

Abstract
The growth factor receptor-bound protein (Grb) 14 is an adapter molecule of the Grb7/10/14 family with characteristic BPS domains serving to avidly bind tyrosine kinases. Grb14 inhibits insulin receptor (IR) catalytic activity through interaction with the BPS domain and impedes peptide substrate binding. Members of this Grb family have also been shown to interact with other kinases through their SH2 domain. Here we examined the functional role of Grb14 in thyroid cancer using loss- and gain-of-function approaches. Stable knockdown of Grb14 in thyroid cancer cells facilitated insulin receptor signaling. In contrast, RET phosphorylation was diminished in concert with reduced activation of Akt and STAT3. Loss of Grb14 also resulted in diminished cell proliferation and invasion both in vitro and in mouse flank xenografts. In complementary studies, forced expression of Grb14 interrupted insulin receptor signaling but facilitated RET activation, STAT3, and Akt phosphorylation. Consistent with these findings Grb14 over-expression enhanced cell invasion and resulted in striking metastases in an orthotopic thyroid cancer mouse xenograft model. Primary human thyroid cancer microarrays revealed a positive correlation between Grb14 expression and invasive behavior. Our findings uncover a new role for Grb14 in finely tuning receptor signaling and modulating thyroid cancer progression.
Keywords: Grb14, RET, thyroid cancer, cancer progression
Introduction
Thyroid cancers represent a heterogeneous group of endocrine malignancies. During the development of epithelial thyroid cancers, the RET proto-oncogene plays an important role in tumor initiation and progression (Kondo et al., 2006). RET is responsible for normal development of the neural crest-derived cell lineages (Avantaggiato et al., 1994), regulating cell proliferation, migration and differentiation in response to its activation by a co-receptor–ligand complex (Durbec et al., 1996). Gain of function mutations or rearrangements of RET lead to constitutive activation of this tyrosine kinase receptor, which in turn causes the up-regulation of oncogenic signaling pathways (Asa, 2001) leading to medullary or papillary thyroid cancer development. It has been shown that the level of phosphorylation of the different tyrosine residues outside the kinase domain may also regulate down-stream signaling to influence proliferation, morphogenesis and tumor formation (Buckwalter et al., 2002).
The growth factor receptor-bound (Grb) family of adaptor proteins (Grb7/10/14) has been characterized by a unique between plekstrin and SH2 (BPS) domain that facilitates avid binding to activated insulin receptor tyrosine kinases (Margolis, 1994). Grb14, the most restricted member of this family, is known to interrupt insulin receptor signaling through binding to the activation loop. In so doing, it impedes down-stream insulin receptor signaling (Kasus-Jacobi et al., 1998), playing a crucial role in the regulation of insulin receptor function. However, other RTKs have also been shown to interact with members of this Grb family. In particular, RET is known to interact with Grb7 and Grb10 (Pandey et al., 1995,Pandey et al., 1996). The functional impact of this interaction, however, has not been examined, nor is the role of Grb14 in thyroid cancer progression known.
The aim of this study was to examine the effect of Grb14 on receptor signaling and thyroid cancer progression. To this end, we tested the impact of Grb14 on the insulin receptor as a recognized target, and RET as a putative target of Grb14 action. Using complementary loss-and gain-of-function approaches, we examined the functional impact of Grb14 on thyroid cancer cell growth, invasiveness, and metastasis. To determine the significance of the results in human disease, we used a tissue microarray of human thyroid carcinomas to validate the relationship between Grb14 and behavior of this neoplasm.
Materials and Methods
Cell lines and cell culture
Human thyroid carcinoma-derived cell lines TT (ATCC, Manassas, VA), WRO-82-1 (established by Dr. G. J. F. Juillard, University of California-Los Angeles School of Medicine, Los Angeles, CA), TPC-1 (provided by Dr. S.M. Jhiang, Ohio State University, Columbus, OH), 8505C (Cell Resource Center for Biomedical Research, Tohoku University, Sendai, Japan), and KTC-1 (established by Dr. J. Kurebayashi, Kawasaki Medical School, Okayama, Japan) were maintained in RPMI 1640 with streptomycin sulphate (100 mg/L) and penicillin G sodium (100 mg/L), and supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Burlington, ON, Canada).
Stimulation studies
Following serum deprivation in RPMI 1640, cells were cultured in RPMI supplemented with 10% FBS, as a source of multiple growth factors, or maintained in serum-free growth medium without or with insulin (300 nM) or glial derived nerve factor (GDNF) (R&D Systems; 50 ng/ml) for up to 30 minutes as indicated. Identical volume of vehicle served as a control.
Stable Grb14 knockdown and forced expression
Grb14 was stably down-regulated using the SureSilencing shRNA plasmid system (SABiosciences, Frederick, MD). Control clones were transfected with scrambled sequences according to the manufacturer’s instructions. To establish Grb14 over-expression, cells were transfected with Grb14 or empty vector (generously provided by R. Daly) as described (Daly et al., 1996). Plasmids were introduced using GeneJuice transfection reagent (Novagen, EMD Chemicals Inc, Merck, Darmstadt). Stably-transfected clones were selected using 1000 μg/μl G418 (Multicell Technologies, Woonsocket, RI, USA) and maintained with G418 (500 μg/μl) during the experiments. At least two independent experimental clones and two independent control clones were used in each experiment.
RNA extraction and reverse transcriptase PCR
Total RNA was isolated from cultured cells using RNeasy Kit (Qiagen, Valencia, CA), followed by DNase I treatment. Reverse transcriptase (RT) reactions were performed from 2 μg total RNA in 20-μl final volume using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA). Specific PCR primers for Grb14, extracellular domain of RET (RET-EC), or tyrosine kinase domain of RET (RET-TK) were used (Supplementary Table S1). Amplification was performed using HotStarTaq DNA polymerase kit (Qiagen). The PCR protocol started with a 15-min activation at 95°C, followed by 35 cycles of denaturation at 94 °C (30 s), an annealing step at a specified temperature between 55 and 65 °C (30 s), and extension at 72 °C (1 min). The final extension was performed at 72°C for 10 min. Negative controls omitting RT were included in each PCR reaction.
Cell lysates and Western blotting
Cultured cells were lysed in an immunoprecipitation buffer (0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, and 1x PBS) containing proteinase inhibitors [100 μg/ml phenylmethylsulfonyl fluoride, 13.8 μg/ml Aprotinin (Sigma, St. Louis, MO), and Phosphatase Inhibitor Cocktail 2 (Sigma) 10 μl/ml. Xenografted tissues were lysed in the same lysis buffer after homogenization using a polytron homogenizer. Total cell lysates were incubated on ice for 30 min followed by microcentrifugation at 10,000 × g for 15 min at 4°C. Equal amounts (50 μg) of protein were separated by 10% SDS-polyacrylamide electrophoresis and transferred onto nitrocellulose membranes, which were blocked in blocking buffer (10% nonfat milk and 0.1% Tween 20 in TBS [20 mmol/L Tris-Cl and 500 mmol/L NaCl (pH 7.5)] for 1 h and incubated overnight at 4°C with primary antibodies, at optimized dilutions, as follows: anti-Grb14 (Millipore; 1:1000; Novus Biologicals 1:500; Santa Cruz Biotechnology 1:1000), anti-IR pY972 (In vitrogen, 1:1000), anti-IR pY1158/1162 (Invitrogen, 1:1000) and (Santa Cruz Biotechnology, 1:1000), anti-IR β (Cell Signaling, 1:1000), anti-actin (Sigma, 1:2000), anti-GAPDH (Sigma, 1:2000), anti-RET pY905 (Cell Signaling, 1:500), anti-RET pY1062 (Santa Cruz Biotechnology, 1:1000), anti-RET (Cell Signaling, 1:1000), anti-pMAPK (Sigma, 1:1000), anti-MAPK (Sigma, 1:2000), anti-pAkt S473 (Cell Signaling, 1:500), anti-Akt (Cell Signaling, 1:1000), anti-pY705-STAT3 (Cell Signaling, 1:500), and anti-STAT3 (Cell Signaling, 1:1000). Membranes were washed three times with washing buffer (TBS containing 1% Tween 20 and incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse secondary antibody (Santa Cruz Biotechnology, 1:2000) for 1h at room temperature. Protein bands were visualized using a chemiluminescence detection system (Amersham, Piscataway, NJ). Band intensities were measured by scanning densitometry for quantification of the signals and the phosphorylated/total target ratio was expressed as a percent.
Invasion assay
Invasion assays were performed using BioCoat Matrigel Invasion Chambers (BD Biosciences). After 18 hrs of serum deprivation, 5×104 cells were suspended in 0.5 mL of serum-free RPMI 1640 and plated into each insert, then wells in the plate were filled with RPMI 1640 supplemented with 10% FBS. After 20 hrs of incubation, non-invading cells were removed from the upper surface of the membrane by a cotton swab. Invaded cells on the lower surface of the membrane were stained with Diff-Quick Stain Solutions (BD Biosciences). After air drying, the membranes were removed from the insert; invaded cells were photographed using a Slide Scanner and quantified with APERIO ImagePro software (Vista, CA).
Cell proliferation assay
One ×105 cells were seeded in 10 cm plates in RPMI 1640 media. Four plates were seeded for each clone for complete cell counting. Total cell counts were measured on day 3, 5 and 7 after seeding using a Vi-Cell Cell Viability Analyzer (Beckman-Coulter, Inc, Brea, CA).
Human thyroid cancer cell xenografts
WRO cells (4×106) under- or over-expressing Grb14 or their controls were xenografted in the flanks or orthotopically implanted in the neck of 5 to 6-wk-old severe combined immunodeficient (SCID) mice for assessment of tumor growth and metastasis. Animal handling and treatment protocols were approved by the Ontario Cancer Institute Animal Care and Utilization Committee. Tumor growth and volume were measured using calipers every other day (tumor volume in mm2 = tumor width × tumor length). Mice were sacrificed on day 15 after inoculation, at which point tumors were excised, weighed, and measured (tumor volume in mm3= tumor width × tumor length × tumor depth/2) and full autopsies were performed. Excised tumor, lung and liver tissues and any other abnormal tissues were fixed in formalin and embedded in paraffin for light microscopy and immunohistochemistry.
Immunohistochemistry, TUNEL assay and primary human tissues
Tissue blocks were fixed in formalin and embedded in paraffin for routine histology and immunohistochemistry. Paraffin sections 5 μm thick were de-waxed in xylene and hydrated through graded alcohols (Ezzat et al., 2002,Ezzat et al., 2005). Immunoreaction of primary antibodies was detected by the avidin-biotin technique and visualized with 3,3′-diaminobenzidine. Cell apoptosis was determined using the terminal deoxyncleotidyl transferase TDT-mediated deoxyuridine-triphosphate nick-end labeling (TUNEL) technique (ApopTag kit, Oncor, Integen Company, Purchase, NY).
A human thyroid cancer tissue micro-array (TMA) was built using cases of well-differentiated human thyroid carcinomas, including triplicate cores for every case and tissue type as described previously (Liu et al., 2007,Briese et al., 2010). Immunohistochemical detection of Grb14 was standardized using positive and negative controls according to the manufacturer (Santa Cruz) and assessed under light microscopy by an endocrine pathologist to confirm appropriate tissue reactivity of the stain. Immunoreactivities were then evaluated by automated analysis using specific algorithms for each compartment as previously described (Cheng et al., 2009,Cheng et al., 2011).
Statistical analysis
Continuous data are presented as mean +/− standard error (SE), while binomial variables are presented as absolute numbers and percent frequency. Results were analyzed using non-parametric statistical analysis, Mann-Whitney U-test when appropriate for comparison of median values of two independent groups. Statistical significance was achieved at two-tailed p<0.05.
RESULTS
Establishing stable Grb14 knock-down and over-expressing thyroid cancer cells
To establish a system to examine the impact of Grb14 in thyroid cancer, we screened a panel of human thyroid carcinoma-derived cell lines for their endogenous expression of this adaptor protein (Fig. 1A,B). Further, given the known role of the RET tyrosine kinase in thyroid cancer we also examined co-expression of this kinase. This survey identified WRO and TPC-1 cells as expressing Grb14 and the RET/PTC-1 re-arrangement (Fig. 1A,B,C). Treatment of these cells with GDNF failed to stimulate RET phosphorylation, confirming their lack of the wild-type RET extracellular domain (data not shown). These two cell lines were selected for the creation of multiple clones with stable knockdown of the Grb14 adaptor to understand its functions (Fig. 1D).
Figure 1. Expression of Grb14 and the RET kinase domain in human thyroid cancer cell lines.

A. RT and real-time quantitative qPCR demonstrate the relative endogenous expression levels of Grb14 in thyroid cancer cell lines. Human PGK-I was used as an internal housekeeping loading control. B. Western blotting comparison of Grb14 protein levels in the same thyroid cancer cell lines. Negative (pcDNA) and positive (Grb14-transfected cells) are shown immediately to the right. Corresponding densitometric values are shown immediately below relative to actin ratios. C. RT-PCR demonstrates expression of the extracellular (EC) and tyrosine kinase (TK) domains of RET. TT cells express full length RET, which yields amplification of both the EC and intracellular TK domains of RET. TPC-1 and WRO cells express the RET/PTC1 rearrangement, which yield amplification of the intracellular TK domain but not the EC domain of RET; instead there is a product using RET/PTC1 primers. Human PGK-I served as an internal housekeeping control. D. Grb14 knockdown in WRO cells was achieved through stable expression of plasmid expressing shRNA targeting the adaptor or scramble sequence controls. Western blotting demonstrates effective Grb14 reduction in 3 independent clones (+) compared to scrambled controls (−).
Grb14 knock-down facilitates insulin receptor signaling but impairs RET activation
To examine the functional impact of Grb14 we first examined responsiveness of the insulin receptor (IR) as a well-recognized target of Grb14 action. In WRO cells, reduction of Grb14 predictably enhanced IR phosphorylation evidenced by increased pY972 and within the kinase domain pY1158/1162 of the receptor (Fig 2A). In contrast, RET phosphorylation was diminished when Grb14 was reduced (Fig. 2B)(Suppl. Fig. S1). Consistent with diminished RET signalling, pMAPK (Fig. 2B) was also attenuated in response to Grb14 reduction. To further corroborate the attenuation of RET signalling, we examined two downstream RET targets. Figure 2C demonstrates diminished pY-STAT3 in response to Grb14 reduction. Akt phosphorylation, another RET signaling target, was measurably impaired in Grb14-reduced cells (Fig. 2D). To examine these Grb14 signaling properties in an alternate thyroid cancer cell line, we examined Grb14 down-regulated TPC-1 cells. Again we noted that forced reduction of Grb14 resulted in increased IR phosphorylation. As with WRO cells, RET phosphorylation was diminished in the face of down-regulated Grb14 in TPC-1 cells (Suppl. Fig. S2). Similarly, Akt activation was attenuated in the face of Grb14 reduction in TPC-1 cells (Suppl. Fig. S2).
Figure 2. Impact of Grb14 knock-down on insulin receptor and RET receptor signaling.

A. Loss of Grb14 differentially impacts receptor phosphorylation. Control (WRO/scramble) and Grb14 knock-down WRO (WRO/shGrb14) cells were serum starved for 18 hrs and treated for 10 minutes with vehicle, FBS, or insulin as indicated. Stable reduction of Grb14 reveals hyper-phosphorylation of the insulin receptor at pY972 and at the activation loop pY1158/1162 site. B. Reduction of Grb14 diminishes RET phosphorylation evidenced by attenuated pY1062 and by reduced MAPK activation. C. Grb14 reduction impairs both STAT3 tyrosyl phosphorylation and D. Akt activation as downstream targets of impaired RET activation. The corresponding densitometric findings throughout the indicated time courses from the mean of 3 independent experiments are shown immediately below.
Grb14 knockdown impairs thyroid cancer cell proliferation and invasion in vitro and in vivo
To test the impact of dysregulated receptor signaling, we examined the role of Grb14 reduction on cell proliferation. WRO cells stably down-regulated for Grb14 revealed altered cell polarity as depicted in phase-contrast photomicrographs (Fig. 3A). Consistent with this altered morphologic appearance, WRO/shGrb14 grew more slowly (Fig. 3B) and invaded matrigel membranes with diminished efficiency (Fig. 3C) compared to control cells stably transfected with scrambled shRNA sequence (WRO/scramble). Similar impact of Grb14 reduction on TPC-1 cell proliferation was also noted (data not shown).
Figure 3. Grb14 down-regulation reduces thyroid cancer cell proliferation and invasion.

A. Phase-contrast photomicrographs reveal contracted WRO thyroid cancer cell polarity. The bar represents a 500 μm scale. B. Diminished cell proliferation is noted during growth curve measurements in response to Grb14 reduction. The summation of three independent proliferation assays demonstrates the significant difference between the proliferation rates of scrambled (WRO/scramble) and knock-down clones (WRO/shGrb14) as shown in the panel to the right. The * denotes significance, where p<0.00005; p<0.00005 and p<0.0005 at 3, 5, and 7 days respectively. C. Representative membranes of a Matrigel Invasion assay demonstrate the reduction in invasiveness of Grb14 knock-down cells compared to their scrambled controls to the left. The mean from two independent experiments quantifies the impact of Grb14 reduction on impaired WRO cell invasion with significance depicted by an asterisk as determined by the Mann-Whitney U-test (p=0.028). D. Photographs of flank xenografts demonstrate the slowed progression of tumor growth in SCID mice carrying Grb14 down-regulated WRO cells (WRO/shGrb14) compared to their scramble (WRO/scramble) controls. Control tumors infiltrate and erode overlying skin, resulting in ulceration, whereas tumors with down-regulation of Grb14 have pushing borders that do not erode overlying skin (hematoxylin-eosin stain). E. This time course demonstrates the reduced rate of tumor development in Grb14 down-regulated xenografts compared to scramble controls. The mean from two independent experiments quantifies the difference between tumor sizes which was statistically significant throughout the experiment. The differences in tumor volumes and weights were significantly different as measured at the time of sacrificing (*p<0.001; **p<0.01 on both panels).
To assess the impact of Grb14 reduction on tumor progression, we used a flank xenograft model in SCID mice that allows accurate analysis of tumor growth (Fig. 3D). Reduced Grb14 impaired thyroid cancer progression wherein WRO/shGrb14 tumors appeared later and grew significantly more slowly than controls (WRO/scramble)(Fig. 3E). Histologic examination revealed less invasive growth and lower proliferation as determined by lower mitotic counts (6% vs. 16%) and Ki-67 labeling indices (18% vs. 47%). No impact on apoptosis was noted by TUNEL staining. Western blotting of lysates from excised tumors confirmed diminished p-Akt levels in response to Grb14 knockdown (data not shown). TPC-1 cells did not develop efficient xenografts and hence mouse studies could not be performed using these cells.
Grb14 over-expression impedes insulin receptor but promotes RET signaling
In complementary experiments we forced Grb14 expression in multiple clones of WRO and TPC-1 cells (Suppl. Fig. S3A) which resulted in higher degree of phosphorylation at the IR-Y1158/1162 (Fig. 4A) consistent with the ability of the adaptor to protect the IR activation loop from dephosphorylation by displacing PTP1b towards the IR-Y972 which showed reduced phosphorylation (Fig. 4A). Additionally and consistent with the effect of Grb14 reduction on reduced RET phosphorylation, forced Grb14 expression facilitated RET signalling and MAPK activation (Fig. 4B) and supported STAT3 (Fig. 4C) and Akt phosphorylation (Fig. 4D)(Suppl. Fig. S3B).
Figure 4. Impact of Grb14 over-expression on insulin receptor and RET receptor signaling.

A. The impact of forced Grb14 over-expression on insulin receptor phosphorylation. WRO/Grb14 cells demonstrate higher phosphorylation at the IR-Y1158/1162 activation loop consistent with protection from dephosphorylation but decreased phosphorylation at the juxtamembrane IR-Y972 site. B. Grb14 over-expressing (WRO/Grb14) cells facilitate RET signaling evident by increased phosphorylation of the receptor and its target MAPK. C. Grb14 over-expression promotes STAT3 phosphorylation and D. enhances Akt activation. Corresponding densitometric changes in phosphorylated/total signals were derived from the mean of 3 independent experiments are shown immediately below.
Grb14 over-expression enhances cell proliferation and promotes cell invasiveness in vitro and in vivo
In line with the impact of Grb14 reduction on cell polarity, over-expression of the adaptor resulted in altered cell architecture (Fig. 5A). Consistent with this phenotypic change, cell proliferation was significantly augmented as determined by direct counting (Fig. 5B). To examine the impact on tumor progression, we used an orthotopic mouse model that provides an opportunity to examine thyroid cancer invasion and metastasis as well as growth (Fig. 5C left). These experiments demonstrated increased tumor invasiveness (Fig. 5C right) and accelerated tumor progression evidenced by significantly increased tumor volumes (Fig, 5D left) and tumor weights (Fig. 5D right). Importantly, complete necropsies revealed a strikingly metastatic phenotype acquired through Grb14 over-expression (Fig. 5E). In control mice, 5/10 animals developed small lung metastases (total of 26 foci) that collectively measured 102880 μm2. Complete autopsies identified no evidence of any other organ involvement. In contrast, 10/10 mice in the WRO/Grb14 group developed multifocal pulmonary metastases (total of 70 metastatic foci) that collectively measured 612057 μm2. In addition, calcified liver metastases were evident in 2/10 animals in the Grb14 over-expressing group. Massive malignant pericardial involvement was also seen in response to forced Grb14 expression (Fig. 5E far right). Additionally, Western blotting of tumor lysates confirmed increased Akt phosphorylation in response to Grb14 over-expression (data not shown).
Figure 5. Grb14 over-expression promotes thyroid cancer cell growth in vitro and in vivo.

A. Forced Grb14 over-expression accentuates thyroid cancer cell architecture, as shown in phase contrast photomicrographs. B. The impact of Grb14 over-expression on cell proliferation is shown in a representative growth curve (left); a summary of two independent proliferation assays depicts the significant difference between control (WRO/control) and Grb14 over-expressing (WRO/Grb14) cells as indicated. Proliferation rate was determined every day as a percentage of the average of controls. The * symbol denotes a highly significant Mann-Whitney test (p<0.000001); ** significant with Mann-Whitney test (p=0.0325). C. An orthotopic mouse xenograft model demonstrates the rapid progression of disease in mice inoculated with Grb14 over-expressing cells compared to their controls. Tumors over-expressing Grb14 (T) infiltrate into surrounding structures including salivary gland (S) and skeletal muscle (M) (hematoxylin-eosin stain). D. The growth curve displays the significantly faster growing tumors in Grb14 over-expressing clones compared to empty vector controls. The differences between tumor xenografted tissues shows larger volumes and greater weights in the Grb14 over-expressing group compared to empty vector clones. The * symbol denotes highly significant differences (p<0.001 at days 5, 9, 11 and tumor volume at sacrificing); ** denotes significance (p<0.01 at days 7, 13, 15, and tumor weights at sacrificing) with Mann-Whitney test. E. At autopsy, the lungs of animals bearing control cells exhibited small foci of metastatic carcinoma (left, arrow) whereas those with tumors over-expressing Grb14 (middle) had huge metastatic deposits with tumor thrombi in vessels (V) and metastatic malignant pericardial disease surrounding the entire heart (right, arrows), a feature that was not seen in any other animals. (Hematoxylin-eosin staining).
Grb14 expression correlates with human thyroid cancer invasiveness
To explore the relevance of our findings to clinical thyroid cancer behavior, we examined expression of this adaptor in primary tissue microarrays. Primary human thyroid samples of well-differentiated thyroid cancer (n=203), their respective normal tissues, and lymph node metastases (n=35) were used for TMA construction and immunohistochemical expression of Grb14 (Fig. 6A,B). Staining intensity and distribution scores were derived from software image analyses as previously described (Cheng et al., 2009,Cheng et al., 2011). This approach identified a positive staining score correlating Grb14 positivity and the presence of lymph node metastases (Fig. 6C). The relationship between Grb14 and behavior was evident in both classic papillary thyroid carcinoma (PTC; n=162) and in follicular variant-papillary thyroid carcinoma (FV-PTC; n=41). Additionally, Grb14 expression correlated positively with the thyroid cancer biomarker HBME-1 (r=0.192; p=0.042).
Figure 6. Tissue microarray analysis demonstrates a correlation between Grb14 expression and human thyroid cancer invasiveness.
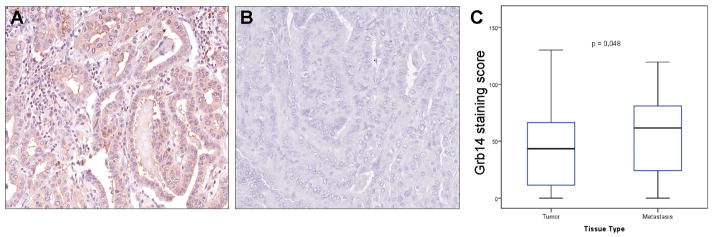
A tissue microarray (TMA) analysis of human thyroid carcinomas included 162 papillary thyroid carcinomas (PTC) and 41 follicular variant FV-PTC were stained for Grb14. A. Two representative papillary carcinomas exhibit markedly different staining for Grb14; the tumor on the left exhibited extra-thyroidal extension and multiple lymph node metastases, whereas the tumor on the right (B) was localized to thyroid. C. Image analysis confirmed a significant correlation between expression of the Grb14 adaptor and the presence of lymph node metastases (LNM). Comparisons were performed using the Wilcoxon rank sum test. The X-values represent the mean Grb14 staining intensity scores.
Discussion
Grb14 is an adaptor protein recognized for its ability to interact with tyrosine kinase receptors including the insulin receptor (Bereziat et al., 2002,Nouaille et al., 2006), the fibroblast growth factor receptor 1 (FGFR1) (Reilly et al., 2000), and the platelet-derived growth factor (PDGF) receptor (Daly et al., 1996). Nevertheless, most functional studies have focused on the ability of Grb14 to interrupt insulin receptor signaling through binding to its activation loop. This interaction protects the IR from de-phosphorylation but also impairs peptide substrate recruitment to diminish insulin-mediated signaling (Han et al., 2001). Consistent with this model, Grb14-deficient mice are noted for their ability to evade insulin resistance (Cooney et al., 2004). Considerably less, however, is known about the role of Grb14 in neoplastic conditions. Here we show for the first time the ability of Grb14 to modulate the RET pathway to significantly control thyroid cancer behavior.
Grb 7 and 10, the two other members of the Grb7/10/14 family associate with RET through their SH2 domains (Pandey et al., 1995,Pandey et al., 1996). Of note, transcriptomic analyses of NIH-3T3 cells harboring activating RET mutations revealed up-regulation of Grb14 expression (Engelmann et al., 2009). These findings and the recognized structural predictions for Grb14 interactions prompted us to investigate the role of this adaptor using thyroid cancer as a model. The key importance of RET in thyroid cancer pathogenesis (de Groot et al., 2006) supported our focus on this disease as a model of Grb14 action in cancer behavior. Indeed, our examination of primary human thyroid carcinoma tissue microarrays identified a positive correlation between the expression of Grb14 and tumor metastasis into the lymphatics. Clearly, additional studies will now be required to determine the extent to which Grb14 expression promotes the behavior of different types of thyroid cancers that rely on distinct signaling pathways.
To investigate the role of Grb14 levels on thyroid tumor behavior, we stably introduced Grb14 knock-down or over-expression to study cell proliferation and invasion in vitro and in vivo. As our first observations, we detected changes in cell polarity and architecture in response to Grb14 reduction. This was associated with diminished cell proliferation and impaired invasion into matrigel chambers. When introduced in mouse xenografts, thyroid cancer cells with reduced Grb14 grew significantly more slowly and displayed attenuated invasive properties. Conversely, Grb14 over-expression resulted in strikingly enhanced oncogenic behavior evidenced by multiple measures. In particular, Grb14 gain resulted in an invasive phenotype which included not only accelerated tumor growth but marked metastatic features. Reminiscent of the human form of the disease, Grb14 excess promoted metastasis into the lungs and surrounding structures. Grb7 and 10 have been reported to similarly facilitate tumor formation in ovarian and breast cancer models (Wang et al., 2010,Bai and Luoh, 2008,Sullivan et al., 2008). Moreover, over-expression of Grb14 has been described in clear cell ovarian cancer (Marchini et al., 2008) and RET-mutation associated thyroid tumor xenografts (Engelmann et al., 2009), but the molecular mechanism remains unclear. Further, the metastatic phenotype acquired through Grb14 shown here has not, to our knowledge, been previously described.
The molecular basis underlying the impressive phenotypic features associated with Grb14 remains poorly understood. We first examined the phosphorylation state of the insulin receptor in the context of Grb14 manipulations. Consistent with the high affinity of Grb14 binding through its unique BPS domain to the activation loop of the insulin receptor (Nouaille et al., 2006), we showed that Grb14 knockdown predictably facilitates signaling through the IR. In contrast and consistent with the key role of RET in thyroid cancer (Kondo et al., 2006), Grb14 knockdown diminished RET phosphorylation at Y905 (corresponding to Y294 in RET/PTC1). This finding is of functional relevance for several reasons. Firstly, RET is a recognized docking site for Grb7 and Grb10 (Pandey et al., 1996,Pandey et al., 1995). Indeed, mutation of this interaction site impairs RET/PTC1-mediated tumor formation in transgenic mice carrying the rearrangement (Buckwalter et al., 2002). These RET mutant mice also show diminished Akt phosphorylation (Buckwalter et al., 2002), a prominent feature we also identified in response to Grb14 manipulation on pAkt in the high-expressing WRO cells. Grb14 also attenuated pAkt responses in TPC-1 cells which harbor this RET rearrangement. Akt is a key survival node in cell cycle progression and in RET-mediated transformation in thyroid cancer cells (Saji and Ringel, 2010). Conversely, Grb14 over-expression supported activation of RET, an effect associated with augmented Akt activation. While the involvement of other RTKs cannot be excluded, our data are consistent with Grb14 as one key regulator of Akt phosphorylation and function that is linked with thyroid cancer behavior.
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor which is classically activated through tyrosyl phosphorylation (Yu et al., 2009). It is also a known target of RET signaling (de Groot et al., 2006). Activation of STAT3 by RET/PTC1 results in enhanced cell proliferation and transformation (Hwang et al., 2003). Thus, we have examined the STAT3 phosphorylation as a down-stream measure of Grb14-controlled RET signaling. Indeed, Grb14 knockdown impaired STAT3 activation in concert with diminished RET phosphorylation. Consistent with this finding Grb14 over-expression resulted in strong STAT3 activation. Taken together, our findings implicate Grb14 in modulating multiple tyrosine kinases. This leads to a signaling repertoire that impedes or augments an oncogenic phenotype. Importantly, the restricted pattern of expression and the strong phenotypic impact of Grb14 predict an important role for this adaptor in modulating cancer progression.
In summary, we have identified that the growth receptor-bound protein 14 is an important regulator of thyroid cancer behavior. We have shown that loss of Grb14 slows down the proliferation of thyroid tumor cells and interrupts their invasive properties. In contrast, over-expression of this adaptor protein significantly enhances thyroid cancer growth, invasiveness, and metastatic behavior. We also show, for the first time, the ability of Grb14 to alter RET signaling and that forced under or over-expression of Grb14 modulates cell proliferation and invasion in an Akt/STAT3-coordinated manner. The contrasting impact of Grb14 on receptor signaling suggests an important role for this adaptor in modulating the balance over multi-RTK-mediated responses. Given the increasingly recognized role of docking proteins in modulating the response to pharmacologic RTK inhibition (Saji and Ringel, 2010), it remains to be seen if this feature is shared with members of the Grb7/10/14 adaptor family.
Supplementary Material
Grb14 knockdown in WRO thyroid cancer cells. Independent WRO cells stably down-regulated through Grb14 shRNA were serum-deprived overnight followed by 10% FBS stimulation as indicated. Western blotting shows diminished RET phosphorylation and Akt responses.
Grb14 knockdown in TPC-1 thyroid cancer cells. Stably down-regulated Grb14 TPC-1 or their scrambled control cells were serum-deprived overnight followed by stimulation as indicated. Western blotting demonstrates increased IR phosphorylation but diminishes RET and Akt activation.
A. Grb14 over-expression in WRO thyroid cancer cells. Stable over-expression is demonstrated by western blotting with corresponding densitometric values shown immediately below. Human breast MDA-231 cells transfected with empty vector or Grb14-expressing vector are shown to the right as negative and positive controls respectively. B. Grb14 over-expression in WRO cells alters MAPK and Akt responses. Independent stably over-expressing WRO cells show enhanced MAPK and Akt phosphorylation compared with empty vector controls.
Acknowledgments
The authors thank Kelvin So for technical assistance. This work was supported by the Canadian Institutes of Health Research (CIHR)(Grant MOP-86493), the Princess Margaret Hospital Foundation, and the Ontario Ministry of Health and Long Term Care (OMOHLTC). The views expressed do not necessarily reflect those of the OMOHLTC.
Footnotes
Disclosure statement: The authors have nothing to disclose.
Conflict of Interest
The authors declare no conflicts of interest.
References
- Asa SL. How familial cancer genes and environmentally induced oncogenes have changed the endocrine landscape. Mod Pathol. 2001;(14):246–253. doi: 10.1038/modpathol.3880293. [DOI] [PubMed] [Google Scholar]
- Avantaggiato V, Dathan NA, Grieco M, Fabien N, Lazzaro D, Fusco A, Simeone A, Santoro M. Developmental expression of the RET protooncogene. Cell Growth Differ. 1994;(5):305–311. [PubMed] [Google Scholar]
- Bai T, Luoh SW. GRB-7 facilitates HER-2/Neu-mediated signal transduction and tumor formation. Carcinogenesis. 2008;(29):473–479. doi: 10.1093/carcin/bgm221. [DOI] [PubMed] [Google Scholar]
- Bereziat V, Kasus-Jacobi A, Perdereau D, Cariou B, Girard J, Burnol AF. Inhibition of insulin receptor catalytic activity by the molecular adapter Grb14. J Biol Chem. 2002;(277):4845–4852. doi: 10.1074/jbc.M106574200. [DOI] [PubMed] [Google Scholar]
- Briese J, Ezzat S, Liu W, Winer D, Wagener C, Bamberger AM, Asa SL. Osteopontin expression in thyroid carcinoma. Anticancer Res. 2010;(30):111–122. [PubMed] [Google Scholar]
- Buckwalter TL, Venkateswaran A, Lavender M, La Perle KM, Cho JY, Robinson ML, Jhiang SM. The roles of phosphotyrosines-294, -404, and -451 in RET/PTC1-induced thyroid tumor formation. Oncogene. 2002;(21):8166–8172. doi: 10.1038/sj.onc.1205938. [DOI] [PubMed] [Google Scholar]
- Cheng S, Liu W, Mercado M, Ezzat S, Asa SL. Expression of the melanoma-associated antigen is associated with progression of human thyroid cancer. Endocr Relat Cancer. 2009;(16):455–466. doi: 10.1677/ERC-09-0002. [DOI] [PubMed] [Google Scholar]
- Cheng S, Serra S, Mercado M, Ezzat S, Asa SL. A high-throughput proteomic approach provides distinct signatures for thyroid cancer behavior. Clin Cancer Res. 2011;(17):2385–2394. doi: 10.1158/1078-0432.CCR-10-2837. [DOI] [PubMed] [Google Scholar]
- Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE, Daly RJ. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J. 2004;(23):582–593. doi: 10.1038/sj.emboj.7600082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly RJ, Sanderson GM, Janes PW, Sutherland RL. Cloning and characterization of GRB14, a novel member of the GRB7 gene family. J Biol Chem. 1996;(271):12502–12510. doi: 10.1074/jbc.271.21.12502. [DOI] [PubMed] [Google Scholar]
- de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;(27):535–560. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, Sariola H, Pachnis V. GDNF signalling through the RET receptor tyrosine kinase. Nature. 1996;(381):789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- Engelmann D, Koczan D, Ricken P, Rimpler U, Pahnke J, Li Z, Putzer BM. Transcriptome analysis in mouse tumors induced by Ret-MEN2/FMTC mutations reveals subtype-specific role in survival and interference with immune surveillance. Endocr Relat Cancer. 2009;(16):211–224. doi: 10.1677/ERC-08-0158. [DOI] [PubMed] [Google Scholar]
- Ezzat S, Zheng L, Zhu XF, Wu GE, Asa SL. Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest. 2002;(109):69–78. doi: 10.1172/JCI14036. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ezzat S, Mader R, Yu S, Ning T, Poussier P, Asa SL. Ikaros integrates endocrine and immune system development. J Clin Invest. 2005;(115):1021–1029. doi: 10.1172/JCI22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DC, Shen TL, Guan JL. The Grb7 family proteins: structure, interactions with other signaling molecules and potential cellular functions. Oncogene. 2001;(20):6315–6321. doi: 10.1038/sj.onc.1204775. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Kim DW, Suh JM, Kim H, Song JH, Hwang ES, Park KC, Chung HK, Kim JM, Lee TH, Yu DY, Shong M. Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: roles in specific gene regulation and cellular transformation. Mol Endocrinol. 2003;(17):1155–1166. doi: 10.1210/me.2002-0401. [DOI] [PubMed] [Google Scholar]
- Kasus-Jacobi A, Perdereau D, Auzan C, Clauser E, Van Obberghen E, Mauvais-Jarvis F, Girard J, Burnol AF. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J Biol Chem. 1998;(273):26026–26035. doi: 10.1074/jbc.273.40.26026. [DOI] [PubMed] [Google Scholar]
- Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;(6):292–306. doi: 10.1038/nrc1836. [DOI] [PubMed] [Google Scholar]
- Liu W, Wei W, Winer D, Bamberger AM, Bamberger C, Wagener C, Ezzat S, Asa SL. CEACAM1 impedes thyroid cancer growth but promotes invasiveness: a putative mechanism for early metastases. Oncogene. 2007;(26):2747–2758. doi: 10.1038/sj.onc.1210077. [DOI] [PubMed] [Google Scholar]
- Marchini S, Mariani P, Chiorino G, Marrazzo E, Bonomi R, Fruscio R, Clivio L, Garbi A, Torri V, Cinquini M, Dell’Anna T, Apolone G, Broggini M, D’Incalci M. Analysis of gene expression in early-stage ovarian cancer. Clin Cancer Res. 2008;(14):7850–7860. doi: 10.1158/1078-0432.CCR-08-0523. [DOI] [PubMed] [Google Scholar]
- Margolis B. The GRB family of SH2 domain proteins. Prog Biophys Mol Biol. 1994;(62):223–244. doi: 10.1016/0079-6107(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Nouaille S, Blanquart C, Zilberfarb V, Boute N, Perdereau D, Roix J, Burnol AF, Issad T. Interaction with Grb14 results in site-specific regulation of tyrosine phosphorylation of the insulin receptor. EMBO Rep. 2006;(7):512–518. doi: 10.1038/sj.embor.7400668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Duan H, Di Fiore PP, Dixit VM. The Ret receptor protein tyrosine kinase associates with the SH2-containing adapter protein Grb10. J Biol Chem. 1995;(270):21461–21463. doi: 10.1074/jbc.270.37.21461. [DOI] [PubMed] [Google Scholar]
- Pandey A, Liu X, Dixon JE, Di Fiore PP, Dixit VM. Direct association between the Ret receptor tyrosine kinase and the Src homology 2-containing adapter protein Grb7. J Biol Chem. 1996;(271):10607–10610. doi: 10.1074/jbc.271.18.10607. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Mickey G, Maher PA. Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. Characterization of a new receptor binding partner. J Biol Chem. 2000;(275):7771–7778. doi: 10.1074/jbc.275.11.7771. [DOI] [PubMed] [Google Scholar]
- Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol. 2010;(321):20–28. doi: 10.1016/j.mce.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan I, Chopra A, Carr J, Kim TS, Cohen EP. Immunity to growth factor receptor-bound protein 10, a signal transduction molecule, inhibits the growth of breast cancer in mice. Cancer Res. 2008;(68):2463–2470. doi: 10.1158/0008-5472.CAN-07-5685. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chan DW, Liu VW, Chiu P, Ngan HY. Differential functions of growth factor receptor-bound protein 7 (GRB7) and its variant GRB7v in ovarian carcinogenesis. Clin Cancer Res. 2010;(16):2529–2539. doi: 10.1158/1078-0432.CCR-10-0018. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;(9):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Grb14 knockdown in WRO thyroid cancer cells. Independent WRO cells stably down-regulated through Grb14 shRNA were serum-deprived overnight followed by 10% FBS stimulation as indicated. Western blotting shows diminished RET phosphorylation and Akt responses.
Grb14 knockdown in TPC-1 thyroid cancer cells. Stably down-regulated Grb14 TPC-1 or their scrambled control cells were serum-deprived overnight followed by stimulation as indicated. Western blotting demonstrates increased IR phosphorylation but diminishes RET and Akt activation.
A. Grb14 over-expression in WRO thyroid cancer cells. Stable over-expression is demonstrated by western blotting with corresponding densitometric values shown immediately below. Human breast MDA-231 cells transfected with empty vector or Grb14-expressing vector are shown to the right as negative and positive controls respectively. B. Grb14 over-expression in WRO cells alters MAPK and Akt responses. Independent stably over-expressing WRO cells show enhanced MAPK and Akt phosphorylation compared with empty vector controls.