

Abstract
Cathepsin G is a major secreted serine peptidase of neutrophils and mast cells. Studies in Ctsg-null mice suggest that cathepsin G supports antimicrobial defenses but can injure host tissues. The human enzyme has unusual “Janus-faced” ability to cleave peptides at basic (tryptic) as well as aromatic (chymotryptic) sites. Tryptic activity has been attributed to acidic Glu226 in the primary specificity pocket and underlies proposed important functions such as activation of pro-urokinase. However, most mammals, including mice, substitute Ala for Glu226, suggesting that human tryptic activity may be anomalous. To test this hypothesis, human cathepsin G was compared with mouse wild type and humanized active site mutants, revealing that mouse primary specificity is markedly narrower than that of human cathepsin G, with much greater Tyr activity and selectivity and near absence of tryptic activity. It also differs from human in resisting tryptic peptidase inhibitors (e.g., aprotinin), while favoring angiotensin destruction at Tyr4 over activation at Phe8. Ala226Glu mutants of mouse cathepsin G acquire tryptic activity and human ability to activate pro-urokinase. Phylogenetic analysis reveals that the Ala226Glu missense mutation appearing in primates 31–43 million years ago represented an apparently unprecedented way to create tryptic activity in a serine peptidase. We propose that tryptic activity is not an attribute of ancestral mammalian cathepsin G, which was primarily chymotryptic, and that primate-selective broadening of specificity opposed the general trend of increased specialization by immune peptidases and allowed acquisition of new functions.
Introduction
Cathepsin G is a non-classical endopeptidase of granulated immune cells. It is highly expressed in mast cells and neutrophils and to lesser extents in monocytes and dendritic cells (1, 2). In human mast cells, cathepsin G is abundant in secretory granules that contain histamine, chymase and tryptases (3, 4). In neutrophils, it resides with elastase in azurophil granules where it is released into phagolysosomes and helps to kill microbes (5, 6). Cathepsin G and elastase also are released extracellularly, where they can resist anti-peptidases, neutralize toxins, and kill bacteria (7, 8). High concentrations of uninhibited peptidase are present where neutrophils accumulate in large numbers, as in Pseudomonas-infected airway in humans with cystic fibrosis (9–11).
Cathepsin G belongs to a family of immune serine peptidases, with closest relatives in humans being granzyme B and mast cell chymase. Genes for these enzymes arose by duplication and divergence of an ancestral chymase-like gene and are clustered on chromosome 14q11.2 (12, 13). Cathepsin G- and granzyme B-like genes separated early in evolution of placental mammals (13). Although phylogenetically related, these peptidases differ radically in substrate specificity. Human granzyme B has caspase-like affinity for substrate Asp residues (14) and chymase is chymotryptic, hydrolyzing after Phe, Tyr, Trp, or Leu (15). Cathepsin G is less selective, with unusual combined ability to hydrolyze chymotryptic and tryptic (especially Lys-containing) substrates (16–18). Tryptic activity is thought to be responsible for activating proteinase-activated receptors (19), C3 (17) and pro-urokinase plasminogen activator (20). Despite its tryptic activity, cathepsin G has an active site that differs from known tryptic serine peptidases, most of which, like trypsin and tryptase, have a highly conserved “specificity triad” configuration of Asp189, Gly216 and Gly226 (chymotrypsinogen numbering). Crystal-derived structures of human cathepsin G (21) suggest that accommodation of basic side chains in tryptic substrates and inhibitors is due to Glu226 at the base of the primary specificity pocket. The side chain carboxylate of Glu226, which is nearly unique at this site among known peptidases, serves the charge-charge-coupling function of Asp189 in classic tryptic peptidases. In similar fashion, with flip-flopping of charges, Arg226 determines specificity for acidic side chains (asp-ase activity) in granzyme B (14). Although human cathepsin G activity is exceptionally broad, it is also comparatively weak, with specificity constants (kcat/Km) towards its best substrates being much lower than those of chymase (16, 22). Some functions may be independent of proteolytic activity; indeed, cathepsin G-derived peptides have anti-bacterial properties (23). Nonetheless, peptidase-dependent properties include secretagogue, angiotensin II-generating, metallopeptidase-activating and hepatocyte growth factor-inactivating functions (24–28).
Studies in Ctsg −/− mice suggest that cathepsin G is important for surviving fungal and bacterial infections, as from intravenous Aspergillus fumigatus (29) and Staphylococcus aureus (5). Immune deficits are even more pronounced when cathepsin G deficiency is combined with deficiency of neutrophil elastase and may be due to ineffective microbial killing by neutrophils (5). Cathepsin G-deficient mouse neutrophils also respond defectively to activation by immune complexes (30). On the other hand, compared to wild type mice, Ctsg −/− mice have lower bacterial counts in a model of bacterial bronchitis produced by tracheal inoculation with Pseudomonas aeruginosa-embedded beads (31), suggesting that cathepsin G interferes with airway defenses (although without a demonstrated effect on inflammation or mortality). In a non-infectious model of renal ischemia-reperfusion injury, Ctsg −/− mice are protected from death and have less inflammation and tissue damage (32). Thus, mouse models suggest that cathepsin G contributes to antimicrobial defenses and to survival after infection, but can harm the host and actually increase mortality. Recent data reveal that dual chymase-cathepsin G inhibitors reduce pathology associated with allergic and neutrophilic inflammation (33, 34). Insights concerning contributions of human cathepsin G to host defense derive mostly from in vitro data. These studies suggest that the enzyme can contribute to host defense by cleaving bacterial virulence factors (35) or detract from host defense by inactivating antimicrobial collectins (36) and neutrophil chemokine receptors (11). Humans with Papillon-Lefevre syndrome, which is characterized by hyperkeratosis and destructive periodontitis, are deficient in active cathepsin G and other immune peptidases (37, 38) due to defects in dipeptidyl peptidase I, which is the major activator of cathepsin G and related peptidases from pro-enzyme forms (39, 40).
The present study was prompted by structural comparisons of mouse and human cathepsin G suggesting that the mouse enzyme differs from the “dual-specificity” tryptic-chymotryptic human enzyme in key specificity-determining residues, instead resembling chymotryptic mast cell chymases. These predictions regarding functions of the heretofore uncharacterized mouse enzyme were tested by comparing substrate preferences of recombinant wild type and humanized mutant forms of mouse cathepsin G with those of human cathepsin G, and by tracking evolution of primary specificity-determining amino acids in mammals from inferred ancestral forms. The findings suggest that mouse and ancestral mammalian cathepsin G, unlike the human enzyme, are purely chymotryptic, and that a small number of active-site mutations in primate ancestors of human cathepsin G dramatically altered activity, inhibitor sensitivity and substrate specificity—causing, most notably, acquisition of trypsin-like ability to hydrolyze substrates after lysine residues and new targeting functions.
Materials and Methods
Phylogenetic analysis
Full amino acid sequences of cathepsin G not already published or annotated were obtained by data mining, including Basic Local Alignment Search Tool searches of high-throughput genome sequence and whole genome shotgun databases at the National Center for Biotechnology Information using human and mouse cathepsin G genes and cDNAs as query sequences. Marmoset sequence (Callithrix jacchus, Contig 30.531) was obtained from a search of genomic sequence archived at the Washington University Genome Sequencing Center (http://genome.wustl.edu). Previously unreported amino acid sequences of cathepsin G were predicted from genomic DNA using existing cathepsin G gene structures as a template following standard rules for placement of intron-exon boundaries. Cathepsin G amino acid sequences were aligned using Geneious software (Biomatters). A likely evolutionary path was constructed from mutations in specificity triad codons using established branching dates for primate ancestors and presuming that mutations accumulated in stepwise fashion. Specificity triad codons in ancestral mammalian cathepsin G were predicted by identifying the most parsimonious path from putative ancestral sequence to known extant mammalian cathepsin G sequences.
Production of wild type mouse cathepsin G
Mouse cathepsin G catalytic domain cDNA was PCR-amplified from Integrated Molecular Analysis of Genomes and their Expression Consortium (http://image.hudsonalpha.org) clone BC125513 with addition of overhang sequence to permit ligation-independent cloning into pIEx-3 (Novagen). The original expressed construct containing an N-terminal GST Tag was insoluble. Therefore, the GST sequence was removed from the plasmid by restriction digestion and the signal sequence was ligated in-frame with the downstream enterokinase cleavage site using a linker. The resulting expression plasmid, after confirmation of sequence, was transfected into adherent Sf-9 cells with Insect GeneJuice transfection reagent (Novagen). Serum-free conditioned medium supernatant was collected 72 h after transfection. Secreted recombinant pro-cathepsin G was purified from supernatant by NaCl gradient elution from a 75 × 750 mm Heparin-5 PW column (Toso-Haas) equilibrated in 20 mM Tris-HCl (pH 7.4) containing 50 mM NaCl and 2 mM CaCl2, with pro-cathepsin G eluting at ~450 mM NaCl. Peak fractions were desalted and concentrated to ~0.05 mL with 5k NMWL centrifugal filters (Biomax) and incubated for 4 h at 25 °C with 0.16 U of enterokinase (Novagen), which was then removed on a Mono S 5/5 HR cation exchange column eluted with a gradient of NaCl. Activated cathepsin G eluted at ~900 mM NaCl.
Production of humanized mutants of mouse cathepsin G
Specificity site mutants were generated from the modified pIEx-3 wild type mouse pro-cathepsin G plasmid using QuikChange II site-directed mutagenesis kits (Stratagene). Single-site mutant Ala226Glu was constructed using QuikChange-designed primers (http://www.stratagene.com/sdmdesigner). Mutant plasmid sequence was verified in both directions. A Ser189Ala/Ala226Glu double mutant was then generated using primers targeting the Ser189 codon on the Ala226Glu plasmid. Primers were as follows: Ala226Glu forward CAACAATGGT AACCCTCCAG AGGTATTCAC CAAAATCCAG AG, reverse AAGCAACAAT GGTAACCCTC CAGCTGTATT CACCAAAATC CAGAGCTT; Ser189Ala forward: CCGAGAGAAA GGAAGGCTGC CTTCAGGGGT G, reverse CACCCCTGAA GGCAGCCTTC CTTTCTCTCG G. Both mutant proteins were expressed, purified and activated as for the wild-type recombinant enzyme.
Determination of active peptidase concentration
To determine level of active peptidase, recombinant, purified wild-type mouse cathepsin G was titrated with human α1-proteinase inhibitor (α1-antitrypsin; Calbiochem) in PBS containing 0.01% Triton X-100 and 0.05% DMSO. Residual activity was measured by addition of cathepsin G substrate suc-L-Val-Pro-Phe-4NA (1 mM) and monitoring change in absorbance at 410 nm on a Synergy 2 microplate spectrophotometer (BioTek). Active concentration of α1-proteinase inhibitor was determined by titrating against human leukocyte elastase (Elastin Products), the active site concentration of which was measured by specific activity assay (41). Glu226 mutants of mouse cathepsin G could not be titrated against α1-proteinase inhibitor or other common inhibitors because of non-stoichiometric inhibition. Instead, active enzyme concentration was based on protein content of purified preparations of highest specific activity. Human cathepsin G (MP Biomedicals) active concentration was determined by assay with 1 mM suc-L-Val-Pro-Phe-4NA from specific activity derived from published kinetic values of active site-titrated enzyme (42).
Determination of P1 substrate preferences
To probe the influence of the P1 residue at the site of hydrolysis, human (0.8 μM) and recombinant mouse cathepsin G (0.03 μM) were used to screen a P1-diverse library of tetrapeptide substrates divided into 20 wells, each containing a fixed P1 residue (standard amino acids plus norleucine, excluding Cys), as described in connection with human mast cell chymase (15). Each P1-fixed well contains an equi-molar mixture of 8,000 peptides representing all combinations of residues at P2, P3 and P4, with each peptide containing the fluorogenic leaving group 7-amino-4-carbamoylmethylcoumarin. The total number of peptides in the P1-diverse library is 160,000, each at a concentration of 31 nM. Assays were performed at 37 °C in 150 mM Tris (pH 8.0), 150 mM NaCl, 0.01% Tween 20, and 1% Me2SO. Hydrolysis was initiated by addition of enzyme and monitored fluorometrically with excitation at 380 nm and emission at 450 nm.
Hydrolysis of synthetic substrates, angiotensin, casein and pro-uPA
Chymotryptic and tryptic activity of the enzymes first were compared using peptidic, colorimetric substrates. Chymotryptic activity was assessed using N-terminally blocked peptidyl 4NAs with Phe in the P1 position at the site of hydrolysis. Standard assays monitored increase in absorbance at 410 nM of 1 mM substrate at 37 °C in PBS containing 0.01% Triton X-100 and 0.05% DMSO spectrophotometrically in 96-well plates. Turnover number kcat and Michaelis constant Km were calculated using Prism 5 software (GraphPad) from substrate hydrolysis rates determined over a range of substrate concentrations bracketing Km. For selected substrates, kinetic constants also were determined in high ionic strength conditions (0.45 M Tris-HCl (pH 8.0), 1.8 M NaCl, 10% DMSO). Tryptic activity was assessed using N-carbobenzyloxy-L-Lys-thiobenzylester (0.1 mM) at 37 °C in PBS containing 0.01% Triton X-100 and 0.05% DMSO. Generation of free thiol by substrate hydrolysis was detected by inclusion of 0.1 mM 5, 5′-dithio-bis (2-nitrobenzoate) in the reaction mixture and monitoring change in absorbance at 412 nm. Angiotensin-cleaving activity of wild type mouse and human cathepsin G was compared with that of recombinant human mast cell chymase (produced as described (43)) by incubating enzymes with 19 μM angiotensin I for 40 min at 37 °C in PBS containing 0.01% Triton X-100. Hydrolyzed fragments were detected by reverse phase-HPLC as in this laboratory’s published work (44, 45). General proteinase activity of human chymase and wild type and mutant cathepsin G was assessed by incubation of purified enzymes with bovine casein in PBS for 1 hour at 37 °C, followed by SDS-PAGE and staining with Coomassie Blue. Human pro-urokinase plasminogen activator (pro-uPA; Landing Biotech) activation by cathepsin G was detected via change in absorbance at 410 nM for 2 hours at 37 °C in PBS containing 0.01% Triton X-100, 0.05% DMSO, 10 mg/ml heparin (bovine lung; Sigma) and 1 mM benzoyl-L-Val-Gly-Arg-4NA (Sigma), which is cleaved by active uPA but not by the forms of cathepsin G in this study.
Determination of inhibitor susceptibility
Inhibitory constants (Ki) for wild type (Lys15) aprotinin with mouse cathepsin G mutants and human cathepsin G were determined by assaying hydrolysis of 0.037–1 mM suc-L-Val-Pro-Phe-4NA in 0.45 M Tris-HCl (pH 8.0) containing 1.8 M NaCl and 10% DMSO in the presence of 12.5 and 25 μM aprotinin for mouse mutants and 1 and 2 mM Lys15-aprotinin for human cathepsin G. Observed Km in the presence of each inhibitor was derived using Prism 5 software (GraphPad) by nonlinear regression fit of the velocity versus substrate concentration curve. Ki was calculated from observed Km (Km observed) and Km obtained without inhibitor according to the equation , where I is inhibitor concentration.
Results
Prediction of mammalian cathepsin G protein sequence
Twenty mammalian sequences containing the complete catalytic domain were identified by in silico translation of genomic sequence. No sequences identifiable as cathepsin G were found in DNA from non-mammalian genomes. GenBank accession numbers of source DNA sequence for cathepsin G sequences identified and compared in this work (Fig. 1) are as follows: human NP_001902; chimpanzee (Pan troglodytes) XP_522810; gorilla (Gorilla gorilla gorilla) CABD01205422; rhesus (Macaca mulatta) XP_001114339; galago (Otolemur garnettii) AAQR01659072; mouse (Mus musculus NP_031826; Norway rat (Rattus norvegicus) NP_001099511; kangaroo rat (Dipodomys ordii) ABRO01262720; dolphin (Tursiops truncates) ABRN01151994; cattle (Bos taurus) XM_581980 (1) and XM_587026 (2); pig (Sus scrofa) XP_001926799; little brown bat (Myotis lucifugus) AAPE01464601; cat (Felis catus) ACBE01202053; dog (Canis lupus familiaris) AAEX02019056; giant panda (Ailuropoda melanoleuca) ACTA01131456; tenrec (Echinops telfairi) AAIY01076445; rocky hyrax (Procavia capensis) ABRQ01191013; armadillo (Dasypus novemcinctus) AAGV020373271; and elephant (Loxodonta africana) AAGU03084767.
FIGURE 1. Mammalian cathepsin G specificity triad sequences.

Aligned portions of the catalytic domains (amino acids 185–220 using standard chymotrypsinogen numbering) bracket specificity-determining “triad” residues 189, 216, and 226 (shaded). Ser195 involved in hydrolysis is marked with an asterisk. Hyphens indicate identity with human sequence. Human cathepsin G differs from consensus at two of three (Ala189 and Glu226) specificity triad residues (58). Mouse cathepsin G and most other mammalian proteases match consensus sequence at these residues (59).
Human cathepsin G specificity triad is doubly mutated compared to ancestral cathepsin G
As shown by the mammalian sequences in Fig. 1, two of the three primary specificity-determining (“specificity triad” (46)) residues differ in human cathepsin G relative to non-primate cathepsin Gs. In comparison to mouse cathepsin G specifically, human cathepsin G replaces Ser with Ala at position 189 and Ala with Glu at position 226. The third triad residue (Gly216) is invariant. The mouse triad (Ser189/Gly216/Ala226) is typical in that it matches cathepsin G consensus sequence at all three positions. It also matches the human chymase triad, even though chymase is less closely related in overall sequence than human cathepsin G and belongs to a distinct clade distinct from mammalian cathepsin G (45). As shown in Fig. 2, residue 189 and 226 codons reveal origins of differences between humans and extant mammals in these specificity-determining amino acids. The Ser189Ala change is absent in our closest mammalian relative, the chimpanzee, and thus occurred within the past 7 million years, which is when humans and chimpanzees are thought last to have shared a common ancestor (47). The Ser189Ala conversion resulted from a single change (T→G) in the first base of the Ser189 codon, which is otherwise highly conserved. The T→A change at the same site in marmoset DNA, which yields a conservative Ser189Thr change, appears to have arisen independently after separation of ape and new world monkey lineages. The more dramatic Ala226Glu change occurred earlier in primate evolution than the Ser189Ala change because although the former mutation is observed in humans, chimpanzee and old world monkey (rhesus macaque), it is absent from more distantly related primates (marmoset and galago) and from non-primates. Compared to marmoset and galago, humans have a single change (C→A) in the second base of the Ala226 codon causing missense conversion to Glu. Thus, Ala226Glu conversion arose by point mutation in the primate lineage 31–43 million years ago, between the time humans last shared a common ancestry with old and new world monkeys, respectively (47). Mouse and rat cathepsin G have the same residues in positions 189 and 226 as most mammals, excepting humans, chimpanzees and marmosets, as noted. However, the base in the wobble position for Ala226 has undergone an A→T synonymous change (see Fig. 2). Mice and rats form the only the group of mammals with such a change, which thus appears to have arisen independently. Similarly, domestic dogs and cats have a synonymous, independent change in the Ser189 codon. From the data summarized in Fig. 2, we conclude that ancestral mammalian cathepsin G had the following specificity triad amino acid sequence and codons: Ser189 (TCT)/Gly216 (GGA)/Ala226 (GCA). Thus, the human specificity triad, compared to ancestral sequence, is more extensively modified than in any other identified mammal, with changes in amino acid sequence arising from two nucleotide mutations, each of which was introduced at a different time point in primate evolution.
FIGURE 2. Evolution of specificity-determining Ala189 and Glu226 in human cathepsin G.

Codon sequence is shown below the corresponding residue. Mammalian consensus and predicted ancestral specificity determining Ser189 (TCT) and Ala226 (GCA) sequence differ from human at both amino acids and at two nucleotides. Nucleotides and amino acids differing from predicted ancestral sequence are indicated in boldface and italics. Nucleotide changes in dog, cat, giant panda, cattle 2, rat, and mouse codons are synonymous. The likely evolutionary path (based on stepwise accumulation of missense mutations, as indicated) predicts that Ala189 and Glu226 mutations in humans arose up to 7 million years ago (Mya) and between 31 and 43 Mya, respectively.
Mouse cathepsin G is more active than human cathepsin G towards chymotryptic substrates and lacks tryptic activity
As shown in Fig. 3, the preferences of mouse cathepsin for substrate residues in the P1 position at the site of hydrolysis are markedly restricted compared to the human enzyme. Human cathepsin G has broad chymotryptic activity, cleaving after Trp, Phe, and Tyr, with little preference for Tyr versus Phe. It also has substantial Leu-ase and Met-ase activity, with little or no ability to cleave after P1 acidic residues (granzyme B-like asp-ase activity) and small aliphatic residues (neutrophil elastase-like activity). Its most unique feature, however, is tryptic activity, which is as strong as its Tyr-cleaving chymotryptic activity and is largely Lys-specific. By contrast, the mouse enzyme lacks the tryptic, Leu-ase and Met-ase activity of the human enzyme and even its chymotryptic profile is narrower, showing preference for Tyr over Phe and little inclination to cleave after Trp. Furthermore, the maximal activity of mouse cathepsin G is much higher than that of human cathepsin G. After normalization for differences in enzyme concentration used in the combinatorial assay, the P1 Tyr substrate-hydrolyzing activity is 19-fold greater for mouse versus human cathepsin G, even though maximal activity for both enzymes was achieved with substrates containing Tyr in the P1 position. As revealed in Figs. 3 and 4, wild type human and mouse cathepsin G both cleave a variety of peptidyl-4NA chymotryptic substrates with Phe at the site of cleavage. Suc-L-Val-Pro-Phe-4-NA and suc-L-Phe-Pro-Phe-4NA are the best identified substrates for both enzymes, suggesting that sub-site P3 is similar for both enzymes. As shown in Table 1Km for suc-L-Val-Pro-Phe-4-NA is substantially and significantly lower for mouse than for human cathepsin G at each of the two buffer conditions (P = .0001 and P < .01 in 1.8 M NaCl and PBS, respectively), possibly because the mouse enzyme better accommodates binding of the P1 Phe side chain, as suggested by modeling of the primary specificity pocket (not shown). Neither enzyme shows much activity towards the human chymase-optimized substrate RETF (48), indicating that the mouse enzyme, although it has a chymase-like specificity triad lining its primary (P1) specificity pocket, does not resemble chymase in its extended substrate-binding fold. Although mouse cathepsin G is more active than human cathepsin G towards most of the surveyed chymotryptic substrates, the mouse enzyme is remarkable for reduced or absent tryptic activity, as reflected by minimal to no ability to cleave tetrapeptide substrates with basic amino acids in the P1 position (Fig. 3) and by lack of Lys-thioesterase activity (Fig. 5).
FIGURE 3. Comparison of mouse and human cathepsin G P1 substrate specificity.
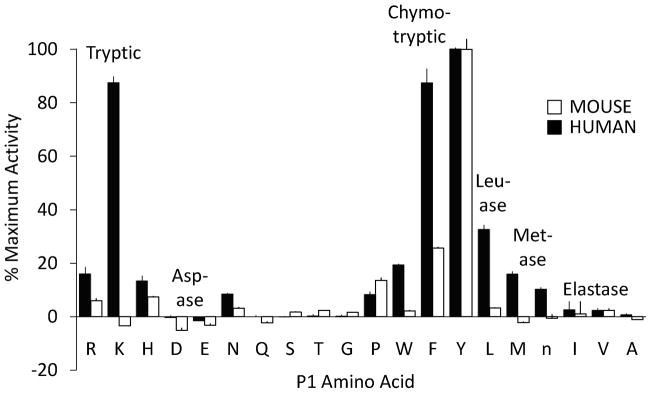
Purified mouse and human enzymes were profiled using a P1-diverse combinatorial library of fluorogenic tetrapeptide substrates containing all combinations of standard amino acids (except Cys) plus norleucine (n) at positions P2-P4, with each library fixed at P. Individual assays assess overall ability to hydrolyze tetrapeptides containing a particular P1 amino acid with an equi-molar mixture of 8,000 peptides varying in 20 residues each in positions P2, P3 and P4 on the N-terminal side of cleavage site P1. To facilitate comparison between human and mouse enzyme, output in fluorescence units is normalized to percentage of maximum output for each enzyme, which was achieved for both enzymes by the P1 Tyr well. The human enzyme has tryptic, chymotryptic, leu-ase and met-ase activity, without asp-ase or elastase-like activity, whereas mouse specificity is much narrower, and mainly chymotryptic. Error bars show standard deviation (N=3).
FIGURE 4. Comparison of chymotryptic activity of mouse vs. human cathepsin G.

The graph compares hydrolytic rates using peptidyl-4NA substrates with P1 Phe, normalized for concentration of active peptidase (nM). White and black bars represent activity of human and mouse cathepsin G, respectively. All substrates were studied in PBS at 1 mM. Substrates are as follows: suc-L-Val-Pro-Phe-4NA (VPF), suc-L-Ala-Ala-Pro-Phe-4NA (AAPF), suc-L-Ala-Glu-Pro-Phe-4NA (AEPF), suc-L-Phe-Pro-Phe-4NA (FPF), acetyl-L-Arg-Glu-Thr-Phe-4NA (RETF), and benzoyl-L-Phe-4NA (F). Results represent mean ± SD of two independent experiments.
Table I.
Comparison of kinetics of chymotryptic (suc-L-Val-Pro-Phe-4NA-hydrolyzing) activity
| Enzyme | Buffer | kcat (s−1)a | Km (mM)a | kcat/Km |
|---|---|---|---|---|
| Mouse Cathepsin G | 1.8 M NaCl | 41 ± 2 | 0.12 ± 0.02 | 330 |
| PBS | 33 ± 10 | 0.20 ± 0.12 | 170 | |
| A226E Mouse Cathepsin G | 1.8 M NaCl | 22 ± 2 | 3.10 ± 0.35 | 7.1 |
| PBS | 12 ± 2 | 0.63 ± 0.05 | 19 | |
| S189A/A226E Mouse Cathepsin G | 1.8 M NaCl | 16 ± 2 | 1.88 ± 0.29 | 8.5 |
| PBS | 9 ± 0 | 0.37 ± 0.05 | 24 | |
| Human Cathepsin G | 1.8 M NaCl | 100 ± 7 | 1.73 ± 0.19 | 58 |
| PBSb | 14 ± 0 | 0.69 ± 0.12 | 20 | |
| Human chymase | 1.8 M NaCl | 230 ± 35 | 0.32 ± 0.12 | 720 |
| PBSb | 17 ± 0 | 0.29 ± 0.07 | 59 |
Mean ± SD, derived from 3 independent measurements at each of 4 substrate concentrations;
as previously determined (48)
FIGURE 5. Comparison of tryptic (Lys-containing) inhibitor and substrates in wild type human cathepsin G (hCG), wild type mouse cathepsin G (mCG) and humanized mutants Ala226Glu (A226E) and Ala226Glu/Ser189Ala (A226E/S189A) mCG.

The left panel compares strength of inhibition, as reflected by the ratio of Km obtained in presence (Km observed) and absence (Km) of 0.27 μM Lys15-aprotinin, using substrate suc-L-Val-Pro-Phe-4NA. Ki units are given in mM. N.I. = no inhibition. The middle panel compares rates of hydrolysis of synthetic tryptic substrate carbobenzyloxy-L-Lys-thiobenzylester (100 μM) normalized for peptidase concentration. The right panel compares rates of hydrolysis of human pro-uPA, which is activated by cleavage at Lys158, as assessed by hydrolysis of uPA substrate suc-L-Val-Gly-Arg-4NA.
Humanized mutants of mouse cathepsin G acquire tryptic activity
As shown in Fig. 5, Lys-thioesterase activity appears in the humanized mutants of mouse cathepsin G, suggesting particularly that the Ala226Glu mutation contributes to tryptic activity in the human enzyme. Key modifications of the active site in human cathepsin G proposed to result in tryptic activity are modeled in Fig. 6.
FIGURE 6. Specificity pocket mutations leading to acquisition of tryptic activity.

The diagram depicts amino acids of the specificity triad lining the primary specificity pocket of cathepsin G and related serine peptidases, with examples of side chains of substrate P1 residues at the site of hydrolysis. The Ala226Glu mutation converted a chymase-like chymotryptic enzyme into a tryptic enzyme capable of hydrolyzing peptides after Lys, as shown, in part due to introduction of the negatively charged Glu side chain, which attracts the positive charge of P1 Lys ε amino group. Configurations of classic chymotryptic and tryptic peptidases plus bovine duodenase, which uses a different configuration to accommodate tryptic and chymotryptic substrates, are shown for comparison. “Tryptase” refers to mast cell β-like tryptases, rather than to human α tryptase, which has an active site blocked by a Gly216Asp mutation (60, 61).
Human but not wild type mouse cathepsin G activates pro-uPA
As shown in Fig. 5, mouse cathepsin G has no detectable pro-uPA-activating activity, which requires tryptic cleavage at Lys158. However, wild type human cathepsin G and the humanized mutants possess this activity, suggesting that the human enzyme’s ability to influence fibrinolysis was acquired with the active site Ala226Glu mutation.
Mouse cathepsin G with humanizing mutations acquires human-like sensitivity to Lys15-aprotinin
As shown in Fig. 5, wild type mouse cathepsin G completely resists inactivation by Lys15-aprotinin, which is a broad-spectrum inhibitor of trypsin-like serine peptidases. However, wild type human cathepsin G and humanized mouse mutants are sensitive, consistent with their tryptic activity, including hydrolysis of the Lys-thioester.
Mouse cathepsin G with humanizing mutations loses sensitivity to α1-proteinase Inhibitor
Wild type mouse cathepsin G is highly sensitive to α1-proteinase inhibitor, which, therefore, was used to titrate active sites in the enzyme preparations. However, the Glu226 mutants—and the wild type human cathepsin G (consistent with prior reports (49))—are resistant (not shown).
Mouse cathepsin G preferentially destroys but human cathepsin G activates angiotensin I
As revealed by chromatograms in Fig. 7a, wild type human chymase and human and mouse cathepsin G exhibit distinct patterns of angiotensin I hydrolysis. Recombinant human chymase, as reported (43), cleaves angiotensin I rapidly and highly selectively at Phe8 to generate vasoactive angiotensin II. Human cathepsin G also hydrolyzes selectively at Phe8, exhibiting very low-level hydrolysis at Tyr4, but is much weaker than human chymase overall in attacking angiotensin I. In contrast, mouse cathepsin G preferentially hydrolyzes at Tyr4, which is an inactivating event in that it prevents angiotensin I from being activated to angiotensin II by hydrolysis at Phe8. Under these conditions, Phe8/Tyr4 selectivity for mouse cathepsin G, human cathepsin G, and human chymase is 0.31, 12 and 460, respectively. Although mouse cathepsin G hydrolyzes angiotensin at Tyr4 3.2 times as often as at Phe8, it is 8.5-fold more active than human cathepsin G in generating angiotensin II, because it is more active overall, as also observed with chymotryptic 4NA substrates (Fig. 4 and Table 1). The mouse enzyme’s preference for cleaving angiotensin at Tyr4 is consistent with its observed preference for tetrapeptide substrates with P1 Tyr versus Phe (Fig. 3).
FIGURE 7. Peptidase and general proteinase activity.

Panel a contains HPLC chromatograms of angiotensin (Ang) I after incubation with human chymase, human cathepsin G, and wild type mouse cathepsin G. Peaks (as detected by monitoring absorbance at 210 nm) correspond to dipeptide HL and active octapeptide product Ang II (both resulting from hydrolysis at Phe8), inactive tetrapeptide DRVY and hexapeptide IHPFHL (resulting from hydrolysis at Tyr4), and uncleaved Ang I, as indicated. Panel b shows results of SDS-PAGE of casein after incubation with peptidases mouse cathepsin G (mCG), Ala226Glu and Ser189Ala/Ala226Glu mutants, and human cathepsin G (hCG). Size in kDa and elution positions of marker proteins are indicated.
Mouse and human cathepsin G are general proteinases
As shown by SDS-PAGE in Fig. 7b, wild type mouse and human cathepsin G, as well as humanized mouse mutants, exhibit general caseinolytic activity. This contrasts with human mast cell chymase, which cleaves casein more selectively, suggesting that the general proteinase activity is not specifically a function of its specificity triad mutations, even though they broaden specificity.
Discussion
This work reveals that cathepsin G substrate specificity underwent major changes during evolution of primates. These changes broadened specificity to include one of human cathepsin G’s most unusual characteristics (namely, tryptic activity) while reducing catalytic efficiency towards chymotryptic substrates. The phylogenetic analysis reveals that these transformations were generated by successive missense mutations in codons for key residues in the primary specificity pocket accommodating the substrate side chain at the site of hydrolysis. Consequently, as reflected by the identity of specificity triad residues 189 and 226, the human cathepsin G active site deviates substantially from that of the last ancestral cathepsin G shared by humans and new world monkeys—and, indeed, its triad differs from that of any known serine peptidase. Mouse cathepsin G, which supports immune function as suggested by phenotypes of mice lacking cathepsin G (5, 29, 50), differs from the human enzyme in two of three triad residues. Instead, it has the predicted ancestral configuration. Thus, substrate preferences of the mouse enzyme, including lack of tryptic activity, more likely represent those of the ancestral form.
The mutagenesis results suggest that acquisition of tryptic activity was mainly due to the Ala226Glu change. Leaving aside the question of whether this change was beneficial, the tryptic anomaly in human cathepsin G is biochemically intriguing because it is a novel solution to the problem of generating tryptic specificity. The crux of that problem is to accommodate positively charged tips of basic amino acid side chains in the generally hydrophobic interior of the globular catalytic domain. In serine peptidases (including immune peptidases) specialized to cleave substrates exclusively at tryptic sites, the specificity triad configuration is universally Asp189/Gly216/Gly226 (see Fig. 6). In classic tryptic peptidases, the anionic Asp189 side chain carboxylate at the base of the primary specificity pocket forms a charge-charge complex with the substrate Lys or Arg side chain. Probably the closest parallel with the human cathepsin G configuration is that of bovine duodenase, which also has dual tryptic and chymotryptic specificity but with specificity triad Asn189/Gly216/Asp226, with Asp226 proposed to serve the function of Asp189 in classic tryptic serine peptidases (51). Duodenase is related to the chymase-cathepsin G-granzyme B family but forms its own clade, is absent in humans and mice, and is prominent in ruminant mammals, which also contain chymase and cathepsin G. The specificity triad of both types of cattle (Bos taurus) cathepsin G is identical to that of mouse and predicted ancestral cathepsin G, and thus is expected to lack tryptic activity. The ancestors of duodenase thus achieved dual specificity independently, but since duodenase and cathepsin G are thought to share a chymotryptic ancestor (13, 46), the starting point was apparently the same. It is tempting to consider that duodenase in cattle plays a role similar to cathepsin G in humans, but not enough is known about roles and sources of these enzymes to inform speculation about functional parity. It may be significant that duodenase and cathepsin G (along with chymases and granzyme B-related peptidases) belong to the subset of immune peptidases that form only three disulfide linkages within the catalytic domain (13)—the fewest such linkages known in the extended trypsin/chymotrypsin family. It is proposed that absence of particular disulfide pair (Cys191-Cys220) in the vicinity of the active site (and otherwise highly conserved in serine peptidases) allows structural plasticity (46, 52) such that changes in residues lining the pocket are better tolerated than in peptidases with the disulfide pair. This may explain how primate cathepsin G and perhaps duodenase were able to acquire tryptic activity with as little as a single amino acid change, even though changing chymotrypsin into a tryptic enzyme requires exchange of many more residues, including loops (53).
Although the mouse Ser189Ala/Ala226Glu double mutant specificity triad matches human, the Ala226Glu single mutant specificity triad matches rhesus macaque (an old world monkey) and ‘the great apes gorilla and chimpanzee. Because tryptic activity of the single mutant lies between that of the mouse wild type and the double mutant, as reflected by Lys15-aprotinin Ki and by rates of Lys-thioester hydrolysis (Fig. 5), tryptic activity of macaque and chimpanzee cathepsin G may be intermediate between that of mouse and human wild-type enzymes. On the other hand, pro-uPA activation, which occurs at Lys158 (20) is less robust in the double than the single mutant. We speculate that the Ser189Ala mutation allows fuller expression of tryptic activity in human cathepsin G for some substrates because the Ala side chain is shorter, therefore partially offsetting loss of space in the primary specificity pocket caused by the Glu for Ala exchange. Ala is also more hydrophobic than Ser, and may increase hydrophobic interactions with the hydrophobic neck (rather than the charged tip) of substrate Lys side chains, thereby improving binding efficiency and Km. Possibly, the Ser189Ala mutation conferred an evolutionary advantage due to more efficient tryptic activity towards some substrates or more effective control by inhibition. Because neither of the humanized mouse cathepsin G mutations generated an enzyme with tryptic activity matching that of the human enzyme, additional changes in the vicinity of the active site affecting Lys substrate binding or turnover likely occurred to increase tryptic activity, perhaps under selective pressure. Potentially, other residues differing between mouse and human cathepsin G make as great a contribution as the Ala/Glu226 difference to acquisition of tryptic specificity, although results of modeling and the duodenase example suggest that this is unlikely.
Broadening of specificity in primate cathepsin G was associated with loss in catalytic efficiency towards chymotryptic substrates, as revealed by the ~19-fold difference between mouse and human cathepsin G in hydrolysis rates of pooled P1 Tyr tetrapeptides in the combinatorial assay, and as shown with various P1 Phe 4NA substrates in Fig. 4. This may be inevitable, because ability to accommodate a variety of substrates likely means that the substrate binding site—and particularly the primary specificity pocket hosting the P1 residue at the site of hydrolysis—is no longer optimized for a particular side chain, resulting in looser binding and higher Km for a given substrate. The importance of interactions involving P4-P2 residues N-terminal to P1, especially for the human enzyme, is illustrated by the observed large differences in rates of hydrolysis of tetra- and tri-peptidyl substrates with invariant P1 Phe. For both enzymes, this is also revealed by very low rates of hydrolysis of benzoyl-L-Phe-4NA, for which there are few contacts outside of the primary specificity pocket. These considerations likely explain why mouse cathepsin G, despite having a primary specificity pocket more similar to human chymase than to human cathepsin G, lacks the selectivity of angiotensin II-generating (Phe8-hydrolyzing) activity that is prominent in both human enzymes, for which surface topography, charge and hydrophilicity outside of the primary specificity pocket are important determinants of angiotensin hydrolysis (44, 54). The finding that mouse cathepsin G preferentially destroys angiotensin I may explain why mast cell protease 4 chymase appears to be the major source of serine peptidase-derived angiotensin II-generating activity in uninflamed mouse tissues (43, 55). On the other hand, our in vitro assays show that mouse cathepsin G generates about one molecule of active angiotensin II for every three molecules of angiotensin I it inactivates. In settings where cathepsin G is released in high concentrations, as in pneumonia, local generation of angiotensin II by cathepsin G may be important.
The comparisons of caseinolytic activity suggest that despite differences in ability to cleave peptidic substrates, all of the tested forms of cathepsin G have general proteinase activity and may be less selective than human chymase, which cleaves casein (and albumin (15)) at fewer sites. It should be noted that the recent acquisition of tryptic activity via the Ala226Glu mutation, with retention of broad specificity, in a sense reverses the proposed general course of evolution of granule-associated immune peptidases of the chymase-cathepsin G-granzyme B family. All of these peptidases, despite their current specificities, are suggested to have evolved originally from tryptic peptidases (with classic triad Asp189/Gly216/Gly226) followed by despecialization (46), resulting in broad but weak activity. Subsequent mutations led to acquisition of specialized activity and higher catalytic efficiency. More generally, these results reveal how small changes in structure in this family of immune peptidases can cause large changes in function, as also occurred in mouse, rat and hamster α-chymase (56, 57), which switched from chymotryptic to elastolytic activity, and in guinea pig α-chymase, which switched from chymotryptic to leu-ase activity (45). Although cathepsin G appears to be microbicidal in mice (6), in the context of chronic airway infection in humans with cystic fibrosis cathepsin G interferes with killing of certain bacteria (11). Potentially, differences in the nature of immune deficits in mice and humans with dipeptidyl peptidase I deficiency/Papillon-Lefevre syndrome are in part due to differences in specificity and immune roles of cathepsin G in the two species. Whether the changes in substrate preferences caused by specificity triad mutations in cathepsin G fundamentally altered host defense functions in humans remains to be determined. Similarly, the importance to homeostasis of novel mutation-enabled capabilities, such as modulation of fibrinolysis by activating pro-uPA, will require further investigation and may be inadequately modeled by mice.
The abbreviations used are
- uPA
urokinase-type plasminogen activator
- 4NA
4-nitroanilide
Footnotes
This work supported in part by National Institute of Health Grant HL024136 to W.W.R and G.H.C. and a Veterans Affairs Career Development Award to N.N.T.
References
- 1.Campbell EJ, Silverman EK, Campbell MA. Elastase and cathepsin G of human monocytes. Quantification of cellular content, release in response to stimuli, and heterogeneity in elastase-mediated proteolytic activity. J Immunol. 1989;143:2961–2968. [PubMed] [Google Scholar]
- 2.Stoeckle C, Sommandas V, Adamopoulou E, Belisle K, Schiekofer S, Melms A, Weber E, Driessen C, Boehm BO, Tolosa E, Burster T. Cathepsin G is differentially expressed in primary human antigen-presenting cells. Cell Immunol. 2009;255:41–45. doi: 10.1016/j.cellimm.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Schechter NM, Irani AM, Sprows JL, Abernethy J, Wintroub B, Schwartz LB. Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J Immunol. 1990;145:2652–2661. [PubMed] [Google Scholar]
- 4.Benyon RC, Enciso JA, Befus AD. Analysis of human skin mast cell proteins by two-dimensional gel electrophoresis: identification of tryptase as a sialylated glycoprotein. J Immunol. 1993;151:2699–2706. [PubMed] [Google Scholar]
- 5.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 6.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 7.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol. 1995;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein W, Doring G. Lysosomal enzymes from polymorphonuclear leukocytes and proteinase inhibitors in patients with cystic fibrosis. Am Rev Respir Dis. 1986;134:49–56. doi: 10.1164/arrd.1986.134.1.49. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Lazarus SC, Caughey GH, Fahy JV. Neutrophil elastase and elastase-rich cystic fibrosis sputum degranulate human eosinophils in vitro. Am J Physiol. 1999;276:L28–34. doi: 10.1152/ajplung.1999.276.1.L28. [DOI] [PubMed] [Google Scholar]
- 11.Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, Krauss-Etschmann S, Koller B, Reinhardt D, Roscher AA, Roos D, Griese M. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- 12.Caughey GH, Schaumberg TH, Zerweck EH, Butterfield JH, Hanson RD, Silverman GA, Ley TJ. The human mast cell chymase gene (CMA1): mapping to the cathepsin G/granzyme gene cluster and lineage-restricted expression. Genomics. 1993;15:614–620. doi: 10.1006/geno.1993.1115. [DOI] [PubMed] [Google Scholar]
- 13.Gallwitz M, Reimer JM, Hellman L. Expansion of the mast cell chymase locus over the past 200 million years of mammalian evolution. Immunogenetics. 2006;58:655–669. doi: 10.1007/s00251-006-0126-1. [DOI] [PubMed] [Google Scholar]
- 14.Waugh SM, Harris JL, Fletterick R, Craik CS. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat Struct Biol. 2000;7:762–765. doi: 10.1038/78992. [DOI] [PubMed] [Google Scholar]
- 15.Raymond WW, Waugh Ruggles S, Craik CS, Caughey GH. Albumin is a substrate of human chymase: prediction by combinatorial peptide screening and development of a selective inhibitor based on the albumin cleavage site. J Biol Chem. 2003;278:34517–34524. doi: 10.1074/jbc.M304087200. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T, Minematsu Y, Reilly CF, Travis J, Powers JC. Human leukocyte cathepsin G. Subsite mapping with 4-nitroanilides, chemical modification, and effect of possible cofactors. Biochemistry. 1985;24:2040–2047. doi: 10.1021/bi00329a036. [DOI] [PubMed] [Google Scholar]
- 17.Maison CM, Villiers CL, Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J Immunol. 1991;147:921–926. [PubMed] [Google Scholar]
- 18.Polanowska J, Krokoszynska I, Czapinska H, Watorek W, Dadlez M, Otlewski J. Specificity of human cathepsin G. Biochim Biophys Acta. 1998;1386:189–198. doi: 10.1016/s0167-4838(98)00085-5. [DOI] [PubMed] [Google Scholar]
- 19.Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- 20.Drag B, Petersen LC. Activation of pro-urokinase by cathepsin G in the presence of glucosaminoglycans. Fibrinolysis. 1994;8:192–199. [Google Scholar]
- 21.Hof P, Mayr I, Huber R, Korzus E, Potempa J, Travis J, Powers JC, Bode W. The 1.8 A crystal structure of human cathepsin G in complex with Suc-Val-Pro- PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996;15:5481–5491. [PMC free article] [PubMed] [Google Scholar]
- 22.Powers JC, Tanaka T, Harper JW, Minematsu Y, Barker L, Lincoln D, Crumley KV, Fraki JE, Schechter NM, Lazarus GG, Nakajima K, Nakashino K, Neurath H, Woodbury RG. Mammalian chymotrypsin-like enzymes. Comparative reactivities of rat mast cell proteases, human and dog skin chymases, and human cathepsin G with peptide 4-nitroanilide substrates and with peptide chloromethyl ketone and sulfonyl fluoride inhibitors. Biochemistry. 1985;24:2048–2058. doi: 10.1021/bi00329a037. [DOI] [PubMed] [Google Scholar]
- 23.Shafer WM, Pohl J, Onunka VC, Bangalore N, Travis J. Human lysosomal cathepsin G and granzyme B share a functionally conserved broad spectrum antibacterial peptide. J Biol Chem. 1991;266:112–116. [PubMed] [Google Scholar]
- 24.Reilly CF, Tewksbury DA, Schechter NB, Travis J. Rapid conversion of angiotensin I to angiotensin II by neutrophil and mast cell proteinases. J Biol Chem. 1982;257:8619–8622. [PubMed] [Google Scholar]
- 25.Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J Clin Invest. 1990;85:682–689. doi: 10.1172/JCI114492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: novel cathepsin G-mediated catalytic activity that is resistant to inhibition. J Immunol. 1998;160:1436–1443. [PubMed] [Google Scholar]
- 27.Shamamian P, Schwartz JD, Pocock BJ, Monea S, Whiting D, Marcus SG, Mignatti P. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- 28.Raymond WW, Cruz AC, Caughey GH. Mast cell and neutrophil peptidases attack an inactivation segment in hepatocyte growth factor to generate NK4-like antagonists. J Biol Chem. 2006;281:1489–1494. doi: 10.1074/jbc.M511154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–210. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 30.Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, Pham CT. Serine protease cathepsin G regulates adhesion-dependent neutrophil effector functions by modulating integrin clustering. Immunity. 2005;22:679–691. doi: 10.1016/j.immuni.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Sedor J, Hogue L, Akers K, Boslaugh S, Schreiber J, Ferkol T. Cathepsin-G interferes with clearance of Pseudomonas aeruginosa from mouse lungs. Pediatr Res. 2007;61:26–31. doi: 10.1203/01.pdr.0000250043.90468.c2. [DOI] [PubMed] [Google Scholar]
- 32.Shimoda N, Fukazawa N, Nonomura K, Fairchild RL. Cathepsin G is required for sustained inflammation and tissue injury after reperfusion of ischemic kidneys. Am J Pathol. 2007;170:930–940. doi: 10.2353/ajpath.2007.060486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tausch L, Henkel A, Siemoneit U, Poeckel D, Kather N, Franke L, Hofmann B, Schneider G, Angioni C, Geisslinger G, Skarke C, Holtmeier W, Beckhaus T, Karas M, Jauch J, Werz O. Identification of human cathepsin G as a functional target of boswellic acids from the anti-inflammatory remedy frankincense. J Immunol. 2009;183:3433–3442. doi: 10.4049/jimmunol.0803574. [DOI] [PubMed] [Google Scholar]
- 34.Maryanoff BE, de Garavilla L, Greco MN, Haertlein BJ, Wells GI, Andrade-Gordon P, Abraham WM. Dual inhibition of cathepsin G and chymase is effective in animal models of pulmonary inflammation. Am J Respir Crit Care Med. 2010;181:247–253. doi: 10.1164/rccm.200904-0627OC. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Boado YS, Espinola M, Bahr S, Belaaouaj A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J Immunol. 2004;172:509–515. doi: 10.4049/jimmunol.172.1.509. [DOI] [PubMed] [Google Scholar]
- 36.Hirche TO, Crouch EC, Espinola M, Brokelman TJ, Mecham RP, DeSilva N, Cooley J, Remold-O’Donnell E, Belaaouaj A. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem. 2004;279:27688–27698. doi: 10.1074/jbc.M402936200. [DOI] [PubMed] [Google Scholar]
- 37.de Haar SF, Jansen DC, Schoenmaker T, De Vree H, Everts V, Beertsen W. Loss-of-function mutations in cathepsin C in two families with Papillon-Lefevre syndrome are associated with deficiency of serine proteinases in PMNs. Hum Mutat. 2004;23:524. doi: 10.1002/humu.9243. [DOI] [PubMed] [Google Scholar]
- 38.Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. 2004;173:7277–7281. doi: 10.4049/jimmunol.173.12.7277. [DOI] [PubMed] [Google Scholar]
- 39.Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. 2001;276:18551–18556. doi: 10.1074/jbc.M100223200. [DOI] [PubMed] [Google Scholar]
- 40.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakajima K, Powers JC, Ashe BM, Zimmerman M. Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the α1-protease inhibitor reactive site. J Biol Chem. 1979;254:4027–4032. [PubMed] [Google Scholar]
- 42.Rehault S, Brillard-Bourdet M, Juliano MA, Juliano L, Gauthier F, Moreau T. New, sensitive fluorogenic substrates for human cathepsin G based on the sequence of serpin-reactive site loops. J Biol Chem. 1999;274:13810–13817. doi: 10.1074/jbc.274.20.13810. [DOI] [PubMed] [Google Scholar]
- 43.Caughey GH, Raymond WW, Wolters PJ. Angiotensin II generation by mast cell α- and β-chymases. Biochim Biophys Acta. 2000;1480:245–257. doi: 10.1016/s0167-4838(00)00076-5. [DOI] [PubMed] [Google Scholar]
- 44.Muilenburg DJ, Raymond WW, Wolters PJ, Caughey GH. Lys40 but not Arg143 influences selectivity of angiotensin conversion by human α-chymase. Biochim Biophys Acta. 2002;1596:346–356. doi: 10.1016/s0167-4838(02)00224-8. [DOI] [PubMed] [Google Scholar]
- 45.Caughey GH, Beauchamp J, Schlatter D, Raymond WW, Trivedi NN, Banner D, Mauser H, Fingerle J. Guinea pig chymase is leucine-specific: a novel example of functional plasticity in the chymase/granzyme family of serine peptidases. J Biol Chem. 2008;283:13943–13951. doi: 10.1074/jbc.M710502200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wouters MA, Liu K, Riek P, Husain A. A despecialization step underlying evolution of a family of serine proteases. Mol Cell. 2003;12:343–354. doi: 10.1016/s1097-2765(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 47.Steiper ME, Young NM. Primate molecular divergence dates. Mol Phylogenet Evol. 2006;41:384–394. doi: 10.1016/j.ympev.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 48.Raymond WW, Su S, Makarova A, Wilson TM, Carter MC, Metcalfe DD, Caughey GH. α2-Macroglobulin capture allows detection of mast cell chymase in serum and creates a reservoir of angiotensin II-generating activity. J Immunol. 2009;182:5770–5777. doi: 10.4049/jimmunol.0900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Travis J, Bowen J, Baugh R. Human α1-antichymotrypsin: interaction with chymotrypsin-like proteinases. Biochemistry. 1978;17:5651–5656. doi: 10.1021/bi00619a011. [DOI] [PubMed] [Google Scholar]
- 50.Raptis SZ, Pham CT. Neutrophil-derived serine proteases in immune complex-mediated diseases. Immunol Res. 2005;32:211–216. doi: 10.1385/IR:32:1-3:211. [DOI] [PubMed] [Google Scholar]
- 51.Pletnev VZ, Zamolodchikova TS, Pangborn WA, Duax WL. Crystal structure of bovine duodenase, a serine protease, with dual trypsin and chymotrypsin-like specificities. Proteins. 2000;41:8–16. [PubMed] [Google Scholar]
- 52.Zamolodchikova TS, Smirnova EV, Andrianov AN, Kashparov IV, Kotsareva OD, Sokolova EA, Ignatov KB, Pemberton AD. Cloning and molecular modeling of duodenase with respect to evolution of substrate specificity within mammalian serine proteases that have lost a conserved active-site disulfide bond. Biochemistry (Mosc) 2005;70:672–684. doi: 10.1007/s10541-005-0168-2. [DOI] [PubMed] [Google Scholar]
- 53.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 54.Sanker S, Chandrasekharan UM, Wilk D, Glynias MJ, Karnik SS, Husain A. Distinct multisite synergistic interactions determine substrate specificities of human chymase and rat chymase-1 for angiotensin II formation and degradation. J Biol Chem. 1997;272:2963–2968. doi: 10.1074/jbc.272.5.2963. [DOI] [PubMed] [Google Scholar]
- 55.Lundequist A, Tchougounova E, Abrink M, Pejler G. Cooperation between mast cell carboxypeptidase A and the chymase mouse mast cell protease 4 in the formation and degradation of angiotensin II. J Biol Chem. 2004;279:32339–32344. doi: 10.1074/jbc.M405576200. [DOI] [PubMed] [Google Scholar]
- 56.Kunori Y, Koizumi M, Masegi T, Kasai H, Kawabata H, Yamazaki Y, Fukamizu A. Rodent α-chymases are elastase-like proteases. Eur J Biochem. 2002;269:5921–5930. doi: 10.1046/j.1432-1033.2002.03316.x. [DOI] [PubMed] [Google Scholar]
- 57.Kervinen J, Abad M, Crysler C, Kolpak M, Mahan AD, Masucci JA, Bayoumy S, Cummings MD, Yao X, Olson M, de Garavilla L, Kuo L, Deckman I, Spurlino J. Structural basis for elastolytic substrate specificity in rodent α-chymases. J Biol Chem. 2008;283:427–436. doi: 10.1074/jbc.M707157200. [DOI] [PubMed] [Google Scholar]
- 58.Salvesen G, Farley D, Shuman J, Przybyla A, Reilly C, Travis J. Molecular cloning of human cathepsin G: structural similarity to mast cell and cytotoxic T lymphocyte proteinases. Biochemistry. 1987;26:2289–2293. doi: 10.1021/bi00382a032. [DOI] [PubMed] [Google Scholar]
- 59.Heusel JW, Scarpati EM, Jenkins NA, Gilbert DJ, Copeland NG, Shapiro SD, Ley TJ. Molecular cloning, chromosomal location, and tissue-specific expression of the murine cathepsin G gene. Blood. 1993;81:1614–1623.1. [PubMed] [Google Scholar]
- 60.Huang C, Li L, Krilis SA, Chanasyk K, Tang Y, Li Z, Hunt JE, Stevens RL. Human tryptases alpha and beta/II are functionally distinct due, in part, to a single amino acid difference in one of the surface loops that forms the substrate-binding cleft. J Biol Chem. 1999;274:19670–19676. doi: 10.1074/jbc.274.28.19670. [DOI] [PubMed] [Google Scholar]
- 61.Marquardt U, Zettl F, Huber R, Bode W, Sommerhoff C. The crystal structure of human α1-tryptase reveals a blocked substrate-binding region. J Mol Biol. 2002;321:491–502. doi: 10.1016/s0022-2836(02)00625-3. [DOI] [PubMed] [Google Scholar]