

Abstract
Objectives
The PTEN pseudogene, PTENP1, was recently shown to play a role in cell proliferation in a prostate cancer model. In the present study, we sought to determine whether PTENP1 is expressed in endometrial cancer (EMCA) cell lines and primary tumors along with the microRNAs (miRNAs) that are predicted to regulate PTEN and PTENP1 transcript levels.
Methods
RNA was prepared from six EMCA cell lines, three normal endometrial samples, and 61 primary tumors. TaqMan® RT-PCR was used to quantitate PTEN expression in all specimens and PTENP1 expression in cell lines, and normal endometrial (NE) samples. PTENP1 expression was evaluated using conventional RT-PCR in primary tumors. MicroRNA profiling was undertaken using NanoString™ technology in AN3CA and KLE cell lines. The relationship between PTEN transcript levels, PTENP1 expression, and PTEN mutation status were investigated.
Results
All NE samples, cell lines, and primary tumors expressed PTEN. PTENP1 transcript was expressed in NE samples, cell lines, and 34 of 61 (56%) primary tumors. The median relative PTEN level was 2.9 arbitrary expression units in PTENP1-positive tumors and 2.3 in PTENP1-negative tumors (p = 0.09). PTEN levels in wild-type and haploinsufficient tumors were variable compared to PTEN-null tumors (p = 0.015). Four microRNAs predicted to bind to PTEN/PTENP1 ranked in the top 20 most abundant microRNA subtypes in the AN3CA and KLE cell lines.
Conclusions
PTENP1 is expressed in NE and EMCA cell lines, as are PTEN/PTENP1 targeting inhibitory miRNAs (cell lines). Further studies are needed to evaluate the impact of PTEN/PTENP1/miRNA interactions on tumorigenesis regulation in EMCA.
Keywords: Endometrial cancer, microRNAs, tumorigenesis, PTENP1, PTEN
Introduction
Endometrial carcinoma (EMCA) is the most common female genital tract neoplasm in the United States. This malignancy accounted for six percent of all carcinomas diagnosed among in US women in 2010 [1]. While patients with localized disease exhibit a five-year survival of up to 96%, this rate drops to 67% and 17% for the patients with regional and distant spread of disease, respectively [1, 2]. To date, limited options are available for the treatment of metastatic and recurrent disease [3, 4]. An improved understanding of the molecular pathogenesis in endometrial carcinomas holds promise of advancing our ability to design effective therapeutics.
Mutations in the PTEN (phosphatase and tensin homologue deleted on chromosome ten) tumor suppressor gene are common in diverse human carcinomas [5, 6]. PTEN is the most frequently altered gene in endometrioid endometrial carcinomas. Reports on PTEN mutation frequency in primary endometrial carcinoma ranges between 40-80% [5-11].
There is an emerging body of literature characterizing expression and potential role of microRNAs (miRNAs) in gynecologic malignancies [12]. A recent study by Cohn et al. showed that PTEN targeting miRNAs are overexpressed in endometrial carcinoma samples compared to normal endometria. Overexpression of miR-183 and miR-200C correlates with decreased PTEN protein levels, suggesting that PTEN is targeted by miRNAs in endometrial cancer [13]. In addition, the mir-200 family has higher expression in endometrial endometrioid carcinomas relative to normal endometria [14].
PTEN and its noncoding pseudogene, PTENP1 share extensive sequence homology [15]. Historically, pseudogenes, such as PTENP1, were believed to be nonfunctional largely because they are untranslated or do not produce full-length protein products [16-19]. Over the last several years, evidence has emerged that supports of a regulatory role for pseudogenes [19-22].
Although thePTENP1 transcript is not translated, it may modulate PTEN transcript levels. Recent work by Poliseno et al. [20] showed that PTENP1 serves as a decoy target for PTEN-binding inhibitory miRNAs. PTENP1's ability to competitively bind miRNAs that alter PTEN function in part explains PTENP1s effect on prostate cancer cell line growth.
The prostatic utricle and the uterus are homologous structures. Rare reports of endometrial carcinoma in the prostatic utricle further highlight the shared embryologic origin [23-25]. We hypothesized that homology would be conserved at the molecular level, and PTENP1 would be expressed in the endometrial tissues, as has been described for the prostate. Once PTENP1 expression was confirmed in these tissues, we compared cohorts of PTENP1-positive and PTENP1-negative tumors for differences in PTEN transcript levels and clinico-pathologic factors.
Methods
Patient tissues, clinical and molecular data
All research subjects were consented to Washington University Medical Center Human Research Protection Office protocols, HRPO-930828 and -040949. All enrolled participants were consented to long term clinical follow-up monitoring as part of ongoing Washington University HRPO–approved research protocols. Clinico-pathologic features, tumor microsatellite instability (MSI), and PTEN mutation status data were abstracted from our electronic research database. MSI determination and PTEN mutation evaluation were performed as previously described [26, 27].
Cancer cell lines
The AN3CA, HEC1A, KLE, and RL952 endometrial cancer cell lines were purchased from American Type Culture Collection (ATCC). Ishikawa was a gift from Dr. Stuart Adler (Washington University School of Medicine in St. Louis) and MFE296 was kindly provided by Dr. Pamela Pollock (TGen, Phoenix, AZ). The DU145 and PC3 prostate cancer cell lines used as reference controls were kindly provided by Dr. Adam Kibel (Washington University School of Medicine in St. Louis).
MicroRNA profiling
KLE and AN3CA cell lines were subjected to global microRNA profiling with Nanostring™ technology (Seattle, WA). Seven hundred and forty-nine microRNAs were evaluated using the nCounter Human miRNA Panel CodeSet®. This was performed similar to previously described mRNA analysis protocol [28].
PTENP1 and PTEN expression studies
Frozen primary tumor and normal tissues were stored at −70°C. Total cellular RNA was extracted utilizing the Trizol® method (Invitrogen, Carlsbad, CA). Tumors tissues (>66% neoplastic cellularity) were pulverized in liquid N2 prior to addition of the Trizol® reagent. Cell lines were lysed in situ. RNA concentration was determined with the NanoDrop spectrophotometry (Thermoscientific, Wilmington, DE). Complementary DNA (cDNA) was generated using 1 μg total RNA and QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Expression of PTEN and PTENP1 was assessed using real-time RT-PCR TaqMan® assays (Applied Biosystems, Foster City, CA) and the Applied Biosystems 7500 Fast real-time PCR system and software. Human β-actin protein was used as the endogenous control as previously described [20]. All qPCR assays were run for 40 cycles. Ct values of < 35 cycles were considered indicative of product expression for all qPCR experiments. Relative expression levels were calculated using the delta-delta Ct method [29]. Primer and probe sequences used were as described by Poliseno et al. [20] (Table 1).
Table 1.
Primers and FAM probe sequences used for the quantitative and conventional RT-PCR assays of PTENP1 expression.
| PTEN | PTENP1 | |
|---|---|---|
| Forward primer TaqMan® qPCR assay | 5′-CCTTCTCCATCTCCTGTGTAATCAA-3′ | 5′-AGTCACCTGTTAAGAAAATGAGAAGACAAA-3′ |
| Reverse primer TaqMan® qPCR & conventional RT-PCR assays | 5′-GTTGACTGATGTAGGTACTAACAGCAT-3′ | 5′-CTGTCCCTTATCAGATACATGACTTTCAA-3′ |
| FAM probe TaqMan® assay | CCAGTGCTAAAATTCA | AAGCAGGGAGAAATT |
| Forward primer conventional RT-PCR | 5′-TGGCATACACCAAATATAAGAGCA-3′ | 5′-TCCCTTATCAGATACATGACTTTCA-3′ |
qPCR assays were performed in triplicate and repeated with the same cDNA. All qPCR analyses were replicated beginning with new cDNA synthesis. Minus RT controls (reverse transcriptase negative cDNA synthesis reaction) were included for both PTEN and PTENP1 assays.
Conventional RT-PCR for PTENP1 analyses
PTENP1 expression in cell lines and normal endometria (NE) specimens was verified with the conventional RT-PCR assay [30]. All assays were repeated in triplicate. Minus RT assays were carried out for each of the cell line specimens and for one of the two normal endometrial specimens that were identified as PTENP1 positive.
The reverse RT-PCR assay primer was identical to the TaqMan® assay PTENP1 primer (Table 1). The forward primer sequence was selected using Primer3Plus software. All conventional RT-PCR assays were run for 35 cycles. PCR products were resolved on 10% polyacrylamide gels and scored for presence of the 246 bp band that corresponded with the expected product size for the PTENP1 product.
Primary tumor samples were considered positive for PTENP1 expression if three or more of the four conventional RT-PCR assays yielded the expected PCR product. Minus RT controls were analyzed to rule out genomic DNA contamination (i.e. false-positive results).
Statistical analysis
Median PTEN levels of the PTENP1-negative and PTENP1-positive tumors were compared using the Mann-Whitney test. The unpaired t-test was used to compare the log10-transformed average PTEN levels between the PTENP1-positive and PTENP1-negative tumors. Associations between clinical features and PTENP1 expression were evaluated with the two-tailed Chi-square test. A test for equality of variances was carried out to compare the variances of the PTEN wild-type (+/+)/haploinsufficient (+/-) tumors vs. PTEN-null (-/-) tumors. All statistical analyses were performed using the GraphPad Prism software (GraphPad software, La Jolla, CA).
Results
Expression of candidate miRNA regulators of PTEN and PTENP1
Global profiling of miRNA expression in the AN3CA and KLE cell lines revealed that all eight miRNAs predicted to inhibit PTEN and PTENP1 are present in both cell lines. These include miR-17, miR-19b, miR-20a, miR-21, miR-26a, miR-214, miR-216, and miR-217. Of these, miR-21, miR-19b, miR-26a, and miR-20a ranked in the top 20 most abundant miRNAs in both cell lines. Of note, miR-21 was the most abundant miRNA subtype in KLE, and the sixth most abundant subtype in the AN3CA cell line. Once we confirmed the presence of these putative PTEN/PTENP1 inhibitors in EMCA cell lines, we proceeded with further analysis of PTENP1 expression.
PTEN and PTENP1 expression in endometrial cancer cell lines and normal endometrium
Quantitative RT-PCR analysis revealed that all six EMCA cell lines and the three NE samples expressed PTEN transcript. Relative fold expression ranged between 0.6 and 4.4 arbitrary expression units (aeu) in EMCA cell line specimens and between 0.04 and 1 aeu in normal endometria. Log10 transformed relative expression values, normalized to DU145, are present in Figure 1A.
Fig. 1.
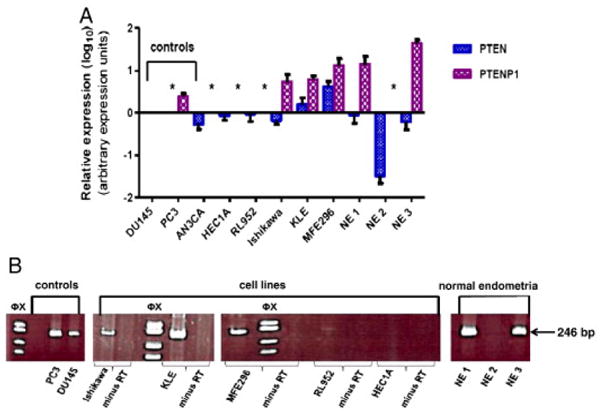
PTEN and PTENP1 expression in endometrial carcinoma cell lines and normal endometrium. A. Quantitative expression of PTEN and PTENP1. Log10-transformed averages of replicate experiments and standard errors of the mean are presented. Relative expression values are normalized to DU145 (prostate cancer cell line). *: PTEN or PTENP1 not expressed (exponential amplification cut off Ct ≥ 35). B. Representative RT-PCR analysis. PCR products (conventional RT-PCR 35 cycles) resolved on 10% polyacrylamide gels. The 246 bp PTENP1 PCR product is present in positive controls (DU145 and PC3 cell lines) and the Ishikawa, KLE, and MFE296 cell lines. Minus RT controls confirm that the PCR product is derived from the PTENP1 transcript. NE1 and NE3 samples are also positive for the 246 bp amplimer. Φx: Φx174 RF DNA/Hae III molecular size marker.
PTENP1 was expressed in three of the six EMCA cell lines (Ishikawa, KLE, and MFE296) and two of the three NE specimens (Figure 1A). AN3CA, HEC1A, and RL952 cell lines and NE specimen #2 did not express PTENP1. Relative PTENP1 fold expression ranged between 6.4 and 15.2 aeu in cell lines, and 17.1 and 45 aeu in normal endometria (Figure 1A).
The PTENP1 transcript levels were overall less abundant than PTEN levels in both cell lines and normal endometria (mean Ct values of 31.92 vs. 23.52 (cell lines); mean Ct values of 34 vs. 28.9 (normal endometria). PTENP1 levels appear to be higher in the primary tissues than the cell lines, while PTEN levels are higher in the cell lines than the normal tissues.
Relative expression of PTEN transcript levels were positively correlated with the PTENP1 transcript levels in the cell lines and normal endometrial specimens. KLE and MFE cell lines, the highest PTENP1 expressers, had the highest PTEN levels in the cell line cohort. The two normal tissues that expressed PTENP1 also expressed PTEN at high levels. PTEN levels were lowest in the NE specimen that did not express PTENP1 (Figure 1A). Because of relatively small number of specimens in the NE and EMCA cohorts, we are unable to statistically analyze the relationship between the PTEN and PTENP1 expression levels.
PTENP1 expression was confirmed by conventional RT-PCR analysis (Figure 1B). The findings for conventional RT-PCR and qPCR analyses were concordant overall. One cell line, RL952, that was classified as negative for PTENP1 expression by qRT-PCR (Ct values >35 cycles) showed a very faint PCR product of the expected 246 bp size in two of six cDNA syntheses. Given the variable expression as measured by conventional RT-PCR amplification, RL952 was classified as negative for PTENP1 expression.
PTEN and PTENP1 expression in primary tumors
PTEN and PTENP1 expression was evaluated in 61 primary endometrioid EMCA specimens. All tumors expressed PTEN, with an approximate 135 fold difference between the lowest and the highest expresser (0.17 and 23.3 relative expression units, Figure 2A).
Fig. 2.
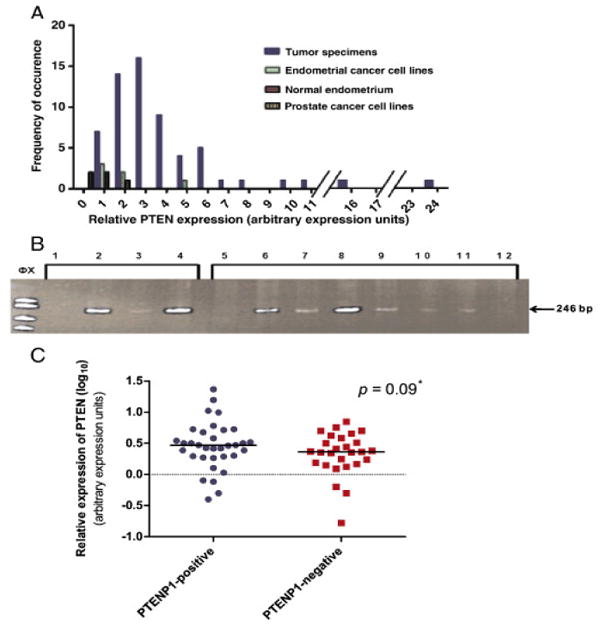
PTEN and PTENP1 expression in primary endometrial adenocarcinomas. A. Frequency distribution of average PTEN levels (expressed in arbitrary expression units) in primary tumors, normal endometria, and endometrial cancer cell lines. Relative expression normalized to DU145. Primary carcinomas show ∼ 135-fold range of expression. B. Representative RT-PCR PTENP1 expression analysis. The 246 bp PTENP1 PCR product is present in the positive control cell lines DU145, PC3 and Ishikawa (lanes 2–4) and in six primary tumors (lanes 6–11). The PTENP1 246 bp RT-PCR product is absent in two tumors that do not express PTENP1 (lanes 5 and 12). No template control is shown in lane 1. PCR products resolved on 10% polyacrylamide gels with Φx174 RF DNA/Hae III size standards. Variable intensity of the PCR product in positive controls and tumors may reflect differences in transcript levels. C. Relative PTEN expression in PTENP1- postivie and PTENP1-negative primary tumors. Graph of log10 transformed mean relative PTEN expression values of PTENP1-positive and PTENP1-negative tumors as assessed by real time PCR normalized to the DU145 cell line. Data points represent log10 transformed averages of three replicate experiments. Median PTEN levels for each group are indicated with horizontal bars. Dashed line: DU145 reference. *: Two tailed Mann–Whitney test.
PTENP1 transcripts were much less abundant than those of PTEN. The qRT-PCR Ct thresholds for detection of PTENP1 were at or near 35 cycles for many tumors. Given the low abundance of PTENP1 transcript (and high number of cycles required for detection) it was not possible to reliably quantify PTENP1 expression in our cohort of tumors. Conventional RT-PCR was therefore used to determine PTENP1 expression in primary tumors (presence/absence of the expected RT-PCR amplicon). Thirty-four of 61 (56%) tumors expressed PTENP1. Mock cDNA synthesis (no reverse transcriptase) proved that the amplification seen in all the 34 PTENP1-positive tumors was PTENP1 transcript derived (Figure 2B).
The relationship between PTEN and PTENP1 expression was investigated by comparing PTEN levels in the 34 PTENP1-positive and 27 PTENP1-negative tumors. PTEN levels ranged from 0.17 to 23.3 aeu. PTEN transcripts were more abundant in PTENP1-positive tumors than in PTENP1-negative tumors (mean and median expression values 4.18 and 2.93 aeu vs. 2.7 and 2.3 aeu, respectively). The difference in medians between the PTENP1-positive and PTENP1-negative tumors did not, however, reach statistical significance (p = 0.19, two-tailed Mann-Whitney test performed using log10-transformed PTEN expression values; p = 0.09, one-tailed Mann-Whitney test, Figure 2C). Given the data in cell lines and normal endometrial tissues suggesting a positive correlation between PTEN and PTENP1, as well as similar findings in prostate tumors [20], a one-tailed statistical test is likely to be an appropriate modality to evaluate the relationship between PTEN and PTENP1.
PTEN mutation data were available for 60 of the 61 tumors investigated. PTENP1 expression was not associated with PTEN mutation status. Five of 27 PTENP1-negative tumors were PTEN wild-type (+/+), 16 were PTEN haploinsufficient (+/-, carrying a single PTEN mutation), and six were PTEN null (-/-). Eight of 33 PTENP1-positive tumors were PTEN wild-type, 15 were haploinsufficient for PTEN mutations, and 10 carried two PTEN mutations (Table 2). When tumor specimens were further stratified by PTENP1 expression and mutation status, there was no difference in PTEN levels between the sub-groups. PTEN levels, however, were more variable in tumors lacking PTEN mutation or carrying a single mutation compared to PTEN-null tumors (p = 0.015, test for equality of variances, Figure 3A). We hypothesized that PTEN levels would be higher in PTENP1-positive tumors if PTENP1 acts as a decoy for miRNAs that normally downregulate PTEN transcript levels. Given the strong selection for reduced PTEN activity in endometrial cancers, PTENP1 effects should be most pronounced in tumors that carry a single PTEN mutation (haploinsufficient) or lack PTEN mutations. We found a trend towards higher PTEN levels in the PTENP1-positive wild-type/haploinsufficient group vs. PTENP1-negative wild-type/haploinsufficient group, but the difference did not reach statistical significance (p = 0.08, unpaired one-tailed t-test, Figure 3B).
Table 2.
PTEN expression levels, mutation status, and PTENP1 expression status for primary endometrioid endometrial cancers.
| Sample # | PTEN mutation | Mutation status | PTEN levels | PTENP1 expression |
|---|---|---|---|---|
| 1574 | c.[131−137del7(+)204C>G] | −/− | 1.766667 | N |
| 1411 | c.[139A>G(+)493G>A] | −/− | 2.3 | N |
| 1287 | c.[367C>T(+)376G>T] | −/− | 3.833333 | N |
| 1120 | c.[388C>G(+)389delG] | −/− | 3.166667 | N |
| 1599 | c.[388C>G(+)511C>T] | −/− | 3.233333 | N |
| 1562 | c.[639−663del25insGGCAA(+)898delA] | −/− | 2.266667 | N |
| 1717 | c.176C>A | +/− | 4.5 | N |
| 1236 | c.209+1del2GT | +/− | 1.333333 | N |
| 1424 | c.371G>C | +/− | 5 | N |
| 1492, 1501 | c.388C>G | +/− | 2.2, 1.23 | N |
| 1576, 1990 | c.389G>A | +/− | 0.63, 4.2 | N |
| 1669 | c.389G>T | +/− | 2.77 | N |
| 1664 | c.46dupT | +/− | 7.03 | N |
| 1656 | c.750–751dupTG | +/− | 2.33 | N |
| 2027 | c.800DelA | +/− | 2.57 | N |
| 1897 | c.867delA | +/− | 1.53 | N |
| 1570 | c.883–884delCT | +/− | 0.17 | N |
| 1482, 1504 | c.955–958DelACTT | +/− | 1.4, 5.7 | N |
| 1977 | c.968dupA | +/− | 1.73 | N |
| 1119, 1474, 1555, 1569, 1639 | None | +/+ | 2.27 5.03, 1.47, 0.5, 2.4 | N |
| 1441 | c.[367C>T(+)389G>A] | +/+ | 3.47 | Y |
| 1557 | c.[384G>T(+)428delG] | +/+ | 3.3 | Y |
| 1484, 1507 | c.[388C>G(+)T166G] | +/+ | 1.07, 0.4 | Y |
| 1539 | c.[407G>A(+)598−599delTT] | +/+ | 3.8 | Y |
| 1744 | c.[60delA(+)G198T] | +/+ | 5.33 | Y |
| 1184 | c.[713dupT(+)867dupA] | +/+ | 2.43 | Y |
| 1637 | c.[950−954delTACTT(+)c.968dupA] | +/+ | 2.63 | Y |
| 1226 | c.[A71G(+)388C>G] | +/+ | 2.43 | Y |
| 1193 | c.T48A(+)386G>A | +/+ | 10.53 | Y |
| 1316 | c.334C>G | +/− | 5.33 | Y |
| 1330 | c.367C>T | +/− | 5.17 | Y |
| 1251, 1513 | c.388C>G | +/− | 2.97, 2.97 | Y |
| 1221, 1727 | c.389G>A | +/− | 3.17, 4.73 | Y |
| 1611 | c.389G>C | +/− | 2.67 | Y |
| 1112 | c.410–414del5 | +/− | 23.33 | Y |
| 1556 | c.520T>G | +/− | 2 | Y |
| 1673 | c.662delA | +/− | 2.9 | Y |
| 1984 | c.741–742InsA | +/− | 0.8 | Y |
| 1652 | c. 802−2A>T | +/− | 1.87 | Y |
| 1685 | c.955–958DelACTT | +/− | 9.87 | Y |
| 1391 | c.C733T | +/− | 1.93 | Y |
| 1609 | del 1bp A 800 | +/− | 1.27 | Y |
| 1087, 1289, 1352, 1419, 1500, 1644, 1792, 1856 | None | +/+ | 15.77, 3.17, 2.6, 0.77, 1.83, 0.5, 1.97, 6.03 | Y |
| 1674 | No data | No data | 3.17 | Y |
Fig. 3.
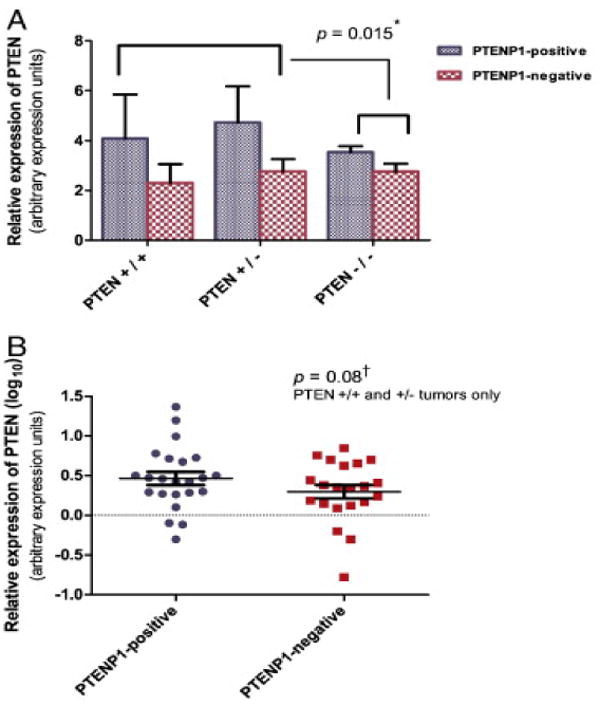
PTENP1 expression and PTEN transcript levels in primary tumors stratified by PTEN mutation status. A. Relative PTEN expression in PTEN wild type (+/+), haploinsufficient (+/−), and PTEN null (−/−) tumors. PTEN levels were more variable in PTEN wild-type (+/+) and haploinsufficient (+/−) tumors than in tumors with two mutations (−/−). *: Test for equality of variances. B. Log10-transformed averages of PTEN expression in wild-type (+/+) and haploinsufficient (+/−) PTENP1-positive and PTENP1-negative tumors are presented. Bars represent mean PTEN transcript values. Error bars represent standard errors of the mean. Dashed line: DU145 reference. †: One-tailed t-test.
Relationship between PTENP1, and clinicopathologic variables and MSI status
PTENP1 expression was not associated with stage, tumor grade, disease recurrence/progression or tumor MSI status (Table 3). Patients with PTENP1-positive tumors exhibited a trend towards lower disease recurrence/progression, compared to the PTENP1-negative group (p = 0.11, OR 0.44, 95% CI 0.81-6.5, two-sided Chi-square analysis).
Table 3.
Relationship between clinico-pathologic features, MSI status, and PTENP1 expression.
| PTENP1 expression | ||||
|---|---|---|---|---|
|
|
|
|
|
|
| Total (n) | Positive (%) | Negative (%) | p value | |
|
| ||||
| Number of patients | 61 | 34 (56%) | 27 (44%) | |
|
| ||||
| Stage | 0.97a | |||
|
| ||||
| I and II | 25 | 14 (56%) | 11 (44%) | |
|
| ||||
| III and IV | 36 | 20 (56%) | 16 (44%) | |
|
| ||||
| Grade | 0.73a | |||
|
| ||||
| 1 | 20 | 12 (60%) | 8 (40%) | |
|
| ||||
| 2 | 22 | 11 (50%) | 11 (50%) | |
|
| ||||
| 3 | 18 | 11 (61%) | 7 (39%) | |
|
| ||||
| Recurrence/progression | 0.11a | |||
|
| ||||
| Yes | 27 | 12 (44%) | 15 (56%) | |
|
| ||||
| No | 34 | 22 (65%) | 12 (35%) | |
|
| ||||
| MSI | 0.76a | |||
|
| ||||
| Yes | 28 | 16 (57%) | 12 (43%) | |
|
| ||||
| No | 33 | 18 (55%) | 15 (45%) | |
Two-tailed Chi-square test.
Discussion
A recent report that the PTEN pseudogene PTENP1 may function as a tumor suppressor in prostate cancer and indirectly impact PTEN activity prompted our investigation of PTENP1 expression in endometrial cancer [20]. We believe our study to be the first report describing PTENP1 expression in normal endometria, endometrial carcinoma cell lines, and primary human carcinoma. We also demonstrated that PTEN/PTENP1 inhibitory miRNAs are present in both EMCA cell lines. Our findings support the hypothesis that the PTEN/PTENP1/miRNA driven tumorigenesis mechanism described in prostate adenocarcinomas by Poliseno and colleagues could be conserved in EMCA [20] but our work does not provide functional evidence for such a mechanism in endometrial cancer.
Poliseno et al. surveyed a broad range of tissues for PTENP1 expression and reported a very low PTENP1 transcript level in the uterus relative to the 47 other primary tissues investigated. Our analysis of endometrial cancer cell lines, normal endometrium, and primary endometrial endometrioid cancers revealed that PTENP1 is frequently expressed in uterine epithelium and endometrial carcinomas. PTENP1 expression levels in the cell lines and normal endometria were comparable to those in prostate adenocarcinoma cell lines (Figure 1A). It is possible that Poliseno and colleagues analyzed the specimens comprised of homogenized uteri (containing a large fraction of smooth muscle tissue and a small fraction of endometrium) and detected low PTENP1 transcript levels. In the current study, we analyzed specimens derived from the endometrium tissue only. The difference in specimen composition may explain the variability in PTENP1 transcript levels between our study and that of Poliseno and colleagues.
PTEN mutation is one of the most frequently observed genetic abnormalities in endometrial endometrioid cancers [31]. Although PTEN's role in tumorigenesis has been firmly established, the observation that tumors frequently carry a single mutation (haploinsufficient rather than PTEN-null) raises the possibility that reduced PTEN function may be sufficient to promote tumor phenotypes [32]. It is probable that for some human tumors with one mutated allele and intact wild-type PTEN allele(s), PTEN is reduced to a critically low level by epigenetic silencing. Methylation of the PTEN promoter has been described many times, and recent papers document the important roles for miRNAs in PTEN regulation [12, 13, 33-35]. The work from Poliseno et al. suggests that PTENP1 expression impacts PTEN activity. Marsit et al. [36] investigated epigenetic modifications of PTEN and PTENP1 in lung cancers. They showed that the PTENP1 promoter is hypermethylated in 66% of the examined lung carcinoma samples, but in 0% of normal blood lymphocytes. The study did not investigate the relationship between the PTENP1 promoter hypermethylation and abundance of PTEN mRNA/protein expression. In light of the recent findings suggesting that PTENP1 may contribute to modulation of PTEN levels [20], and prior reports of PTEN promoter methylation in endometrial cancers [37], further investigation of the effects of PTENP1 promoter hypermethylation may be warranted.
We have demonstrated that there is significant variation in PTEN levels of the wild-type and haploinsufficient tumors as compared with PTEN-null tumors. Although PTENP1-positive-tumors exhibited a trend towards increased PTEN transcript levels, the difference was not statistically significant.. Of note, the transcripts of PTEN-null specimens are non-functional and undergo nonsense-mediated decay before being translated into protein product. Variance in PTEN levels may in part be due to selective methylation of the PTEN promoter in a fraction of wild-type/haploinsufficient tumors. Methylated tumors may express lower PTEN transcript levels than their unmethylated counterparts. When examined as a group, methylated and unmethylated tumors would exhibit high variance in PTEN transcript levels, as observed in our study. We did not, however, assess PTEN methylation in our interrogation of primary tumors. Understanding the relative contributions that promoter methylation, miRNA, and PTENP1 expression play in endometrial tumorigenesis will require large scale studies with comprehensive mRNA and miRNA profiling in combination with PTEN mutation and PTEN/PTENP1 promoter methylation analyses.
We did not identify any clinico-pathologic factors associated with PTENP1 expression in EMCA specimens. Given the limited number of samples that we investigated, our ability to detect clinicopathologic associations of modest size was limited. The association between PTENP1 expression and disease outcome warrants further investigation in a larger study cohort along with analysis of expression on the microRNAs that may regulate the PTEN/PTENP1 expression.
In summary, the current study serves as a springboard for further investigation of the role of PTENP1 and PTEN/PTENP1 inhibitory miRNAs in EMCA. The next logical step would entail the investigation of PTEN/PTENP1 inhibitory miRNAs in EMCA tissues and their impact on tumor growth in vitro. Further understanding of PTENP1's role in tumorigenesis may lead to the discovery of novel therapeutic targets.
Acknowledgments
We would like to thank Dominic Thompson, Jr., MA and Amy Schmidt, BS for technical assistance. We would like to thank Dr. Kim Trinkaus for assistance with statistical analyses.
Grant support: RO1 CA71754-13 (PJG), P50CA134254-1 (PJG), BJF 6863-33 (DGM), and a grant from the Foundation for Barnes-Jewish Hospital.
Footnotes
Disclosure Statement: To the best of our knowledge, the authors of this manuscript have no conflicts of interest.
References
- 1.ACS. Cancer facts and figures 2010. Atlanta: American Cancer Society Inc.; 2010. [Google Scholar]
- 2.Ellis PE, GhaemMaghami S. Molecular characteristics and risk factors in endometrial cancer: what are the treatment and preventative strategies? Int J Gynecol Cancer. 2010;20:1207–16. doi: 10.1111/igc.0b013e3181f1a400. [DOI] [PubMed] [Google Scholar]
- 3.Ray M, Fleming G. Management of advanced-stage and recurrent endometrial cancer. Semin Oncol. 2009;36:145–54. doi: 10.1053/j.seminoncol.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Lee NK. Adjuvant treatment of advanced-stage endometrial cancer. Clin Obstet Gynecol. 2011;54:256–65. doi: 10.1097/GRF.0b013e318218c659. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 6.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 7.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 8.Kong D, Suzuki A, Zou TT, Sakurada A, Kemp LW, Wakatsuki S, et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nature Genetics. 1997;17:143–4. doi: 10.1038/ng1097-143. [DOI] [PubMed] [Google Scholar]
- 9.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daikoku T, Jackson L, Besnard V, Whitsett J, Ellenson LH, Dey SK. Cell-specific conditional deletion of Pten in the uterus results in differential phenotypes. Gynecol Oncol. 2011 doi: 10.1016/j.ygyno.2011.04.022. Epub ahead of print 5/17/2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, et al. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619–27. doi: 10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchini S, Cavalieri D, Fruscio R, Calura E, Garavaglia D, Nerini IF, et al. Association between miR-200c and the survival of patients with stage I epithelial ovarian cancer: a retrospective study of two independent tumour tissue collections. Lancet Oncol. 2011;12:273–85. doi: 10.1016/S1470-2045(11)70012-2. [DOI] [PubMed] [Google Scholar]
- 13.Cohn DE, Fabbri M, Valeri N, Alder H, Ivanov I, Liu CG, et al. Comprehensive miRNA profiling of surgically staged endometrial cancer. Am J Obstet Gynecol. 2010;202:656 e1–8. doi: 10.1016/j.ajog.2010.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JW, Park YA, Choi JJ, Lee YY, Kim CJ, Choi C, et al. The expression of the miRNA-200 family in endometrial endometrioid carcinoma. Gynecol Oncol. 2011;120:56–62. doi: 10.1016/j.ygyno.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 15.Fujii GH, Morimoto AM, Berson AE, Bolen JB. Transcriptional analysis of the PTEN/MMAC1 pseudogene, psiPTEN. Oncogene. 1999;18:1765–9. doi: 10.1038/sj.onc.1202492. [DOI] [PubMed] [Google Scholar]
- 16.Hirotsune S, Yoshida N, Chen A, Garrett L, Sugiyama F, Takahashi S, et al. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature. 2003;423:91–6. doi: 10.1038/nature01535. [DOI] [PubMed] [Google Scholar]
- 17.Mighell AJ, Smith NR, Robinson PA, Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–14. doi: 10.1016/s0014-5793(00)01199-6. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Carriero N, Gerstein M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet. 2004;20:62–7. doi: 10.1016/j.tig.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–8. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piehler AP, Hellum M, Wenzel JJ, Kaminski E, Haug KB, Kierulf P, et al. The human ABC transporter pseudogene family: Evidence for transcription and gene-pseudogene interference. BMC Genomics. 2008;9:165. doi: 10.1186/1471-2164-9-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou M, Baitei EY, Alzahrani AS, AlMohanna F, Farid NR, Meyer B, et al. Oncogenic activation of MAP kinase by BRAF pseudogene in thyroid tumors. Neoplasia. 2009;11:57–65. doi: 10.1593/neo.81044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melicow MM, Tannenbaum M. Endometrial carcinoma of uterus masculinus (prostatic utricle). Report of 6 cases. J Urol. 1971;106:892–902. doi: 10.1016/s0022-5347(17)61430-7. [DOI] [PubMed] [Google Scholar]
- 24.Walther MM, Nassar V, Harruff RC, Mann BB, Jr, Finnerty DP, HewenLowe KO. Endometrial carcinoma of the prostatic utricle: a tumor of prostatic origin. J Urol. 1985;134:769–73. doi: 10.1016/s0022-5347(17)47433-7. [DOI] [PubMed] [Google Scholar]
- 25.Das S. Endometrial carcinoma of prostate. Urology. 1986;27:543–5. doi: 10.1016/0090-4295(86)90338-9. [DOI] [PubMed] [Google Scholar]
- 26.Zighelboim I, Schmidt AP, Gao F, Thaker PH, Powell MA, Rader JS, et al. ATR mutation in endometrioid endometrial cancer is associated with poor clinical outcomes. J Clin Oncol. 2009;27:3091–6. doi: 10.1200/JCO.2008.19.9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA, et al. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68:6902–7. doi: 10.1158/0008-5472.CAN-08-0770. [DOI] [PubMed] [Google Scholar]
- 28.Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol. 2011;Chapter 25 doi: 10.1002/0471142727.mb25b10s94. Unit25B 10. [DOI] [PubMed] [Google Scholar]
- 29.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 30.Mei M, Liu D, Dong S, Ingvarsson S, Goodfellow PJ, Chen H. The MLH1 -93 promoter variant influences gene expression. Cancer Epidemiol. 2010;34:93–5. doi: 10.1016/j.canep.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 32.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato K, Tamura G, Tsuchiya T, Endoh Y, Sakata K, Motoyama T, et al. Analysis of genetic and epigenetic alterations of the PTEN gene in gastric cancer. Virchows Arch. 2002;440:160–5. doi: 10.1007/s004280100499. [DOI] [PubMed] [Google Scholar]
- 34.Salvesen HB, MacDonald N, Ryan A, Jacobs IJ, Lynch ED, Akslen LA, et al. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int J Cancer. 2001;91:22–6. doi: 10.1002/1097-0215(20010101)91:1<22::aid-ijc1002>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 35.Ho CM, Lin MC, Huang SH, Huang CJ, Lai HC, Chien TY, et al. PTEN promoter methylation and LOH of 10q22-23 locus in PTEN expression of ovarian clear cell adenocarcinomas. Gynecol Oncol. 2009;112:307–13. doi: 10.1016/j.ygyno.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 36.Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–76. doi: 10.1016/j.humpath.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol. 2006;24:4783–91. doi: 10.1200/JCO.2006.06.7173. [DOI] [PubMed] [Google Scholar]