWCO at 10 kDa, was from PALL (Port Washington, NY). Competent E. coli (DH5α) was from ZYMO Research (Irvine, CA). 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was from JT Baker (Central Valley, PA). Competent E. coli BL21-T1R, protease inhibitor cocktail, and other chemicals were from Sigma-Aldrich (St. Louis, MO).
Production of expression constructs
For GST-HE-MAP, two partial complementary single-stranded deoxyribonucleic acid (ssDNA) sequences were synthesized by ValueGene (San Diego, CA). The full-length double-stranded deoxyribonucleic acid (dsDNA) encoding HE-MAP peptide was acquired by one single elongation step after partial annealing of the aforementioned ssDNAs. Following polymerase chain reaction (PCR) amplification, the full sequence was cloned into the pGEX-4T-1 plasmid through BamHI and NotI sites. The inserted sequence was confirmed by sequencing (GENEWIZ, San Diego, CA). For GST-HE or GST-MAP, both sense and anti-sense sequences were synthesized and annealed to obtain full-length dsDNA. The fragment encoding peptide (HE)10 was cloned into pGEX-4T-1 plasmid through BamHI and EcoRI sites. The fragment encoding MAP was cloned into pGEX-4T-1 plasmid through EcoRI and XhoI sites. Both inserts were confirmed after sequencing.
DNA sequences synthesized during cloning:
For GST-HE-MAP:
5’-GGATCCCATGAACACGAACATGAGCATGAACATGAACATGAGCACGAAC ATGAACACGAGCATGAAGGTGGTGGTGGAGGTAAATTAG-3’ (sense)
5’-GCGGCCGCTTAATATGCCAGTTTCAGTGCTGCTTTCAGTGCTTTCAGAGC CAGTTTTAATGCTAATTTACCTCCACCACCACCTTCATGCTC-3’ (anti-sense)
For GST-HE:
5’-GTGTGGATCCCATGAACACGAACATGAGCATGAACATGAACATGAGCAC GAACATGAACACGAGCATGAATTCTCTC-3’ (sense)
5’-GAGAGAATTCATGCTCGTGTTCATGTTCGTGCTCATGTTCATGTTCATGCT CATGTTCGTGTTCATGGGATCCACAC-3’ (anti-sense)
For GST-MAP:
5’-GAGTGAATTCAAATTAGCATTAAAACTGGCTCTGAAAGCACTGAAAGCA GCACTGAAACTGGCATATCTCGAGTCAC-3’ (sense)
5’-GTGACTCGAGATATGCCAGTTTCAGTGCTGCTTTCAGTGCTTTCAGAGCC AGTTTTAATGCTAATTTGAATTCACTC-3’ (anti-sense)
Expression and purification of recombinant proteins
The plasmids with correct insertions were transformed into E. coli expression strain BL21. For expression of recombinant proteins, bacteria were incubated in terrific broth (TB) media with 75 μg/mL ampicillin at 37 °C with 300 rpm shaking speed until the OD600 of the media reached 2.5–3.0. IPTG was added into TB media to a final concentration of 0.2 mM. After 3–4 hours of additional incubation, the bacteria were collected and stored at −80 °C. Expression of the GST-fusion proteins was monitored by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) followed by Coomassie blue staining.
To purify GST-HE or GST-HE-MAP, bacterial pellets were resuspended in phosphate buffered saline (PBS), pH 7.4, and lysozyme was added to reach a final concentration of 0.25 mg/mL. After ~30 min incubation on ice, PMSF was added to 1 mM and Triton X-100 was added to a final concentration of 1% (v/v). The bacteria were lysed by sonication (Misonix Ultrasonic Liquid Processors S-4000, Misonix, Farmingdale, NY) on ice at amplitude 10 for 4–5 min total working time at a 10 sec on/15 sec off working cycle. The lysate was centrifuged at 15000 g for 30 min at 4 °C. The supernatant was loaded on GSH agarose column pre-balanced with PBS. The column was washed with 1% Triton X-100 in PBS and then PBS alone. Fusion protein was eluted with PBS containing 50 mM GSH and 0.5% CHAPS, pH 7.4. The eluted protein was concentrated and exchanged into PBS with Microsep™ centrifugal device, MWCO at 10 kDa. For animal studies, the protein was further purified by HisPur™ Ni-NTA resin according to the manufacturer’s protocol.
To purify GST-MAP, bacterial pellets were resuspended in PBS, pH 7.4. After ~30 min lysozyme treatment on ice, PMSF was added to 1 mM and sarkosyl was added to a final concentration of 1.5% (w/v). After sonication and centrifugation, CHAPS and Triton X-100 were added to the supernatant to final concentrations of 30 mM and 3% (v/v), respectively [24, 25]. The mixture was loaded on a GSH agarose column pre-balanced with PBS. As mentioned above, the column was washed and GST-MAP was eluted, concentrated, and exchanged into PBS.
During purification, GST-fusion proteins were monitored by absorbance at a wavelength of 280 nm, and SDS-PAGE with Coomassie blue staining. The band densities were measured using Quantity One software (BioRad, Hercules, CA) and used to estimate fusion protein purity.
Labeling of purified proteins
The purified GST-fusion proteins were radiolabeled with 125I using the chloramine T method as previously described [26] and 125I-proteins were purified by Sephadex G50. The fractions containing 125I-labeled proteins were determined using a gamma counter (Cobra II Auto-Gamma, Packard, Downers Grove, IL). For animal studies, the fusion proteins were labeled with IRDye 800CW NHS ester according to the manufacturer’s protocol. Briefly, to achieve a ~1:1 modification ratio, the reactions were carried out at room temperature for 2 hours with a molar ratio (dye/protein) of ~4:1. The IR800-labeled proteins were purified by either Sephadex G50 or dialysis (MWCO: 12–14 kDa) and sterilized by passing through 0.22 μm filters. After labeling, the concentrations of 125I-protiens or IR800-proteins were determined by Micro BCA™ protein assay kit (Thermo Fisher Scientific, Waltham, MA)
In vitro assays
HeLa or MDA-MB-231 cells were grown in 6-well plates in RPMI 1640 media supplemented with 10% FBS, 2 mM L-glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin. The cells were incubated at 37 °C at 5% CO2, and were replenished with fresh media the day before confluence. The confluent cell monolayers were first incubated with serum-free media (self-made from RPMI 1640 powder, without NaHCO3, with total 10 mM Na2HPO4 and 10 mM citrate/citric acid, pH 7.2–7.4) for 10 minutes at 37 °C. In most cases, the cells were treated with self-made RPMI 1640 media adjusted to various pHs (pH 6.0, 6.5, 7.0, or 7.5 for HeLa cells; pH 6.5, or 7.4 for MDA-MB-231 cells) containing 150 nM 125I-proteins and protease inhibitor cocktail containing 4 μM AEBSF, 0.6 nM aprotinin, 260 nM bestatin, 2 μM EDTA, 28 nM E-64, and 2 μM leupeptin. For two experimental groups, the MDA-MB-231 cells were treated with similar dosing solution containing 150 nM GST-MAP and 150 nM (HE)10 peptides. After 1 h incubation at 37 °C, the cell monolayers were washed 3 times with 1 mL cold PBS and detached by treatment with trypsin-EDTA for 3 min at 37 °C. The total cell associated radioactivity (i.e. surface bound and internalized) was determined using a gamma counter.
Xenograft mouse model of human breast cancer
All animal studies were performed according to the protocols approved by the University of Southern California Institutional Animal Care and Use Committee. The mouse model was generated according to a protocol similar to a previous study [27]. Female athymic nude mice (Hsd:Athymic Nude-Foxn1nu, 4–6 weeks old) were purchased from Harlan (Livermore, CA). The xenograft model was generated by subcutaneous injection of ~7 × 106 MDA-MB-231 human breast adenocarcinoma cells suspended in 100 μL Matrigel™ into the right flank of mice. Tumor volume was calculated using the formula (S2 × L) where S and L represent the small and large dimensions. Tumors were allowed to grow to 0.5 – 1 cm3.
In vivo and ex vivo infrared fluorescence imaging
All fluorescence imaging studies were performed using the IVIS SPECTRUM pre-clinical in vivo imaging system and analyzed using the IVIS Living Imaging 4.2 software (PerkinElmer, Waltham, MA). An infrared filter set, excitation at 745 nm and emission at 800 nm, was used to acquire the fluorescence of IR800-proteins. All illumination conditions (lamp voltage, filters, f/stop, field of views, binning) were set to the same levels for all imaging within one experiment. Fluorescence emissions were normalized and reported as radiant efficiency, unit = (photon/sec/cm2/sr)/(μW/cm2). For the first experiment, the control mouse was injected intravenously with 8 nmol of free dye, IRDye 800CW carboxylate, while another mouse received 5.75 nmol IR800-labeled GST-HE-MAP (255 nmol/kg). The mice were imaged at 1, 6, 24 h post-injection before ex vivo imaging of collected tumors and organs. To compare GST-HE, GST-MAP, and GST-HE-MAP, each group of mice (n = 3) were injected with 3.65 nmol (160 nmol/kg) of one IR800-labeled protein intravenously and imaged at various time points post-injection.
Statistical analysis
All values are represented as mean ± standard deviation (SD) with n = 3, and significant differences were evaluated using the Student’s t-test. Values of p < 0.05 were accepted as statistical significant.
Results
Production and purification of fusion proteins
A dsDNA fragment encoding the HE-MAP peptide was successfully cloned into the pGEX-4T-1 vector. The resultant fusion protein, GST-HE-MAP, was expressed in E. coli BL21 strain. Similarly, two other constructs were created, which expressed GST-HE or GST-MAP respectively. As shown in Figure 1, all three fusion proteins were purified with GSH agarose to > 90% purity as determined by SDS-PAGE with Coomassie blue staining. Among the three fusion proteins, GST-HE had the highest expression level at 80–100 mg/L, followed by GST-HE-MAP with an expression level at 10–20 mg/L, and the lowest expression level at 5–10 mg/L for GST-MAP. After all purification procedures, the final concentrations of GST-HE, GST-MAP, and GST-HE-MAP were around 20 mg/mL, 1.3 mg/mL, and 5 mg/mL, respectively.
Figure 1.
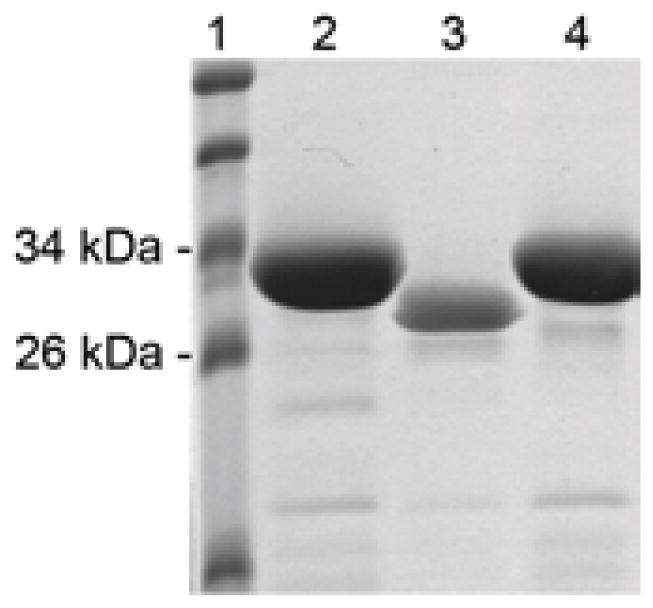
SDS-PAGE analysis and Coomassie blue staining of purified fusion proteins. GST-fusion proteins were purified from bacteria by affinity chromatography according to the procedures described in the methods section. Lane 1: Molecular weight markers. Lane 2: GST-HE, expected MW = 30.3 kDa. Lane 3: GST-MAP, expected MW = 29.6 kDa. Lane 4: GST-HE-MAP, expected MW = 31.3 kDa.
Cell association of 125I-labeled GST-fusion proteins at different pH conditions
Purified GST-HE, GST-MAP, and GST-HE-MAP were radiolabeled with 125I by the chloramine T method. Cell association of the three 125I-labeled fusion proteins was first analyzed in HeLa cells at pH 6.0, 6.5, 7.0, or 7.5 (Figure 2). For 125I-GST-HE, the amount of total cell associated proteins remained low (1–2 pmol/cell monolayer) across the entire pH range. In contrast, the cell association of 125I-GST-MAP was high across the entire pH range, where the amounts were 14.5 – 20.6 pmol/cell monolayer. The total cell associated 125I-GST-HE-MAP showed more significant changes among different pH conditions than the other two fusion proteins. The cell association was low at pH 7.5 to 7.0 (3.3 ± 0.1 and 4.8 ± 0.4 pmol/cell monolayer at pH 7.5 and 7.0, respectively), and then increased 3–6 fold to 16.1 ± 0.3 and 22.4 ± 0.3 pmol/cell monolayer at pH 6.5 and 6.0, respectively.
Figure 2.
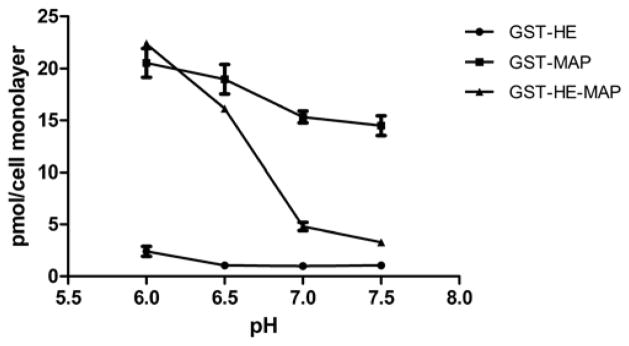
Total cell associated 125I-GST-HE, 125I-GST-MAP, and 125I-GST-HE-MAP in HeLa cells at 4 different pH conditions. HeLa cells were treated with 150 nM 125I-labeled fusion proteins at pH 6.0, 6.5, 7.0, or 7.5 for 1 hour at 37 °C. The amounts of total cell associated proteins were determined according to the protocol described in the methods section. Data were expressed as mean ± SD of triplicates.
The 125I-proteins were also tested for cell association assays in MDA-MB-231 cells at pH 6.5 or 7.4. As shown in Figure 3A, the trend was similar, where 125I-GST-HE had low total cell association at both pH 7.4 and 6.5 (0.7 ± 0.1 and 0.5 ± 0.2 pmol/cell monolayer, respectively), and 125I-GST-MAP had high cell association at both pH 7.4 and 6.5 (14.7 ± 1.0 and 13.4 ± 0.4 pmol/cell monolayer, respectively). The cell association of 125I-GST-HE-MAP was pH sensitive, where the amount was 4.4 ± 0.2 pmol/cell monolayer at pH 7.4 which then increased ~3-fold to 14.6 ± 0.5 pmol/cell monolayer at a lower pH of 6.5. Next, the requirement of direct attachment of (HE)10 to the MAP-fusion protein was evaluated by mixing 125I-GST-MAP with (HE)10 at a 1:1 molar ratio. At pH 7.4, only 125I-GST-MAP-HE showed a reduction of cell associated protein (4.4 ± 0.1 pmol/cell monolayer), while 125I-GST-MAP and 125I-GST-MAP mixed with (HE)10 were similar (15.2 ± 0.7 and 15.0 ± 1.0 pmol/cell monolayer, respectively) (Figure 3B). 125I-GST-MAP, 125I-GST-MAP mixed with (HE)10, and 125I-GST-MAP-HE showed similar amounts of cell association at pH 6.5 (14.1 ± 0.8, 14.4 ± 0.6, and 13.4 ± 1.0 pmol/cell monolayer for three groups, respectively). In summary, pH-dependent cell association was observed only when the (HE)10 peptide was fused with MAP.
Figure 3.

Total cell associated 125I-labeled fusion proteins in MDA-MB-231 cells at pH 6.5 or 7.4. MDA-MB-231 cells were treated at 37 °C for 1 hour with: A. 150 nM 125I-GST-HE, 125I-GST-MAP, or 125I-GST-HE-MAP; B. 150 nM 125I-GST-MAP only, 125I-GST-MAP with (HE)10 peptide (molar ratio= 1:1), or 125I-GST-HE-MAP. The amounts of total cell associated proteins were determined as described in the methods section. Data were expressed as mean ± SD of triplicates. **indicates a statistically significant difference (p < 0.01) between the two groups.
Imaging studies with IR800-labeled fusion proteins in mouse xenograft model of breast cancer
In order to determine if similar pH-dependent cell association could be achieved in vivo, all fusion proteins were labeled with an infrared fluorescent dye. After purification, IR800-labeled proteins were injected intravenously into MDA-MB-231 tumor-bearing nude mice through the tail vein. With a mildly acidic environment around solid tumors [28], this model was ideal for the purposes of this study. First, the distribution of free IR800 dye and IR800-GST-HE-MAP in mice was compared. IR800-GST-HE-MAP showed strong fluorescent signals around tumor with a long retention up until 24 h post-injection (Figure 4A), while the free dye had an extensive distribution at 1 h post-injection and was quickly eliminated before 6 h post-injection. Tumor distribution of GST-HE-MAP was further validated by ex vivo imaging at 24 h post-injection (Figure 4B). The highest level of fluorescence was detected in the tumor and kidneys. IR800-GST-HE-MAP was also found in the liver, with minimal activity in the spleen and no detectable activity in the heart or pancreas.
Figure 4.
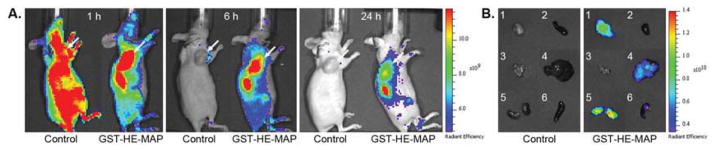
In vivo imaging and organ distribution study for IR800-GST-HE-MAP in MDA-MB-231 tumor-bearing nude mice. Free IR800 dye (control) or IR800-labeled GST-HE-MAP was injected intravenously. Fluorescent images were acquired (A) 1, 6, 24 h post-injection and (B) ex vivo imaging of tumors and other organs after euthanizing the mice at 24 h post-injection: 1. Tumor, 2. Heart, 3. Pancreas, 4. Liver, 5. Kidneys, 6. Spleen. Arrows indicate tumor sites.
The distribution of GST-HE-MAP in mice was further evaluated alongside GST-HE and GST-MAP controls (n = 3). Whole-body distribution of the injected IR800 labeled fusion proteins peaked at 2 h post-injection (Figure 5A), where the resultant fluorescent images for the three groups showed a pattern similar to what was observed in cell assays. The fluorescent signals were low with limited whole body distribution for the IR800-GST-HE group, but high and extensive for the IR800-GST-MAP group. In contrast, the fluorescent signalsin mice injected with IR800-GST-HE-MAP were relatively low in most organs, but enriched around tumor sites. The other regions with high fluorescent levels were due to accumulation of the fusion protein in the kidneys and liver, likely due to the clearance of the protein from the body. Time-dependent accumulation of IR800-GST-HE-MAP in tumors showed enrichment in tumor sites as early as 0.5 h post injection, with peak enrichment at around 1–2 h post injection, and remained in the tumor until 6 h post injection (Figure 5B). The fluorescent proteins inside tumors had slower clearance than most normal tissues and organs except the kidneys and liver.
Figure 5.
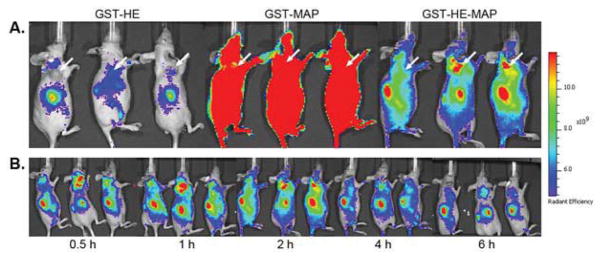
In vivo imaging of MDA-MB-231 tumor-bearing nude mice after intravenous injection of IR800-labeled GST-fusion proteins. Each mouse received one of the three IR800-labeled fusion proteins at a dose of 3.65 nmol. (A) Images at 2 h post-injection for IR800-GST-HE, IR800-GST-MAP, and IR800-GST-HE-MAP. Arrows indicate tumor sites. (B) Images for IR800-GST-HE-MAP from 0.5 to 6 h post-injection.
Discussion
The pH difference between tumor and normal tissues offers a great opportunity to achieve targeted delivery to tumor sites. Since the extracellular pH of tumors is only 0.4–0.8 unit lower than that of normal tissues [29], a tumor-targeting delivery system must exhibit a high sensitivity in a mildly acidic pH range of 6.5 to 7.4. Acid-labile chemical bonds have been used to create pH-sensitive delivery systems [22, 30]. However, the cleavage rate of these chemical bonds at mildly acidic pH may not be efficient enough for systemic application in vivo. Other types of particulate systems including micelles and polymers are able to achieve this mild pH sensitivity [31, 32], but have fallbacks with premature release rates and inadequate depth of tumor penetration due to their large size. An alternative pH-sensitive complex formation was reported from our laboratory over 20 years ago using polylysine and histamine-modified polyglutamate [33]. Based on this idea, a (HE)10 peptide was linked to MAP in order to mask the positive charges at neutral pH. As explained in our concept paper [34], negatively charged glutamate residues in (HE)10 can interact with positively charged lysine residues in MAP at neutral pH, which masks the non-specific cell association of MAP. At mildly acidic pH, some of the histidine residues are protonated, which disrupts the association of (HE)10 and MAP, thus, revealing the strong cell association properties of MAP.
With the intent of adding peptide/protein cargos to the construct for future application in drug delivery, HE-MAP peptide was expressed as a GST-fusion protein in E. coli. In a previous study, this fusion protein showed high cell internalization at mildly acidic pH and minimum cell internalization at neutral pH [34]. To further study this pH sensitivity, both GST-HE and GST-MAP were produced and purified for comparative studies alongside GST-HE-MAP. Three different fusion proteins could be purified easily with GSH agarose although they had quite different yields (Figure 1). The large reduction in expression level of GST-MAP and GST-HE-MAP compared to GST-HE is presumably due to MAP, which is known to have a certain level of antimicrobial activity [35]. The attachment of (HE)10 to MAP increased the yield of GST-HE-MAP, which is indirect evidence that MAP is masked by (HE)10, thus reducing its cytotoxicity. While MAP also showed toxicity in mammalian cells at concentrations over 2 μM [36], the amounts of MAP used in this study were well below this level and showed no cytotoxicity in thiazolyl blue tetrazolium bromide (MTT) assays.
To understand how (HE)10 and/or MAP affected the cell association of GST, three 125I-labeled fusion proteins were first compared at 4 pH conditions in HeLa cells (Figure 2). With ten negative charges from (HE)10 peptide, it was not surprising that GST-HE showed low cell association at pH 6.5–7.5 in HeLa cells. The cell associated GST-HE slightly increased at pH 6.0, presumably because the protonation of histidine residues neutralized some of the negative charges. The addition of (HE)10 peptide to GST does not change its cell association within this pH range [37]. In comparison to GST-HE, GST-MAP exhibited about an 8–18 fold higher cell association at each pH tested. These results support MAP as a potent carrier for proteins beyond neutral pH conditions [4, 23]. GST-MAP was slightly pH sensitive, with a ~40% increase at pH 6 compared to pH 7.5. GST-HE-MAP on the other hand had low cell association, close to GST-HE, at neutral pH but high cell association, comparable to GST-MAP, at mildly acidic pH. The combined contribution of (HE)10 and MAP indeed resulted in a pH-sensitive delivery of the fusion protein, which preferentially bound and entered cells at mildly acidic environment. This pH sensitivity of GST-HE-MAP was also found in MDA-MB-231 cells (Figure 3A). According to our previously published hypothesis [34], the charge interaction between (HE)10 and MAP masks MAP and its association with cells at neutral pH. In order to determine if this interaction requires a chemical linkage of the two peptides, equimolar amounts of GST-MAP and (HE)10 were mixed prior to dosing cells, where it was shown that only the directly linked treatment group (GST-MAP-HE) showed pH sensitivity (Figure 3B).
To evaluate the pH-sensitive cell association in vivo, a mouse xenograft human breast cancer model was generated using MDA-MB-231 cells. The tumors were allowed to grow to a certain size so that they would provide a mildly acidic microenvironment as established in previous publications [29, 38]. As shown in Figure 4A, the kinetics of the free dye was completely different from that of IR800-labeled GST-HE-MAP. This result suggests that the conjugation of dye is stable enough to represent the distribution of fusion protein in mouse and that the increased fluorescence signal in tumors is unlikely due to the property of the dye itself. Additionally, both in vivo imaging and ex vivo organ distribution (Figure 4, A and B) indicate the tumor retention of GST-HE-MAP can be detected for more than 24 hours. To further investigate and confirm the initial findings in vitro, equimolar doses of three IR800-labeled fusion proteins were injected intravenously into mice (n = 3) and imaged serially. At 2 h post-injection, the three groups injected with different IR800 fusion proteins showed distinct patterns of infrared fluorescent signals (Figure 5A), which correlate well with our in vitro results (Figure 3A). Consistent with the in vitro results, GST-HE showed a low signal throughout the body except for the kidneys, liver, and bladder where the protein was presumably processed and/or excreted. For GST-MAP, the fluorescent signal was widespread throughout the body and above the maximum of the scale. This extensive in vivo distribution of CPPs is consistent with a previous finding [13], and serves as additional evidence for CPP’s lack of specificity. GST-HE-MAP exhibited an elevated tumor distribution compared to GST-HE, and had reduced distribution in other normal organs and tissues when compared to GST-MAP. This distribution was consistent with ex vivo imaging (Figure 5B). The comparison among three fusion proteins in vivo clearly supports our notion that (HE)10 can mask MAP at normal pH, which leads to a selective distribution of cargo protein to the mildly acidic tumor site [34]. In addition, the time course data indicate that GST-HE-MAP can target efficiently (within 30 min post injection) to mildly acidic tumor microenvironments in vivo and has a long retention in the tumor site (up to 24 hours) (Figure 4A and 5B).
The data presented in this manuscript establishes GST-HE-MAP as a CPP-based recombinant protein, which targets the mildly acidic tumor environment in vivo. Although the fusion protein is heavily distributed to kidneys, presumably due to its small size [39], this issue could conceivably be significantly improved by increasing the protein size above the renal filtration cutoff of ~60 kDa. After optimization of in vivo distribution, this HE-CPP based system could be used for diagnostic and/or therapeutic purposes for treatment of cancer.
Conclusions
In this report, a targeted CPP, HE-MAP, was designed and produced as a GST-fusion protein. Both in vitro and in vivo data unambiguously supports our prospect that HE-MAP can deliver a cargo protein, GST, to cells or tissues within a mildly acidic environment (~ pH 6.5). This pH-sensitive peptide can be developed as a diagnostic and/or therapeutic tool for acidic tumors. Moreover, the design of HE-MAP offers a simple and effective way to reduce the non-specific binding and uptake of CPPs at normal physiological pH. This design not only brings forward a concise delivery platform for targeting of peptide/protein drugs, but also pushes forward the application of CPPs and potential macromolecular therapeutics.
Acknowledgments
We would like to thank Daisy Shen and Hsin-Fang Lee for their assistance during this study. This work was supported by grants from the NIH National Cancer Institute (R21 CA169841) and the University of Southern California (USC) Ming Hsieh Institute for Research of Engineering-Medicine for Cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–50. [PubMed] [Google Scholar]
- 3.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–7. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 4.Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, et al. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. BBA-Biomembranes. 1998;1414:127–39. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 5.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, et al. Arginine-rich peptides: an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–40. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 6.Perez F, Joliot A, Bloch-Gallego E, Zahiraoui A, Triller A, Prochiantz A. Antennapedia homeobox as a signal for the cellular internalization and nuclear addressing of a small exogenous peptide. J Cell Sci. 1992;102:717–22. doi: 10.1242/jcs.102.4.717. [DOI] [PubMed] [Google Scholar]
- 7.Fawell S, Seery J, Daikh Y, Morre C, Chen LL, Pepinsky B, et al. Tat-mediated delivery of heterologous proteins into cells. P Natl Acad of Sci USA. 1994;91:664–8. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. P Natl Acad of Sci USA. 2001;98:8786–91. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaro J, Shen W. Cytosolic delivery of a p16-peptide oligoarginine conjugate for inhibiting proliferation of MCF7 cells. J Control Release. 2005;108:409–17. doi: 10.1016/j.jconrel.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 10.Mo R, Zaro J, Shen W. Comparison of cationic and amphipathic cell penetrating peptides for siRNA delivery and efficacy. Mol Pharm. 2012;9:299–309. doi: 10.1021/mp200481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Andaloussi S, Jarver P, Johansson HJ, Langel U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem J. 2007;407:285–92. doi: 10.1042/BJ20070507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24:1977–92. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 13.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–72. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 14.Vives E, Schmidt J, Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. BBA-Rev Cancer. 2008;1786:126–38. doi: 10.1016/j.bbcan.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Fei L, Ren L, Zaro JL, Shen WC. The influence of net charge and charge distribution on cellular uptake and cytosolic localization of arginine-rich peptides. J Drug Target. 2011;19:675–80. doi: 10.3109/1061186X.2010.531729. [DOI] [PubMed] [Google Scholar]
- 16.Shamay Y, Adar L, Ashkenasy G, David A. Light induced drug delivery into cancer cells. Biomaterials. 2011;32:1377–86. doi: 10.1016/j.biomaterials.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Mok H, Bae K, Ahn C, Park T. PEGylated and MMP-2 specifically dePEGylated quantum dots: comparative evaluation of cellular uptake. Langmuir. 2009;25:1645–50. doi: 10.1021/la803542v. [DOI] [PubMed] [Google Scholar]
- 18.Zhu L, Kate P, Torchilin V. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. Acs Nano. 2012;6:3491–8. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuai R, Yuan W, Qin Y, Chen H, Tang J, Yuan M, et al. Efficient delivery of payload into tumor cells in a controlled manner by TAT and thiolytic cleavable PEG co-modified liposomes. Mol Pharm. 2010;7:1816–26. doi: 10.1021/mp100171c. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Jiang Y, Wang H, Wang J, Shin MC, Byun Y, et al. Curb challenges of the "Trojan Horse" approach: smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv Drug Deliver Rev. 2013;65:1299–315. doi: 10.1016/j.addr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. P Natl Acad of Sci USA. 2004;101:17867–72. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fei L, Zaro JL, Shen WC. Acid-labile modification of a cell penetrating peptide for use in targeted drug delivery. 2009 AAPS Annual Meeting and Exposition; Los Angeles, USA. 2009. [Google Scholar]
- 23.Kenien R, Zaro JL, Shen WC. MAP-mediated nuclear delivery of a cargo protein. J Drug Target. 2012;20:329–37. doi: 10.3109/1061186X.2011.649481. [DOI] [PubMed] [Google Scholar]
- 24.Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione-S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–87. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 25.Tao H, Liu WJ, Simmons BN, Harris HK, Cox TC, Massiah MA. Purifying natively folded proteins from inclusion bodies using sarkosyl, Triton X-100, and CHAPS. Biotechniques. 2010;48:61–4. doi: 10.2144/000113304. [DOI] [PubMed] [Google Scholar]
- 26.Sonoda S, Schlamow M. Studies of I-125 trace labeling of immunoglobulin G by chloramine-T. Immunochemistry. 1970;7:885–98. doi: 10.1016/0019-2791(70)90051-0. [DOI] [PubMed] [Google Scholar]
- 27.Chen K, Yap LP, Park R, Hui XL, Wu KC, Fan DM, et al. A Cy5.5-labeled phage-displayed peptide probe for near-infrared fluorescence imaging of tumor vasculature in living mice. Amino Acids. 2012;42:1329–37. doi: 10.1007/s00726-010-0827-5. [DOI] [PubMed] [Google Scholar]
- 28.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. J Magn Reson Imaging. 2002;16:430–50. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 29.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194–8. [PubMed] [Google Scholar]
- 30.Kale AA, Torchilin VP. Enhanced transfection of tumor cells in vivo using "Smart" pH-sensitive TAT-modified pegylated liposomes. J Drug Target. 2007;15:538–45. doi: 10.1080/10611860701498203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sethuraman VA, Bae YH. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release. 2007;118:216–24. doi: 10.1016/j.jconrel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J Control Release. 2008;129:228–36. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen WC. Acid-sensitive dissociation between poly(lysine) and histamine-modified poly(glutamate) as a model for drug-releasing from carriers in endosomes. Biochim Biophys Acta. 1990;1034:122–4. doi: 10.1016/0304-4165(90)90163-q. [DOI] [PubMed] [Google Scholar]
- 34.Zaro JL, Fei L, Shen WC. Recombinant peptide constructs for targeted cell penetrating peptide-mediated delivery. J Control Release. 2012;158:357–61. doi: 10.1016/j.jconrel.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 35.Palm C, Netzerea S, Hallbrink M. Quantitatively determined uptake of cell-penetrating peptides in non-mammalian cells with an evaluation of degradation and antimicrobial effects. Peptides. 2006;27:1710–6. doi: 10.1016/j.peptides.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Scheller A, Oehlke J, Wiesner B, Dathe M, Krause E, Beyermann M, et al. Structural requirements for cellular uptake of alpha-helical amphipathic peptides. J Pept Sci. 1999;5:185–94. doi: 10.1002/(SICI)1099-1387(199904)5:4<185::AID-PSC184>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Fei L, Zaro JL, Shen WC. Recombinant cell penetrating peptide construct for targeting mildly acidic pH. The 39th Annual Meeting & Exposition of the Controlled Release Society; Quebec City, Canada. 2012. [DOI] [PubMed] [Google Scholar]
- 38.Raghunand N, Altbach MI, van Sluis R, Baggett B, Taylor CW, Bhujwalla ZM, et al. Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by P-31 magnetic resonance spectroscopy. Biochem Pharmacol. 1999;57:309–12. doi: 10.1016/s0006-2952(98)00306-2. [DOI] [PubMed] [Google Scholar]
- 39.Maack T, Johnson V, Kau ST, Figueiredo J, Sigulem D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: A review. Kidney Int. 1979;16:251–70. doi: 10.1038/ki.1979.128. [DOI] [PubMed] [Google Scholar]