

Abstract
The internal ribosome entry site (IRES) in the 5′ untranslated region (UTR) of the hepatitis C virus (HCV) genome initiates translation of the viral polyprotein precursor. The unique structure and high sequence conservation of the 5′ UTR render the IRES RNA a potential target for the development of selective viral translation inhibitors. Here, we provide an overview of approaches to block HCV IRES function by nucleic acid, peptide and small molecule ligands. Emphasis will be given to the IRES subdomain IIa which currently is the most advanced target for small molecule inhibitors of HCV translation. The subdomain IIa behaves as an RNA conformational switch. Selective ligands act as translation inhibitors by locking the conformation of the RNA switch. We review synthetic procedures for inhibitors as well as structural and functional studies of the subdomain IIa target and its ligand complexes.
1. INTRODUCTION
Synthesis of the HCV polyprotein depends on an internal ribosome entry site (IRES) in the 5′ untranslated region (UTR) of the viral RNA genome.1, 2 The IRES element is responsible for assembling functional ribosomes at the viral start codon in a mechanism that bypasses canonical translation initiation which depends on the 5′ cap modified terminus of eukaryotic mRNA. The HCV IRES first recruits host cell small (40S) ribosomal subunits as well as eukaryotic initiation factor 3 (eIF3), which is a large multi-protein complex required to prevent premature association of 40S and 60S ribosomal subunits. In a complex process that is coordinated by the IRES RNA large (60S) subunits join to assemble complete (80S) ribosomes at the start codon and translation is initiated through a 5′ cap-independent mechanism that obviates the need for other initiation factors.3–6 The IRES RNA is a potential target for HCV translation inhibitors due to its unique function and high conservation in clinical virus isolates.7–10 While drugs directed at the highly conserved viral IRES promise to benefit from rare occurrence and low fitness of resistance mutants in the treatment-naïve population, developing drug-like selective inhibitors for an RNA target is an exceptionally challenging endeavor.11–14
The HCV IRES element extends from position 40 through 372 of the viral RNA genome, spanning the 5′ UTR and 30 nucleotides beyond the start codon at A342 into the polyprotein coding frame.15 The IRES contains three independently folding domains (II–IV) connected by flexible linkers (Figure 1).16 Domain I within the 5′ UTR is not required for IRES-driven translation but participates in viral replication together with the 3′ UTR.17 Ribosomal recruitment and positioning of the viral mRNA start codon is orchestrated by intermolecular interactions across the central domain III which also provides the binding site for the eIF3 complex.18, 19 The hairpin loop of domain IV, which includes the start codon as well as ~ 13 nucleotides of the coding region, unwinds during 40S binding at the IRES to expose the viral translation initiation site in the ribosomal decoding groove.19–21
Figure 1.
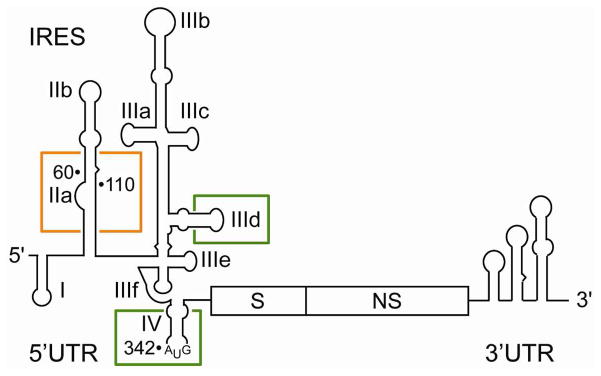
Organization of the HCV RNA genome showing the structured untranslated regions (UTR) including the IRES element in the 5′ UTR. Structural (S) and nonstructural (NS) gene regions are indicated. Boxed subdomains have been extensively studied as targets for small molecule translation inhibitors (IIa) and oligonucleotides as well as peptides (IIId, IV).
In addition to scaffolding the assembly of components in the translational machinery, the IRES RNA participates as an active player in dynamic changes during translation initiation. IRES domains affect the catalytic activity and subunit composition of initiation factors. The IRES domain II promotes both eIF2-catalyzed GTP hydrolysis as well as subsequent eIF2/GDP release from the 48S complex and mediates removal of the eIF3j component before 60S subunit joining.22, 23 The correct orientation of the HCV mRNA at the ribosomal decoding site involves interaction with the IRES subdomain IIb, a completely conserved RNA hairpin loop at the tip of domain II which also contacts ribosomal protein rpS5.23–25 Positioning of the subdomain IIb hairpin in the ribosomal E-site at the subunit interface relies on the folding of the asymmetric internal loop of subdomain IIa which adopts a 90° bent conformation.26–28 The topology of the IRES-ribosome interaction requires subdomain IIb to be removed from the E-site to allow the P-site tRNA to translocate during translation initiation.20, 25 It has been suggested that conformational changes in the subdomain IIa may release the hairpin IIb from the E-site.29, 30 Alteration of the L-shaped architecture of subdomain IIa through mutations or by interference with small molecule ligands that affect the RNA conformation have been shown to disrupt IRES-driven translation and effectively inhibit viral protein synthesis.22, 29–31
Here, we provide a review of approaches to inhibit viral translation by targeting domains of the HCV IRES with oligonucleotides, peptides and small molecules. Emphasis will be given to the subdomain IIa which currently is the most advanced IRES target for small molecule translation inhibitors. The subdomain IIa is an RNA conformational switch capable of forming a deep solvent-excluded binding pocket for small molecule ligands which may serve as leads for the development of IRES-targeting antiviral drugs.
2. STRATEGIES FOR INHIBITION OF THE HCV IRES
HCV translation has been recognized as a potential target for the development of antiviral therapeutics.7–10 The high conservation of the IRES element suggests that inhibitors will potentially benefit from selection of low-fitness resistance mutants which have reduced frequency of occurrence in the treatment-naïve population. IRES ligands that have been considered as potential inhibitors of HCV translation include oligonucleotides, peptides as well as small molecules. The following sections discuss briefly rationale and mechanisms for the various inhibitor classes.
2.1. Oligonucleotide Inhibitors
Soon after the discovery of the IRES as the key driver of HCV translation, antisense oligonucleotides targeting the 5′ UTR were studied as inhibitors of viral gene expression.32–35 In addition to chemically modified oligonucleotides, morpholino, peptide nucleic acid (PNA) and locked nucleic acid (LNA) antisense oligonucleotides were used to suppress IRES function.36–40 Among the IRES hairpin loop motifs, the subdomain IIId and domain IV proved to be the most responsive targets for antisense approaches (Figure 1). A 20 residue phosphorothioate oligodeoxynucleotide, complementary to the region around the start codon in domain IV,41 was evaluated in a Phase I clinical trial. While a related antisense oligonucleotide (ISIS 6547)42, 43 which targeted the same IRES region had an in vitro IC50 value of 0.1–0.2 μM, the ISIS 1480341 drug showed only weak transient activity in human, with plasma HCV RNA reduction of < 2 log(10) at thrice-weekly dosing of 2–3 mg over four weeks.42, 43 The insufficient potency accompanied by side effects eventually led to discontinuation of the development effort.44
In addition to antisense ligands, RNA aptamer inhibitors were developed for all domains (I–IV) of the IRES. Published aptamers of the HCV IRES recognize apical loops in hairpin domains by hybridization with a fully complementary sequence.45–51 Aptamer oligonucleotides for domain I interfere with replication rather than translation, as was expected from the function of this IRES domain. 49, 51 The most potent inhibitors were found among aptamers of the subdomain IIId, with in vitro activities comparable to antisense oligonucleotides.46, 48 In vivo data has not been published for IRES-targeted aptamers.
Inhibition of HCV translation by oligonucleotide-mediated cleavage of the IRES RNA was investigated using ribozymes,52–58 DNAzymes,59, 60 conjugated chemical nucleases61, 62 as well as siRNA and shRNA.54, 63–72 The design principle shared in common by these constructs requires an accessible single stranded sequence in the IRES73 that provide the target for a complementary oligonucleotide ligand which is either associated with or, in the case of siRNA and shRNA, directs a ribonuclease activity. Among ribozymes tested in vitro were hammerhead constructs targeted at the apical loop of subdomain IIIb (HCV-195)52, 53 or the subdomain IIIf (HH363).56–58 Conjugated chemical nucleases consisting of RNA-hydrolyzing imidazole derivatives tethered to guide oligonucleotides were shown to inhibit in vitro IRES-driven translation at sub-μM IC50 values.61 However, testing of the conjugates in HCV-infected cells suggested that the inhibition was due to the antisense effect of the guide oligonucleotide and not through cleavage by the attached imidazole.62
Combination of ribozymes with siRNAs directed at sequences within the IRES resulted in additive inhibition of viral translation.54 Targeting of IRES sequences by siRNA or shRNA has resulted in efficient in vitro knockdown of translation by the RNA interference mechanism.63–72 The inhibition potency of interfering RNAs was essentially related to the accessibility of the target sequences within the IRES structure. For siRNA and shRNA inhibitors as well as other types of oligonucleotide ligands targeting the IRES, efficient cellular delivery remains a confounding challenge.
2.2. Peptide Inhibitors
Only few studies have been reported of targeting the HCV IRES with peptides. A 24 amino acid peptide (LaR2C)74 derived from the RNA recognition motif 2 (RRM2) of the human Lupus autoantigen (La) was shown to moderately inhibit IRES-driven translation by competing with binding of cellular La protein. A similar inhibitory effect was achieved with a peptide (LAP)75, 76 corresponding to the N-terminal 18 amino acids of La.75, 76 La protein is an abundant component in the nucleus where it plays diverse roles in the metabolism of various RNA precursors.77 HCV translation as well as replication requires the La protein which binds to both the IRES domain IV and the 3′ UTR78–80 and which itself has been proposed as a potential therapeutic target.81
Amphiphilic peptides consisting mainly of lysines and leucines were shown to bind with nanomolar affinity to a hairpin RNA derived from domain IV of the HCV IRES albeit with only moderate (up to 2-fold) discrimination against other RNA targets.82 Antiviral activity of the peptides was not tested.
In an attempt to create an artificial metallonuclease that site-selectively cleaves the HCV IRES, a Y(D-R)FK tetrapeptide was linked with a copper-chelating GGH tripeptide.83 Copper(II) cleaves ribose moieties in proximity by an oxidative mechanism which likely involves peroxide generation. While no rationale was provided for the proposed selectivity of the peptide for the IRES subdomain IIb, the artificial metallonuclease did degrade a hairpin loop IIb model oligonucleotide but not two unrelated RNAs. The cleavage mechanism and site selectivity were not further investigated. However, the peptide metallonuclease showed antiviral activity with an IC50 value of 0.58 μM in an HCV replicon assay.
2.3. Sequestration of IRES-Binding Factors
In an approach that does not target the IRES directly but proteins that bind to the HCV 5′UTR, decoy-like nucleic acids have been used to sequester host cell factors that are required for IRES function. A 60 nt RNA (IRNA),76, 84 which originally had been isolated from yeast, was shown to inhibit HCV translation both in extract and cells, likely by sequestration of IRES-binding factors including the La protein. Inspired by these findings, RNA decoys derived from subdomains of the HCV IRES were tested for their ability to inhibit viral translation.85 RNAs corresponding to the full domain III or just the subdomain IIIef inhibited IRES-driven translation in a dose-dependent fashion. The ribosomal protein S5 was identified as one of the host factors interacting with the subdomain IIIef decoy RNA. Even smaller oligodeoxynucleotides containing 9 residues were shown to inhibit IRES-mediated translation by sequestration of two yet to be identified host cell proteins.86
2.4. Small Molecule IRES Inhibitors
Both in vitro translation and direct target binding assays have been used to screen for small molecule inhibitors that affect HCV IRES-driven translation.87–93 High-throughput screening of ~300,000 compounds by electron spray ionization mass spectrometry (ESI/MS) against an oligonucleotide representing the subdomain IIIe hairpin identified six binders with affinities < 50 μM.89 Among the hits were four peptides and two aminoglycosides of undisclosed structure. Biological activity in an IC50 value range of 3–12 μM was reported for the six hits, however, without a description of the biological assay. An affinity assay that identified small molecule ligands based on stabilization of a structured RNA (“SCAN”) had been used to screen ~135,000 compounds against the subdomain IIId hairpin loop.91 The SCAN screen of the IIId hairpin loop identified 12 potential hits including one selective small molecule ligand that bound the target with 0.7 μM affinity. Biological activity and structures of the hits were not disclosed.
Screening attempts for IRES-binding inhibitors have been hampered by complications specifically encountered with RNA targets. Firstly, there is a lack of drug-like molecules for screening that exhibit bias for RNA binding but are not structurally complex natural products. Secondly, challenges arise from the need for assays that identify hits based on functional consequences of ligand binding to an RNA target rather than returning binding affinities only. Assessment of functional inhibition is critical for RNA-binding ligands which typically have binding affinities inferior to potent inhibitors targeting well-defined hydrophobic pockets in proteins. Weak binding affinities observed for RNA-binding antibiotics might be related to their unique mechanisms of action which often do not involve direct competition with a cognate ligand. For example, macrolides and ketolides, which are among the most potent RNA-binding antibiotics, bind to the ribosomal peptidyl transferase active site with low nanomolar affinities (erythromycin: ~14 nM).94 Aminoglycoside antibiotics, including paromomycin and tobramycin, have weak binding affinities ranging around 1μM for their target in the bacterial ribosomal decoding site RNA.95 For oxazolidinones such as linezolid, RNA target dissociation constants in the range of 20–100μM have been measured.96, 97
In vitro translation (IVT) assays test for functional discrimination of compound impact on IRES-driven versus cap-driven translation by using either bicistronic or sets of monocistronic reporters. While IVT assays probe for inhibition of IRES function, ribosomes present in the assay mix at high concentration function both as key functional components as well as a source of complex competitor RNA that may interfere with ligand binding to the target under study. The large number of false positive hits reported in a recent IVT screen of the HCV IRES has been attributed to off-target effects, for example by inhibitors targeting other RNA components of the translation machinery.92 From a cell-based bicistronic reporter screen of ~132,000 compounds, amino-substituted phenazines 1 (Figure 2) were reported as IRES inhibitors, however, which lacked significant selectivity for the IRES target.87 A high throughput IVT screen against a library of ~180,000 compounds identified biaryl guanidines 2, which were moderately active, highly polar translation inhibitors that proved difficult to optimize.88 In what is probably the most elaborate published IVT screening campaign for IRES inhibitors, interrogation of a library of ~430,000 compounds resulted in ~1700 initial hits which in thorough secondary screening turned out to be luciferase or general translation inhibitors.92
Figure 2.

Inhibitors of HCV IRES-driven translation. Phenazines like 1 and biaryl guanidines such as 2 were discovered in IRES-translation inhibition screens.87, 88 Benzimidazoles 3–5, which target the IRES subdomain IIa and inhibit HCV replicon, were initially identified by mass-spectrometric screening at Isis Pharmaceuticals.90 The diaminopiperidine 6 also targets the subdomain IIa but inhibits HCV translation through a mechanism that is distinct from that of the benzimidazole ligands.30 Target binding, translation inhibition and replicon activity of the inhibitors are summarized in Table 1.
In a seminal contribution to the discovery of HCV translation inhibitors, high throughput screening by ESI/MS of a 29-mer oligonucleotide representing the IRES subdomain IIa led to the identification of 3 (Figure 2) which had a target affinity of 100 μM in the mass-spectrometric assay.90 Medicinal chemistry elaboration of the hit compound, which relied on extensive structure-activity relationship data,9, 90, 98 eventually furnished multicyclic derivatives such as 4 and 5 with improved binding to the subdomain IIa fragment at KD values of 0.86 μM and 0.72 μM, respectively. Benzimidazoles 4 and 5 were shown to inhibit HCV replicon at EC50 values of 4–5 μM without cytotoxic effects up to 100 μM in Huh-7 cells. Binding affinity and replicon inhibition were measured using racemic mixtures of 4 and a mixture of cis and trans diastereomers of 5. Subsequent testing of 5 in a replicon translation assay established the benzimidazole derivatives as selective inhibitors of IRES-driven viral translation.29, 99
A second, chemically distinct series of subdomain IIa-binding inhibitors of the HCV replicon was discovered among modular 3,5-diaminopiperidine (DAP) dipeptides such as the lysine derivative 6 (Figure 2).30 The DAP scaffold had been proposed as a simplified structural mimetic of 2-deoxystreptamine (2-DOS) which is a recurring pharmacophore in RNA-binding natural aminoglycoside antibiotics.100
3. THE IRES SUBDOMAIN IIa TARGET
3.1. Structure, Conservation and Function of the IRES Subdomain IIa
The HCV IRES is composed of independently folding domains connected by flexible single-stranded regions. This architecture gives rise to an ensemble of conformers for the isolated full-length RNA.16, 102, 103 A single defined overall fold has been observed for the IRES element in cryo-EM studies of complexes with the 40S subunit and the 80S ribosome.20, 25 Higher resolution three-dimensional structures have been determined by X-ray crystallography and NMR for individual (sub)domains,104 including II,26 IIa,27, 28 IIIabc,105 IIIb,106 IIIc, IIId,107, 108 IIIe107 and, most recently, the central pseudoknot domain IIIe–f.19 Atomic structures of IRES domains in conjunction with cryo-EM and small-angle X-ray scattering data have been used to construct models of the full-length IRES.19, 103
NMR analysis first established that the metal-dependent folding of the subdomain IIa internal loop (Figure 3A) is responsible for the overall bent shape of domain II26 which directs the apical hairpin loop IIb towards the ribosomal E-site.20, 25 Crystal structure analysis of subdomain IIa revealed a high resolution picture of the subdomain IIa architecture, which adopts an L-shaped motif that contains three magnesium ions as an intrinsic part of the RNA fold (Figure 3B).27, 28 The nucleotide arrangement in the internal loop, which introduces a 90° bend in the RNA between base pairs G52-C111 and C58-G110, is stabilized by a combination of stacking, hydrogen bonding and metal ion participation. The bases of A53, A54 and C55 stack continuously on the G52-C111 pair and extend the lower stem into the bend. Connected by the looped out residue U56 the bases of A57 and C58 stack in extension of the upper stem. Coordination with two magnesium ions further reinforces the interfaces of both helical parts with residues of the internal loop. A third structural magnesium site is found locking U106 looped out from the upper stem.
Figure 3.

The HCV IRES subdomain IIa target. (A) Secondary structure of the subdomain IIa RNA. Conservation of residues in clinical isolates is indicated by font color (black: 100%; blue: >90%; red: <90%). Conservation analysis was performed using 5000 unique clinical isolate sequences identified by BLASTn109 search against the NCBI nonredundant nucleotide collection database. As the query sequence we used the HCV genotype 1 Reference Sequence, accession number NC_004102.1. Due to the heterogeneous sequencing coverage within the 5′UTR of HCV clinical isolates, the internal loop of subdomain IIa (residues 53–57) was present in ~2000 isolates while the 3′ region (residues 105–112) was covered in ~4800 sequences. Conservation percentages were calculated relative to the actual number of sequences available for each position. (B) Crystal structure of the subdomain IIa. The RNA fold is stabilized by three structural magnesium ions (cyan spheres).27 Image was prepared from PDB coordinate file 2NOK.
Residues of the subdomain IIa are 99–100% conserved in clinical isolates (Figure 3A), with the exception of A57, C104, U106 and G107, which show conservation of 88%, 93%, 98% and 77%, respectively. For the lowest conserved G107, the exclusively observed exchange is transition to an A (23% of clinical isolates) which retains canonical base pairing with U61. At A57, the impact of the dominating transversion to U (12% of clinical isolates) is not readily explained by the crystal structure. The Hoogsteen edge of the A57 base forms hydrogen bonds to the ribose 2′-OH of C55, which helps to stabilize a right-angled kink in the backbone between residues C55 and A57. Attempts to obtain diffracting crystals for the U57 variant subdomain IIa RNA have failed so far.
While the U57 variant is found in clinical isolates in conjunction with other mutations,110 introduction of this single mutation in genotype 1b reporter replicon led to reduction of IRES-driven translation activity to about 15% of the wild type level.29 Deletion of the subdomain IIa internal loop (ΔA53-A57) or parts thereof (ΔA53-A55) eliminates the bend in domain II that is responsible for directing subdomain IIb to the E-site. In these deletion mutants translation is abolished through stalled assembly of 80S ribosomes after 48S complex formation and decreased levels of eIF2 release as well as GTP hydrolysis.22, 23 The effect of the QIIa mutation is comparable to that observed after deletion of the entire domain II.
3.2. Translation Inhibitors Targeting the IRES Subdomain IIa
Two classes of translation inhibitors targeting the subdomain IIa have been identified, including benzimidazoles and diaminopiperidines (DAP derivatives) (section 2.4. and Figure 2). FRET studies using a fluorescently labeled target model suggested that the benzimidazole inhibitors capture a straightened conformation of the subdomain IIa that compromises IRES function.29 Binding of the benzimidazole compounds was not affected by the presence of excess competitor RNA or salt, including magnesium, which indicates that IIa target interaction with the ligands is specific and not governed by electrostatic components. X-ray crystallographic analysis of subdomain II with bound 4 revealed a fully extended RNA structure for the complex.101 Both, the FRET and crystallography studies are described in sections below.
The original synthetic route to constrained benzimidazoles such as 5 suffered from a low overall yield (Scheme 1).90, 98 The chroman scaffold in 5 was constructed over six steps. Polar amino and hydroxy groups were carried through the sequence and complicated purification, negatively affecting the yield of the original procedure. An improved synthetic route aimed at early generation of the chroman scaffold as well as introduction of polar functional groups in protected form to facilitate normal phase purification of all intermediates (Scheme 2).111 The improved 9 step synthesis furnished 4 in 10% overall yield.
Scheme 1.
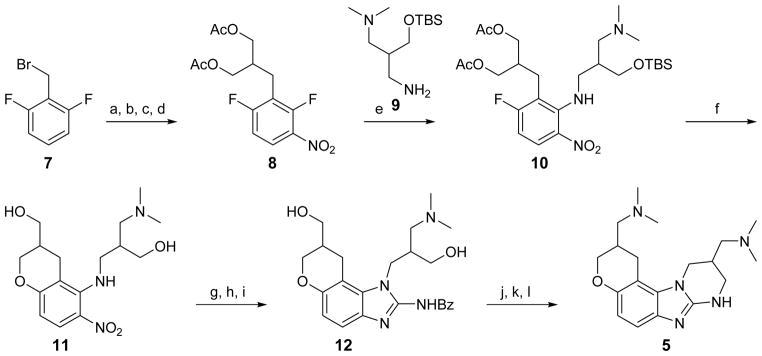
Original synthesis of IRES-binding 5.a,90
a Reagents: (a) NaH, diethyl malonate, THF; (b) LiAlH4, THF; (c) acetyl chloride, DCM, TEA, DMAP; (d) HNO3; (e) 9, DCM, CaCO3; (f) DMSO, MeOH, K2CO3; (g) Pd/C, H2, MeOH; (h) BzNCS, DIPEA, DCM; (i) EDC, DCM; (j) HCl, dioxane; (k) mesyl chloride, TEA, DMAP, DCM; (l) Me2NH/H2O, DMF.
Scheme 2.

Improved synthesis of IRES-binding 4.a,111
a Reagents: (m) acrolein 14, DABCO, CH3CN; (n) Ag2O, NaOH, EtOH/H2O; (o) Na/Hg, NaOH, H2O; (p) EDC, Me2NH.HCl, HOBt, N-Me-morpholine, DCM; (q) NaNO3, TFA; (r) 17, NMP; (s) Pd/C, H2, EtOH; (t) BrCN/CH3CN; (u) LiAlH4, THF.
Discovery of DAP derivatives as ligands of the subdomain IIa relied on a fluorescence assay using target RNA in which A54 in the internal loop was replaced by a fluorescent 2-aminopurine (2AP) base analog.27 Unlike the benzimidazole inhibitors which capture an extended conformation of the subdomain IIa RNA, the DAP ligands seem to stabilize the bent form of the target. Modular RNA-targeting DAP derivatives were readily synthesized by coupling of a protected DAP heterocycle with an α-amino acid followed by a second amide coupling step with a carboxylic acid building block (Scheme 3). Compound 6 and the corresponding arginine derivative bound to the subdomain IIa target with 6–7 μM affinity and inhibited HCV replicon. RNA interaction of the DAP ligands involved a strong electrostatic component as indicated by competition with magnesium binding.
Scheme 3.

Synthesis of IRES-binding 6.a,30
a Reagents: (v) TEA, HATU, HOAT, DCM; (w) Pd/C, H2, MeOH; (x) 19, TEA, HATU, HOAT, DCM; (y) HCl/dioxane, MeOH.
3.3. Subdomain IIa RNA Conformations Captured by Ligand Binding
The bent subdomain IIa plays a key role in positioning the IIb hairpin in the ribosomal E-site. This led us to propose that the subdomain IIa might serve as a target for HCV translation inhibitors that interfere with the conformation of the RNA. To study folding and ligand binding of the subdomain IIa, we developed fluorescence assays which provided insight into the interaction of translation inhibitors with the RNA target. Oligonucleotide models of the subdomain IIa were fluorescently labeled either by replacement of A54 with the base analog 2AP (“2AP54 construct”)27 or by 5′-terminal attachment of a FRET cyanine dye pair (“FRET construct”, Figure 4A).29 Both constructs were used to monitor magnesium-induced folding of the RNA. In the absence of divalent metal ions, the internal loop of the subdomain IIa is unstructured and flexible, giving rise to a preferentially extended overall shape of the RNA. Magnesium binding induces structural organization of the internal loop, resulting in a dose-dependent increase of FRET signal due to the proximity of the cyanine dyes in the folded L-shaped RNA (Figure 4B).
Figure 4.

FRET-based assay for the monitoring of conformational changes in the subdomain IIa target during folding and ligand binding.29, 93 (A) Cyanine dye-labeled RNA construct used for FRET measurements. Nucleotides that deviate from the HCV genotype 1b sequence are in outlined font. (B) FRET monitoring of the Mg2+ induced folding of the IIa RNA. (C) Benzoxazole derivative 23 identified in a FRET screen for subdomain IIa ligands.93
Fluorescence from the 2AP54 construct, however, decreases upon folding which results in stacking of the fluorescent base analog between neighboring residues (Figure 3B). Comparative metal ion binding studies, which exploited the differential preference of magnesium versus manganese ions for oxygen over nitrogen coordination, substantiated the role of metal ions observed in the crystal structure as intrinsic constituents of the folded subdomain IIa RNA in solution. The observation that 2AP54 fluorescence is diminished but not fully quenched in the magnesium-stabilized subdomain IIa indicates dynamic properties of the internal loop which may allow the RNA motif to explore conformations other than the 90° bend found in the crystal structure.
Titration of Isis benzimidazole translation inhibitors such as 4 and 5 to the folded IIa FRET construct resulted in dose-dependent fluorescence quenching which indicated a conformational change in the RNA that separated the cyanine dye labels beyond the Föster radius. The 2AP54 construct, on the other hand, responded with a signal increase suggesting exposure of the fluorescent label upon ligand binding. We concluded that the benzimidazole translation inhibitors either capture or trigger a straightened conformation of the subdomain IIa and thereby disrupt correct positioning of the IIb hairpin at the ribosome. Support for this hypothesis comes from studies of a subdomain IIa variant carrying the A57U mutation which occurs in HCV clinical isolates. The A57U exchange weakens by 7-fold binding of benzimidazole 5 to the target RNA as measured by FRET and, as a consequence, reduces the inhibitory effect of 5 on translation in HCV replicon systems.29
Binding of 6 to the subdomain IIa was originally discovered using the 2AP54 target construct.30 Residual fluorescence of the 2AP label, which likely originates from the dynamic conformation of the internal loop, is quenched upon binding of 6. These data in conjunction with the observed competition of DAP ligands with magnesium ions, led to the proposal that compounds such as 6 bind to the subdomain IIa target by displacing structural metals and rigidifying the dynamic internal loop RNA.
Concluding from the fluorescence studies of conformational dynamics and impact of ligand binding, a model was proposed for the subdomain IIa target as a conformational switch that seeks to integrate the role of RNA flexibility for IRES function as well as the mode of action of HCV translation inhibitors (Figure 5).30 The notion of IRES-targeting inhibitors as allosteric effectors of RNA conformation bears resemblance to the mechanism observed for aminoglycoside antibiotics which target a conformational switch in the decoding site RNA of the bacterial ribosome.
Figure 5.

Model for the mode of action of translation inhibitors on the subdomain IIa conformational switch in the HCV IRES. The subdomain IIa RNA adopts a bent fold that is stabilized by magnesium ions and is required for the correct positioning of the IRES on the ribosome. Release of the ribosome after translation initiation might involve dynamical changes in the L-shaped RNA structure, which might be captured by a cognate ligand such as an arginine of a cellular protein or a guanosine in an RNA (center). Benzimidazole IRES inhibitors such as 5 (left) capture an extended state of the IIa RNA switch which leads to disruption of IRES function and inhibition of viral protein synthesis.29 The diaminopiperidine derivative 6 (right) binds in competition with structural Mg2+ ions and locks the IIa RNA switch in the bent conformation, which inhibits IRES function perhaps by preventing ribosome release.30
3.4. Screening for Translation Inhibitors Targeting the Subdomain IIa
Previously, affinity screening of IRES fragment targets by mass spectrometry had been successful in the discovery of benzimidazole HCV translation inhibitors (section 2.4. and Figure 2).90 Target selectivity of the ligands was tested by differential screening of subdomain IIa RNA in the presence of an unspecified competitor RNA of similar size and, presumably, structural complexity. Detection of selectivity for a specific RNA such as an HCV IRES domain over other cellular nucleic acids may be achieved by monitoring a ligand-induced capture or triggering of a unique event in the target. The paradigm for such a conformational RNA target is the ribosomal decoding site in which a change in the orientation of two adenine residues is triggered by the binding of aminoglycoside antibiotics.112 Outside the ribosome, such conformational RNA targets for small molecule ligands had been largely elusive.
The large conformational change exhibited by the subdomain IIa RNA provides an ideal mechanism for the establishment of a target-selective combined binding and functional screen. The FRET assay outlined in the previous section was readily adapted to a high-throughput screening format in which ligands were identified based on their ability to reduce the FRET signal by induction of a widened interhelical angle in the cyanine dye labeled subdomain IIa target.93 As a control for nonspecific fluorescence quenching, emission from the Cy3 donor was recorded which for true ligands increased concurrently with FRET quenching due to the diminishing resonance energy transfer efficiency at growing distances of the cyanine dyes. Feasibility of the assay format was demonstrated by screening a small set of molecules biased for binding to RNA targets from which 23 (Figure 4C) was identified as a selective ligand for the subdomain IIa RNA. The benzoxazole 23 quenched FRET with an EC50 value of ~100 μM, comparable to the affinity of the hit 1, and selectively inhibited IRES-initiated translation by 30% at 100 μM concentration while not affecting the cap-dependent process. The structural similarity of 23 compared to previously discovered benzimidazoles such as 1 readily explains the activity of the benzoxazole as a ligand of the subdomain IIa target and an IRES inhibitor. The lower basicity as well as reduced hydrophilicity of the benzoxazole scaffold compared to benzimidazoles may lend superior drug-like properties to subdomain IIa binding translation inhibitors emerging from future optimization of the hit 23.
3.5. Structure of a Subdomain IIa Translation Inhibitor Complex
Structural investigations of the subdomain IIa RNA in complex with benzimidazole translation inhibitors have been performed by NMR and X-ray crystallography. An NMR study of the target bound with a racemic benzimidazole derivative related to 4 found an overall extended conformation of the RNA ligand complex, supporting the FRET experiments.31 However, the NMR structure did not reveal details of the binding interaction. An X-ray crystallographic analysis revealed at 2.2 Å resolution the structure of the target IIa RNA bound with 4 (Figure 6).101 In the complex the RNA adopts an extended architecture with the internal loop refolded from its curved conformation in the free target and flanked by coaxially stacking helices (Figure 6A). A deep cavity is formed that encapsulates the ligand which docks by hydrogen bonding to the guanine heterocycle in the C58-G110 base pair and stacking interactions with A53 as well as the G52-C111 pair (Figure 6BC). An additional intramolecular hydrogen bond occurs between the protonated dimethylamino-propyl side chain of the benzimidazole inhibitor and an RNA phosphate group. Formation of the binding pocket upon adaptive ligand recognition further involves RNA base triples which engage in cross-bracing interactions along the RNA helix (Figure 6B). The participation of magnesium ions in the stabilization of the subdomain IIa target is maintained for the RNA ligand complex. The free RNA as well as the complex each contains three magnesium ions as intrinsic structural components which undergo adaptive reorganization upon ligand binding.
Figure 6.

Crystal structure of the subdomain IIa target in complex with 4.101 (A) Overall view of the binding site. The ligand 4 is shown in yellow sticks. Mg2+ ions are indicated by light blue spheres. (B) Interactions in the ligand binding site. Hydrogen bonds are shown as dashed lines. Stacked lines (≡) indicate stacking of bases and intercalation of the ligand. Formation of non-Watson-Crick base pairs is indicated with solid lines and symbols according to Leontis and Westhof. (C) Surface representation of the binding site in a front side view (rotated by 90° from panel A), highlighting the deep ligand pocket. (D) Backside view of the complex showing hydrogen bonding interactions of the A57 base whose exchange to a uridine weakens binding of the benzimidazole translation inhibitors. Images were prepared from PDB coordinate file 3TZR.
While co-crystallization of the subdomain IIa RNA was performed with a racemate of 4, the complex structure did not unambiguously reveal the ligand stereochemistry. However, X-ray crystallographic analysis of racemic 4 by itself showed that the structures of the enantiomers are sufficiently similar to allow for RNA target binding of either form through the observed hydrogen bonding and stacking interactions.113 Titrations of pure enantiomers of a benzimidazole related to 4, in which the tetrahydropyran ring was replaced by a tetrahydrofuran, with a 2AP fluorescently labeled target RNA27 suggested that one isomer bound about 3.5-fold tighter than the other.31 However, the absolute configuration of the tighter binding enantiomer was not determined. Structure-activity relationships for related benzimidazole derivatives of 4 were conclusively explained by the crystal structure.101 For example, benzimidazoles that lack the amino substituent at the 2-position or the N,N-dimethylaminopropyl chain off the N1, both of which participate in hydrogen bonds with the target, did not bind to the subdomain IIa RNA. Improved binding was observed for derivatives carrying modifications at the 6-position with basic substituents producing best activities and larger non-polar groups adding no benefit. Substitution at the 4- and 5-position of the benzimidazole was not tolerated due to the tight fit of the ligand into the substrate binding site and derivatives with such modifications were inactive.
The U57 variant, which conferred HCV replicon resistance to the related translation inhibitor 5, displayed weakened binding for 4 as well, which had a ~3-fold lower affinity for the variant target. While the residue at position 57 is not involved in direct contacts with the bound ligand, A57 which occurs in 88% of HCV clinical isolates plays an important role in stabilizing the back side of the benzimidazole pocket in the complex by forming hydrogen bonds with C111 and coordinating a key structural magnesium ion (Figure 6D). We speculate that transversion to U57 increases the flexibility of the subdomain IIa internal loop and thereby interferes with both the ability of the IRES to initiate translation and benzimidazole inhibitor binding. The position 57, which is one of the less conserved residues in the HCV IRES, may very well represent the Achilles’ heel of the subdomain IIa target. Further structural studies on the U57 variant RNA are required to devise ligand design strategies for inhibitors that retain activity in this mutant.
Key hydrogen bonds of the subdomain IIa benzimidazole complex interaction are solventexcluded inside the binding pocket which, together with the stacking interactions, lends a strong hydrophobic component to ligand recognition. Both the depth and hydrophobicity of the ligand pocket in the subdomain IIa are unusual features for an RNA target which render the benzimidazole docking site more similar to inhibitor binding pockets in protein targets. Hence, development of drug-like HCV translation inhibitors directed at the subdomain IIa RNA seems a feasible endeavour, supported in particular by guidance from the high resolution structure for the RNA target complex.
4. OUTLOOK
Approaches to inhibit HCV translation by targeting the highly conserved IRES RNA have resulted in a diverse set of ligands including oligonucleotdies, peptides as well as small molecules which block IRES function by distinct mechanisms. Currently, the subdomain IIa is the only IRES target for which a binding site for small molecule translation inhibitors has been identified. From investigations of the mechanism of inhibitors targeting the subdomain IIa the picture is emerging that this RNA domain functions as a conformational switch whose state may be locked by ligand binding. Structural studies by electron microscopy of IRES-ribosome complexes have led to the conclusion that after assembly of 80S complexes the bent topology of the domain II would prevent the progression of the ribosome from initiation to elongation.20, 25
Domain II has to be moved out of the E site to make room for deacylated tRNA during the transition to productive translation. Based on findings of ligand-dependent conformational switching in the highly conserved subdomain IIa, it was proposed that a conformational change triggered by adaptive recognition of a cognate ligand may facilitate the release of the ribosome from the IRES-bound complex (Figure 5).101 Benzimidazole translation inhibitors may be fortuitous ligands of the subdomain IIa conformational switch perhaps due to their ability to mimic interactions of arginine or guanosine at the Hoogsteen edge of the G110-C58 base pair while stacking between neighbouring residues G52 and A53 (Figure 7). Arginine side chains have been found in isostructural interaction with G-C pairs in numerous peptide and protein complexes of both RNA and DNA.114–117 Guanosine docking at the Hoogsteen edge of G in a Watson-Crick G-C pair accounts for one of the most common base triples found in RNA architectures.118–121
Figure 7.
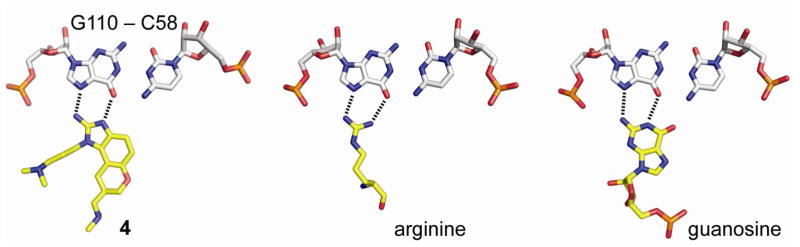
Examples of Hoogsteen edge recognition of G-C pairs by arginine or guanosine compared to the G110-C58 interaction with 4 in the subdomain IIa complex. The image of the benzimidazole complex was prepared from PDB coordinate file 3TZR.
While the high resolution crystal structure of the subdomain IIa benzimidazole complex points the way for structure-guided discovery of HCV translation inhibitors, the hunt for potential cognate triggers of the RNA switch has just begun. At least two proteins, including ribosomal protein S5 (rpS5) and heterogeneous ribonucleoprotein D (hnRNP D) have been identified by UV-crosslinking and immunoprecipitation as direct binding partners of the HCV domain II RNA.24, 122 It remains to be tested, however, whether these candidate proteins, or any other yet to be discovered factors, interact at the subdomain IIa or affect the conformation of the RNA switch. Alternatively, it is conceivable that either the viral mRNA itself or ribosomal RNA may trigger release of the IRES by insertion of a G residue into the subdomain IIa switch. Recently, direct interactions between the IRES domain II and 18S rRNA have been confirmed by chemical probing. It could also be envisioned that the viral mRNA emerging from the ribosome upon translation may provide a guanosine residue for participation in a base triple interaction at the ligand binding pocket of the subdomain IIa switch.
The discovery of the HCV IRES subdomain IIa as a conformational switch that is a target for small molecule translation inhibitors paves the way for the development of drugs directed at the viral RNA. The high conservation of the subdomain IIa in clinical isolates promises that mutations around the ligand binding pocket will be difficult to reconcile with IRES function. Inhibitors directed at this RNA target will potentially benefit from selection of low-fitness resistance mutants with reduced frequency of occurrence. The availability of a target specific functional binding assay combined with high resolution structural data for the free RNA and an inhibitor complex will provide tools for overcoming the Achilles’ heel of the subdomain IIa in the U57 target variant and enable the discovery of drug-like HCV translation inhibitors.
Table 1.
Target Binding and Biological Activity of IRES Inhibitors
| Compound | Subdomain IIa target affinity (μM) | IRES translation inhibition (μM) | IRES/cap selectivity (fold) | Replicon inhibition (μM) | Reference |
|---|---|---|---|---|---|
| 1 | n. a. | 0.08 | 10 | n. d. | 87 |
| 2 | n. a. | 9.6 | 2.4 | n. d. | 88 |
| 3 | 100a/50b,c | 90/a | |||
| 4 | 0.86a/3.4b | 3.9/2.8 | 90/101 | ||
| 5 | 0.72a/0.6b | 5.4/4.0 | 90/29 | ||
| 6 | 6.3b | 75% inhibition at 10 μM | 30 | ||
| 23 | 100b | 30% inhibition at 100 μM | 93 |
Acknowledgments
Funding Sources
HCV research in the T.H. group was supported by faculty startup funds from the University of California, the National Institutes of Health (grant No. R01 AI072012/NIAID) and the National Science Foundation (CRIF grant No. CHE-0741968).
We thank our collaborators Drs. B. M. Bergdahl, S. Dutta, M. A. Parker, D. L. Wyles and undergraduate students who contributed to our HCV research, including C. J. Higginson, B. T. Ho, H. Johnston-Cox and Y.-H. Weng.
ABBREVIATIONS
- 2AP
2-aminopurine
- DABCO
1,4-diazabicylclo[2.2.2]octane
- DAP
diaminopiperidine
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- 2-DOS
2-deoxystreptamine
- EDC
1-ethyl-3- (3-dimethylaminopropyl)carbodiimide
- eIF3
eukaryotic initiation factor 3
- FRET
Föster resonance energy transfer
- GTP
guanosine triphosphate
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HCV
hepatitis C virus
- HOAT
1-hydroxy-7-azabenzotriazole
- HOBt
hydroxybenzotriazole
- IRES
internal ribosome entry site
- mRNA
messenger RNA
- NMP
N-methylpyrrolidone
- rRNA
ribosomal RNA
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- UTR
untranslated region
Biographies
Sergey M. Dibrov attended Voronezh State University, Russia, where he earned his B.Sc. in Chemistry prior to pursuing Ph.D. studies in Chemistry under the direction of Prof. Robert Bachman at Georgetown University, Washington, DC. Following postdoctoral studies with Prof. James Ibers at Northwestern University, IL, and with Prof. Jay Kochi at the University of Houston, TX, he joined the laboratory of Prof. Thomas Hermann at the University of California, San Diego (UCSD), in 2006. As a Project Scientist, he is in charge of both small molecule and biomolecule crystallography in the Hermann group.
Jerod Parsons earned his B.Sc. in Genetics from Texas A&M University and joined the Chemistry Graduate Program at UCSD in 2005. He was the first Ph.D. student performing research under the direction of Prof. Thomas Hermann. For his contributions that established the HCV IRES targeting effort in the laboratory, he received his Ph.D. in Chemistry and Biochemistry in 2010. He currently works as an NRC Postdoc in the NIST Advances in Biomedical Measurement Science Program at Stanford University.
Maia Carnevali received her B.Sc. in Biochemistry and Molecular Biology from the University of California, Santa Cruz, and went on to work as a Scientist at Gryphon Therapeutics, South San Francisco, CA. After entering the Chemistry Graduate Program at UCSD in 2004, she joined the Hermann laboratory in 2006 where she was a founding member of the synthetic chemistry group and worked on the preparation of RNA-binding ligands. She earned her Ph.D. in Chemistry in 2011, and currently does research as a postdoctoral fellow at Aridis Pharmaceuticals, San Jose, CA.
Shu Zhou earned her B.Sc. in Organic Chemistry from Shanghai University, China. In 2006, she joined the Biochemistry Graduate Program at Ohio University, where she developed screening assays to identify T box antiterminator RNA ligands as potential antibacterial agents. After receiving her Ph.D. in Biochemistry she joined the Hermann group at UCSD as a postdoctoral fellow, in 2012, and established FRET technology for screening of the HCV IRES target. She currently performs research as a postdoctoral fellow in the Department of Pharmacology at UCSD.
Kevin D. Rynearson received his B.Sc. in Chemistry from San Diego State University. He entered the Chemistry Graduate Program at UCSD in 2008, where he performs synthetic method development for RNA-binding ligands in the Hermann laboratory. He is expected to defend his Ph.D. thesis in the fall of 2013.
Kejia Ding studied Chemistry at San Jose State University, CA, where he earned his B.Sc. prior to joining the Chemistry Graduate Program at UCSD in 2009. He performs in silico studies as well as synthetic research on RNA-binding ligands in the Hermann laboratory.
Emily Garcia Sega (née Satkiewicz) received her B.Sc. in Chemistry from Seton Hall University, NJ, and joined the Chemistry Graduate Program at UCSD in 2006. She was awarded her Ph.D. in 2011 for synthetic studies on RNA-binding ligands in the Hermann group. After a postdoctoral stint at Ithaca College, NY, she joined the Chemistry Department of Bridgewater State University, MA, as an Assistant Professor, in 2013.
Nicholas D. Brunn received his B.Sc. in Chemistry from San Diego State University. After working as a research assistant at The Scripps Research Institute, La Jolla, CA, he entered the Chemistry Graduate Program at UCSD in 2009 where he joined the Hermann laboratory to perform biochemistry and crystallography research on RNA targets including the HCV IRES and the human thymidylate synthase mRNA. He is expected to defend his Ph.D. thesis in the fall of 2013.
Mark A. Boerneke earned a B.Sc. in Chemistry from Point Loma Nazarene University, CA, and joined the Chemistry Graduate Program at UCSD in 2011. In the Hermann group he studies functional and structural aspects of viral RNA targets including the HCV IRES.
M. Paola Castaldi studied Pharmaceutical Chemistry and received her Laurea degree at the University of Padova, Italy. She then pursued chemistry graduate research at Imperial College, London, UK, under the direction of Prof. Susan E. Gibson. After earning her Ph.D. in 2005, she joined the laboratory of Prof. Thomas Hermann where she was a founding member of the synthetic chemistry group and worked on the preparation of RNA-binding ligands. After joining Prof. John Porco’s laboratory at CMLD-BU, she spent two years in the Chemical Genetics group at Sanofi Oncology in Cambridge, MA. She is currently working as Associate Principal Scientist in the Chemical Biology group at Astra Zeneca, Waltham, MA.
Thomas Hermann earned a Chemistry Diploma from the University of Ulm, Germany. He performed thesis research at the Max-Planck Institute for Biochemistry and received a Ph.D. from the Ludwig-Maximilians University in Munich in 1996. As a postdoc, he worked with Prof. Eric Westhof at the CNRS in Strasbourg, France, and with Prof. Dinshaw Patel at the Sloan-Kettering Cancer Center in New York. Subsequently, he joined Anadys Pharmaceuticals in San Diego in 2001, where he built the structural chemistry group and led discovery teams working on ribosome-targeted antibiotics and HCV polymerase inhibitors, laying the foundation for the development of setrobuvir. In 2005, he joined UCSD where he currently is an Associate Professor and serves as a founding director of the Center for Drug Discovery Innovation.
References
- 1.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J Virol. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hellen CU, Pestova TV. Translation of hepatitis C virus RNA. J Viral Hepat. 1999;6:79–87. doi: 10.1046/j.1365-2893.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- 4.Kieft JS, Grech A, Adams P, Doudna JA. Mechanisms of internal ribosome entry in translation initiation. Cold Spring Harb Symp Quant Biol. 2001;66:277–283. doi: 10.1101/sqb.2001.66.277. [DOI] [PubMed] [Google Scholar]
- 5.Ji H, Fraser CS, Yu Y, Leary J, Doudna JA. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc Natl Acad Sci U S A. 2004;101:16990–16995. doi: 10.1073/pnas.0407402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto GA, Puglisi JD. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Gallego J, Varani G. The hepatitis C virus internal ribosome-entry site: a new target for Antiviral. Res.earch. Biochem Soc Trans. 2002;30:140–145. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 8.Jubin R. Targeting hepatitis C virus translation: stopping HCV where it starts. Curr Opin Investig Drugs. 2003;4:162–167. [PubMed] [Google Scholar]
- 9.Davis DR, Seth PP. Therapeutic targeting of HCV internal ribosomal entry site RNA. Antivir Chem Chemother. 2011;21:117–128. doi: 10.3851/IMP1693. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman B, Liu Q. Hepatitis C viral protein translation: mechanisms and implications in developing antivirals. Liver Int. 2011;31:1449–1467. doi: 10.1111/j.1478-3231.2011.02543.x. [DOI] [PubMed] [Google Scholar]
- 11.Hermann T. Strategies for the Design of Drugs Targeting RNA and RNA-Protein Complexes. Angew Chem Int Ed Engl. 2000;39:1890–1904. doi: 10.1002/1521-3773(20000602)39:11<1890::aid-anie1890>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 12.Gallego J, Varani G. Targeting rna with small-molecule drugs: therapeutic promise and chemical challenges. Acc Chem Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 13.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 14.Guan L, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 15.Honda M, Ping LH, Rijnbrand RC, Amphlett E, Clarke B, Rowlands D, Lemon SM. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31–42. doi: 10.1006/viro.1996.0395. [DOI] [PubMed] [Google Scholar]
- 16.Kieft JS, Zhou K, Jubin R, Murray MG, Lau JY, Doudna JA. The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J Mol Biol. 1999;292:513–529. doi: 10.1006/jmbi.1999.3095. [DOI] [PubMed] [Google Scholar]
- 17.Friebe P, Lohmann V, Krieger N, Bartenschlager R. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol. 2001;75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7:194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011;19:1456–1466. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 21.Filbin ME, Kieft JS. HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit’s decoding groove. RNA. 2011;17:1258–1273. doi: 10.1261/rna.2594011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locker N, Easton LE, Lukavsky PJ. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007;26:795–805. doi: 10.1038/sj.emboj.7601549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filbin ME, Vollmar BS, Shi D, Gonen T, Kieft JS. HCV IRES manipulates the ribosome to promote the switch from translation initiation to elongation. Nat Struct Mol Biol. 2013;20:150–158. doi: 10.1038/nsmb.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K. Ribosomal protein S5 interacts with the internal ribosomal entry site of hepatitis C virus. J Biol Chem. 2001;276:20824–20826. doi: 10.1074/jbc.C100206200. [DOI] [PubMed] [Google Scholar]
- 25.Boehringer D, Thermann R, Ostareck-Lederer A, Lewis JD, Stark H. Structure of the hepatitis C Virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure (Camb) 2005;13:1695–1706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Structure of HCV IRES domain II determined by NMR. Nat Struct Biol. 2003;10:1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 27.Dibrov SM, Johnston-Cox H, Weng YH, Hermann T. Functional architecture of HCV IRES domain II stabilized by divalent metal ions in the crystal and in solution. Angew Chem Int Ed Engl. 2007;46:226–229. doi: 10.1002/anie.200603807. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Q, Han Q, Kissinger CR, Hermann T, Thompson PA. Structure of hepatitis C virus IRES subdomain IIa. Acta Crystallogr D Biol Crystallogr. 2008;64:436–443. doi: 10.1107/S0907444908002011. [DOI] [PubMed] [Google Scholar]
- 29.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat Chem Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carnevali M, Parsons J, Wyles DL, Hermann T. A modular approach to synthetic RNA binders of the hepatitis C virus internal ribosome entry site. Chembiochem. 2010;11:1364–1367. doi: 10.1002/cbic.201000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulsen RB, Seth PP, Swayze EE, Griffey RH, Skalicky JJ, Cheatham TE, 3rd, Davis DR. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc Natl Acad Sci U S A. 2010;107:7263–7268. doi: 10.1073/pnas.0911896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanecak R, Brown-Driver V, Fox MC, Azad RF, Furusako S, Nozaki C, Ford C, Sasmor H, Anderson KP. Antisense oligonucleotide inhibition of hepatitis C virus gene expression in transformed hepatocytes. J Virol. 1996;70:5203–5212. doi: 10.1128/jvi.70.8.5203-5212.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Hanecak R, Brown-Driver V, Azad R, Conklin B, Fox MC, Anderson KP. Antisense oligonucleotide inhibition of hepatitis C virus (HCV) gene expression in livers of mice infected with an HCV-vaccinia virus recombinant. Antimicrob Agents Chemother. 1999;43:347–353. doi: 10.1128/aac.43.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tallet-Lopez B, Aldaz-Carroll L, Chabas S, Dausse E, Staedel C, Toulme JJ. Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids Res. 2003;31:734–742. doi: 10.1093/nar/gkg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinand-Mari C, Lebleu B, Robbins I. Oligonucleotide-based strategies to inhibit human hepatitis C virus. Oligonucleotides. 2003;13:539–548. doi: 10.1089/154545703322860834. [DOI] [PubMed] [Google Scholar]
- 36.McCaffrey AP, Meuse L, Karimi M, Contag CH, Kay MA. A potent and specific morpholino antisense inhibitor of hepatitis C translation in mice. Hepatology. 2003;38:503–508. doi: 10.1053/jhep.2003.50330. [DOI] [PubMed] [Google Scholar]
- 37.Nulf CJ, Corey D. Intracellular inhibition of hepatitis C virus (HCV) internal ribosomal entry site (IRES)-dependent translation by peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) Nucleic Acids Res. 2004;32:3792–3798. doi: 10.1093/nar/gkh706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caldarelli SA, Mehiri M, Di Giorgio A, Martin A, Hantz O, Zoulim F, Terreux R, Condom R, Patino N. A cyclic PNA-based compound targeting domain IV of HCV IRES RNA inhibits in vitro IRES-dependent translation. Bioorg Med Chem. 2005;13:5700–5709. doi: 10.1016/j.bmc.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Alotte C, Martin A, Caldarelli SA, Di Giorgio A, Condom R, Zoulim F, Durantel D, Hantz O. Short peptide nucleic acids (PNA) inhibit hepatitis C virus internal ribosome entry site (IRES) dependent translation in vitro. Antiviral Res. 2008;80:280–287. doi: 10.1016/j.antiviral.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 40.Laxton C, Brady K, Moschos S, Turnpenny P, Rawal J, Pryde DC, Sidders B, Corbau R, Pickford C, Murray EJ. Selection, optimization, and pharmacokinetic properties of a novel, potent antiviral locked nucleic acid-based antisense oligomer targeting hepatitis C virus internal ribosome entry site. Antimicrob Agents Chemother. 2011;55:3105–3114. doi: 10.1128/AAC.00222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witherell GW. ISIS-14803 (Isis Pharmaceuticals) Curr Opin Investig Drugs. 2001;2:1523–1529. [PubMed] [Google Scholar]
- 42.Soler M, McHutchison JG, Kwoh TJ, Dorr FA, Pawlotsky JM. Virological effects of ISIS 14803, an antisense oligonucleotide inhibitor of hepatitis C virus (HCV) internal ribosome entry site (IRES), on HCV IRES in chronic hepatitis C patients and examination of the potential role of primary and secondary HCV resistance in the outcome of treatment. Antivir Ther. 2004;9:953–968. [PubMed] [Google Scholar]
- 43.McHutchison JG, Patel K, Pockros P, Nyberg L, Pianko S, Yu RZ, Dorr FA, Kwoh TJ. A phase I trial of an antisense inhibitor of hepatitis C virus (ISIS 14803), administered to chronic hepatitis C patients. J Hepatol. 2006;44:88–96. doi: 10.1016/j.jhep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Georgopapadakou N. Discontinued drugs in 2005: anti-infectives. Expert Opin Investig Drugs. 2007;16:1–10. doi: 10.1517/13543784.16.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Kikuchi K, Fukuda K, Umehara T, Hwang J, Kuno A, Hasegawa T, Nishikawa S. In vitro selection of RNA aptamers that bind to domain II of HCV IRES. Nucleic Acids Res Suppl. 2002:267–268. doi: 10.1093/nass/2.1.267. [DOI] [PubMed] [Google Scholar]
- 46.Kikuchi K, Umehara T, Fukuda K, Hwang J, Kuno A, Hasegawa T, Nishikawa S. Structure-inhibition analysis of RNA aptamers that bind to HCV IRES. Nucleic Acids Res Suppl. 2003:291–292. doi: 10.1093/nass/3.1.291. [DOI] [PubMed] [Google Scholar]
- 47.Kikuchi K, Umehara T, Fukuda K, Hwang J, Kuno A, Hasegawa T, Nishikawa S. RNA aptamers targeted to domain II of hepatitis C virus IRES that bind to its apical loop region. J Biochem (Tokyo) 2003;133:263–270. doi: 10.1093/jb/mvg036. [DOI] [PubMed] [Google Scholar]
- 48.Kikuchi K, Umehara T, Fukuda K, Kuno A, Hasegawa T, Nishikawa S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III–IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res. 2005;33:683–692. doi: 10.1093/nar/gki215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konno K, Fujita S, Iizuka M, Nishikawa S, Hasegawa T, Fukuda K. Isolation and characterization of RNA aptamers specific for the HCV minus-IRES domain I. Nucleic Acids Symp Ser (Oxf) 2008:493–494. doi: 10.1093/nass/nrn250. [DOI] [PubMed] [Google Scholar]
- 50.Kikuchi K, Umehara T, Nishikawa F, Fukuda K, Hasegawa T, Nishikawa S. Increased inhibitory ability of conjugated RNA aptamers against the HCV IRES. Biochem Biophys Res Commun. 2009;386:118–123. doi: 10.1016/j.bbrc.2009.05.135. [DOI] [PubMed] [Google Scholar]
- 51.Konno K, Iizuka M, Fujita S, Nishikawa S, Hasegawa T, Fukuda K. An RNA aptamer containing two binding sites against the HCV minus-IRES domain I. Nucleosides Nucleotides Nucleic Acids. 2011;30:185–202. doi: 10.1080/15257770.2011.562475. [DOI] [PubMed] [Google Scholar]
- 52.Macejak DG, Jensen KL, Jamison SF, Domenico K, Roberts EC, Chaudhary N, von Carlowitz I, Bellon L, Tong MJ, Conrad A, Pavco PA, Blatt LM. Inhibition of hepatitis C virus (HCV)-RNA-dependent translation and replication of a chimeric HCV poliovirus using synthetic stabilized ribozymes. Hepatology. 2000;31:769–776. doi: 10.1002/hep.510310331. [DOI] [PubMed] [Google Scholar]
- 53.Lee PA, Blatt LM, Blanchard KS, Bouhana KS, Pavco PA, Bellon L, Sandberg JA. Pharmacokinetics and tissue distribution of a ribozyme directed against hepatitis C virus RNA following subcutaneous or intravenous administration in mice. Hepatology. 2000;32:640–646. doi: 10.1053/jhep.2000.16599. [DOI] [PubMed] [Google Scholar]
- 54.Jarczak D, Korf M, Beger C, Manns MP, Kruger M. Hairpin ribozymes in combination with siRNAs against highly conserved hepatitis C virus sequence inhibit RNA replication and protein translation from hepatitis C virus subgenomic replicons. FEBS J. 2005;272:5910–5922. doi: 10.1111/j.1742-4658.2005.04986.x. [DOI] [PubMed] [Google Scholar]
- 55.Romero-Lopez C, Barroso-delJesus A, Puerta-Fernandez E, Berzal-Herranz A. Interfering with hepatitis C virus IRES activity using RNA molecules identified by a novel in vitro selection method. Biol Chem. 2005;386:183–190. doi: 10.1515/BC.2005.023. [DOI] [PubMed] [Google Scholar]
- 56.Romero-Lopez C, Diaz-Gonzalez R, Berzal-Herranz A. Inhibition of hepatitis C virus internal ribosome entry site-mediated translation by an RNA targeting the conserved IIIf domain. Cell Mol Life Sci. 2007;64:2994–3006. doi: 10.1007/s00018-007-7345-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Romero-Lopez C, Diaz-Gonzalez R, Barroso-delJesus A, Berzal-Herranz A. Inhibition of hepatitis C virus replication and internal ribosome entry site-dependent translation by an RNA molecule. J Gen Virol. 2009;90:1659–1669. doi: 10.1099/vir.0.008821-0. [DOI] [PubMed] [Google Scholar]
- 58.Romero-Lopez C, Berzal-Herranz B, Gomez J, Berzal-Herranz A. An engineered inhibitor RNA that efficiently interferes with hepatitis C virus translation and replication. Antiviral Res. 2012;94:131–138. doi: 10.1016/j.antiviral.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 59.Roy S, Gupta N, Subramanian N, Mondal T, Banerjea AC, Das S. Sequence-specific cleavage of hepatitis C virus RNA by DNAzymes: inhibition of viral RNA translation and replication. J Gen Virol. 2008;89:1579–1586. doi: 10.1099/vir.0.83650-0. [DOI] [PubMed] [Google Scholar]
- 60.Kumar D, Chaudhury I, Kar P, Das RH. Site-specific cleavage of HCV genomic RNA and its cloned core and NS5B genes by DNAzyme. J Gastroenterol Hepatol. 2009;24:872–878. doi: 10.1111/j.1440-1746.2008.05717.x. [DOI] [PubMed] [Google Scholar]
- 61.Guerniou V, Gillet R, Berree F, Carboni B, Felden B. Targeted inhibition of the hepatitis C internal ribosomal entry site genomic RNA with oligonucleotide conjugates. Nucleic Acids Res. 2007;35:6778–6787. doi: 10.1093/nar/gkm770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gamble C, Trotard M, Le Seyec J, Abreu-Guerniou V, Gernigon N, Berree F, Carboni B, Felden B, Gillet R. Antiviral effect of ribonuclease conjugated oligodeoxynucleotides targeting the IRES RNA of the hepatitis C virus. Bioorg Med Chem Lett. 2009;19:3581–3585. doi: 10.1016/j.bmcl.2009.04.139. [DOI] [PubMed] [Google Scholar]
- 63.Korf M, Jarczak D, Beger C, Manns MP, Kruger M. Inhibition of hepatitis C virus translation and subgenomic replication by siRNAs directed against highly conserved HCV sequence and cellular HCV cofactors. J Hepatol. 2005;43:225–234. doi: 10.1016/j.jhep.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 64.Wang Q, Contag CH, Ilves H, Johnston BH, Kaspar RL. Small Hairpin RNAs Efficiently Inhibit Hepatitis C IRES-Mediated Gene Expression in Human Tissue Culture Cells and a Mouse Model. Mol Ther. 2005;12:562–568. doi: 10.1016/j.ymthe.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 65.Hamazaki H, Takahashi H, Shimotohno K, Miyano-Kurosaki N, Takaku H. Inhibition of hcv replication in HCV replicon by shRNAs. Nucleosides Nucleotides Nucleic Acids. 2006;25:801–805. doi: 10.1080/15257770600726091. [DOI] [PubMed] [Google Scholar]
- 66.Ray RB, Kanda T. Inhibition of HCV replication by small interfering RNA. Methods Mol Biol. 2009;510:251–262. doi: 10.1007/978-1-59745-394-3_19. [DOI] [PubMed] [Google Scholar]
- 67.Kanda T, Steele R, Ray R, Ray RB. Small interfering RNA targeted to hepatitis C virus 5′ nontranslated region exerts potent antiviral effect. J Virol. 2007;81:669–676. doi: 10.1128/JVI.01496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prabhu R, Garry RF, Dash S. Small interfering RNA targeted to stem-loop II of the 5′ untranslated region effectively inhibits expression of six HCV genotypes. Virol J. 2006;3:100. doi: 10.1186/1743-422X-3-100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Chevalier C, Saulnier A, Benureau Y, Flechet D, Delgrange D, Colbere-Garapin F, Wychowski C, Martin A. Inhibition of hepatitis C virus infection in cell culture by small interfering RNAs. Mol Ther. 2007;15:1452–1462. doi: 10.1038/sj.mt.6300186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ilves H, Kaspar RL, Wang Q, Seyhan AA, Vlassov AV, Contag CH, Leake D, Johnston BH. Inhibition of hepatitis C IRES-mediated gene expression by small hairpin RNAs in human hepatocytes and mice. Ann N Y Acad Sci. 2006;1082:52–55. doi: 10.1196/annals.1348.060. [DOI] [PubMed] [Google Scholar]
- 71.Vlassov AV, Korba B, Farrar K, Mukerjee S, Seyhan AA, Ilves H, Kaspar RL, Leake D, Kazakov SA, Johnston BH. shRNAs targeting hepatitis C: effects of sequence and structural features, and comparision with siRNA. Oligonucleotides. 2007;17:223–236. doi: 10.1089/oli.2006.0069. [DOI] [PubMed] [Google Scholar]
- 72.Khaliq S, Jahan S, Pervaiz A, Ali Ashfaq U, Hassan S. Down-regulation of IRES containing 5′UTR of HCV genotype 3a using siRNAs. Virol J. 2011;8:221. doi: 10.1186/1743-422X-8-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ryu KJ, Lee SW. Identification of the most accessible sites to ribozymes on the hepatitis C virus internal ribosome entry site. J Biochem Mol Biol. 2003;36:538–544. doi: 10.5483/bmbrep.2003.36.6.538. [DOI] [PubMed] [Google Scholar]
- 74.Pudi R, Ramamurthy SS, Das S. A peptide derived from RNA recognition motif 2 of human la protein binds to hepatitis C virus internal ribosome entry site, prevents ribosomal assembly, and inhibits internal initiation of translation. J Virol. 2005;79:9842–9853. doi: 10.1128/JVI.79.15.9842-9853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fontanes V, Raychaudhuri S, Dasgupta A. A cell-permeable peptide inhibits hepatitis C virus replication by sequestering IRES transacting factors. Virology. 2009;394:82–90. doi: 10.1016/j.virol.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dasgupta A, Das S, Izumi R, Venkatesan A, Barat B. Targeting internal ribosome entry site (IRES)-mediated translation to block hepatitis C and other RNA viruses. FEMS Microbiol Lett. 2004;234:189–199. doi: 10.1016/j.femsle.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 77.Maraia RJ, Bayfield MA. The La protein-RNA complex surfaces. Mol Cell. 2006;21:149–152. doi: 10.1016/j.molcel.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 78.Ali N, Siddiqui A. The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc Natl Acad Sci U S A. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. Role of La autoantigen and polypyrimidine tract-binding protein in HCV replication. Virology. 2005;335:72–86. doi: 10.1016/j.virol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 80.Pudi R, Srinivasan P, Das S. La protein binding at the GCAC site near the initiator AUG facilitates the ribosomal assembly on the hepatitis C virus RNA to influence internal ribosome entry site-mediated translation. J Biol Chem. 2004;279:29879–29888. doi: 10.1074/jbc.M403417200. [DOI] [PubMed] [Google Scholar]
- 81.Shirasaki T, Honda M, Mizuno H, Shimakami T, Okada H, Sakai Y, Murakami S, Wakita T, Kaneko S. La protein required for internal ribosome entry site-directed translation is a potential therapeutic target for hepatitis C virus replication. J Infect Dis. 2010;202:75–85. doi: 10.1086/653081. [DOI] [PubMed] [Google Scholar]
- 82.Lee SJ, Hyun S, Kieft JS, Yu J. An approach to the construction of tailor-made amphiphilic peptides that strongly and selectively bind to hairpin RNA targets. J Am Chem Soc. 2009;131:2224–2230. doi: 10.1021/ja807609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bradford S, Cowan JA. Catalytic metallodrugs targeting HCV IRES RNA. Chem Commun (Camb) 2012;48:3118–3120. doi: 10.1039/c2cc17377h. [DOI] [PubMed] [Google Scholar]
- 84.Das S, Ott M, Yamane A, Tsai W, Gromeier M, Lahser F, Gupta S, Dasgupta A. A small yeast RNA blocks hepatitis C virus internal ribosome entry site (HCV IRES)-mediated translation and inhibits replication of a chimeric poliovirus under translational control of the HCV IRES element. J Virol. 1998;72:5638–5647. doi: 10.1128/jvi.72.7.5638-5647.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ray PS, Das S. Inhibition of hepatitis C virus IRES-mediated translation by small RNAs analogous to stem-loop structures of the 5′-untranslated region. Nucleic Acids Res. 2004;32:1678–1687. doi: 10.1093/nar/gkh328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li X, Mueller S, Wimmer E. Inhibition of hepatitis C virus IRES-mediated translation by oligonucleotides. Virus Res. 2009;146:29–35. doi: 10.1016/j.virusres.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 87.Wang W, Preville P, Morin N, Mounir S, Cai W, Siddiqui MA. Hepatitis C viral IRES inhibition by phenazine and phenazine-like molecules. Bioorg Med Chem Lett. 2000;10:1151–1154. doi: 10.1016/s0960-894x(00)00217-1. [DOI] [PubMed] [Google Scholar]
- 88.Jefferson EA, Seth PP, Robinson DE, Winter DK, Miyaji A, Osgood SA, Swayze EE, Risen LM. Biaryl guanidine inhibitors of in vitro HCV-IRES activity. Bioorg Med Chem Lett. 2004;14:5139–5143. doi: 10.1016/j.bmcl.2004.07.066. [DOI] [PubMed] [Google Scholar]
- 89.Gooding KB, Higgs R, Hodge B, Stauffer E, Heinz B, McKnight K, Phipps K, Shapiro M, Winkler M, Ng WL, Julian RK. High throughput screening of library compounds against an oligonucleotide substructure of an RNA target. J Am Soc Mass Spectrom. 2004;15:884–892. doi: 10.1016/j.jasms.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 90.Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, Ranken R, Massire C, Sampath R, Ecker DJ, Swayze EE, Griffey RH. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J Med Chem. 2005;48:7099–7102. doi: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- 91.Baugh C, Wang S, Li B, Appleman JR, Thompson PA. SCAN--a high-throughput assay for detecting small molecule binding to RNA targets. J Biomol Screen. 2009;14:219–229. doi: 10.1177/1087057108330111. [DOI] [PubMed] [Google Scholar]
- 92.Berry KE, Peng B, Koditek D, Beeman D, Pagratis N, Perry JK, Parrish J, Zhong W, Doudna JA, Shih IH. Optimized high-throughput screen for hepatitis C virus translation inhibitors. J Biomol Screen. 2011;16:211–220. doi: 10.1177/1087057110391665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhou S, Rynearson KD, Ding K, Brunn ND, Hermann T. Screening for inhibitors of the hepatitis C virus internal ribosome entry site RNA. Bioorg Med Chem. 2013;21:6139–6144. doi: 10.1016/j.bmc.2013.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Douthwaite S. Structure-activity relationships of ketolides vs. macrolides. Clin Microbiol Infect. 2001;7(Suppl 3):11–17. [PubMed] [Google Scholar]
- 95.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem Int Ed Engl. 2004;43:3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 96.Aoki H, Ke L, Poppe SM, Poel TJ, Weaver EA, Gadwood RC, Thomas RC, Shinabarger DL, Ganoza MC. Oxazolidinone antibiotics target the P site on Escherichia coli ribosomes. Antimicrob Agents Chemother. 2002;46:1080–1085. doi: 10.1128/AAC.46.4.1080-1085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou CC, Swaney SM, Shinabarger DL, Stockman BJ. 1H nuclear magnetic resonance study of oxazolidinone binding to bacterial ribosomes. Antimicrob Agents Chemother. 2002;46:625–629. doi: 10.1128/AAC.46.3.625-629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seth PP, Jefferson EA, Griffey RH, Swayze EE. WO2004050035. Benzimidazoles and analogs thereof as antivirals. 2004
- 99.Liu S, Nelson CA, Xiao L, Lu L, Seth PP, Davis DR, Hagedorn CH. Measuring antiviral activity of benzimidazole molecules that alter IRES RNA structure with an infectious hepatitis C virus chimera expressing Renilla luciferase. Antiviral Res. 2011;89:54–63. doi: 10.1016/j.antiviral.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou Y, Gregor VE, Sun Z, Ayida BK, Winters GC, Murphy D, Simonsen KB, Vourloumis D, Fish S, Froelich JM, Wall D, Hermann T. Structure-guided discovery of novel aminoglycoside mimetics as antibacterial translation inhibitors. Antimicrob Agents Chemother. 2005;49:4942–4949. doi: 10.1128/AAC.49.12.4942-4949.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dibrov SM, Ding K, Brunn ND, Parker MA, Bergdahl BM, Wyles DL, Hermann T. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc Natl Acad Sci U S A. 2012;109:5223–5228. doi: 10.1073/pnas.1118699109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beales LP, Rowlands DJ, Holzenburg A. The internal ribosome entry site (IRES) of hepatitis C virus visualized by electron microscopy. RNA. 2001;7:661–670. doi: 10.1017/s1355838201001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Perard J, Leyrat C, Baudin F, Drouet E, Jamin M. Structure of the full-length HCV IRES in solution. Nat Commun. 2013;4:1612. doi: 10.1038/ncomms2611. [DOI] [PubMed] [Google Scholar]
- 104.Lukavsky PJ. Structure and function of HCV IRES domains. Virus Res. 2009;139:166–171. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kieft JS, Zhou K, Grech A, Jubin R, Doudna JA. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat Struct Biol. 2002;9:370–374. doi: 10.1038/nsb781. [DOI] [PubMed] [Google Scholar]
- 106.Collier AJ, Gallego J, Klinck R, Cole PT, Harris SJ, Harrison GP, Aboul-Ela F, Varani G, Walker S. A conserved RNA structure within the HCV IRES eIF3-binding site. Nat Struct Biol. 2002;9:375–380. doi: 10.1038/nsb785. [DOI] [PubMed] [Google Scholar]
- 107.Lukavsky PJ, Otto GA, Lancaster AM, Sarnow P, Puglisi JD. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat Struct Biol. 2000;7:1105–1110. doi: 10.1038/81951. [DOI] [PubMed] [Google Scholar]
- 108.Klinck R, Westhof E, Walker S, Afshar M, Collier A, Aboul-Ela F. A potential RNA drug target in the hepatitis C virus internal ribosomal entry site. RNA. 2000;6:1423–1431. doi: 10.1017/s1355838200000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 110.Gallegos-Orozco JF, Arenas JI, Vargas HE, Kibler KV, Wilkinson JK, Nowicki M, Radkowski M, Nasseri J, Rakela J, Laskus T. Selection of different 5′ untranslated region hepatitis C virus variants during post-transfusion and posttransplantation infection. J Viral Hepat. 2006;13:489–498. doi: 10.1111/j.1365-2893.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 111.Parker MA, Satkiewicz E, Hermann T, Bergdahl BM. An efficient new route to dihydropyranobenzimidazole inhibitors of HCV replication. Molecules. 2011;16:281–290. doi: 10.3390/molecules16010281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ogle JM, Carter AP, Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci. 2003;28:259–266. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 113.Dibrov SM, Parker MA, Bergdahl BM, Hermann T. Crystal structure of a benzimidazole hepatitis C virus inhibitor free and in complex with the viral RNA target. J Chem Cryst. 2013;43:235–239. doi: 10.1007/s10870-013-0410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Puglisi JD, Chen L, Frankel AD, Williamson JR. Role of RNA structure in arginine recognition of TAR RNA. Proc Natl Acad Sci U S A. 1993;90:3680–3684. doi: 10.1073/pnas.90.8.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weiss MA, Narayana N. RNA recognition by arginine-rich peptide motifs. Biopolymers. 1998;48:167–180. doi: 10.1002/(SICI)1097-0282(1998)48:2<167::AID-BIP6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 116.Hoffman MM, Khrapov MA, Cox JC, Yao J, Tong L, Ellington AD. AANT: the Amino Acid-Nucleotide Interaction Database. Nucleic Acids Res. 2004;32:D174–D181. doi: 10.1093/nar/gkh128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kondo J, Westhof E. Classification of pseudo pairs between nucleotide bases and amino acids by analysis of nucleotide-protein complexes. Nucleic Acids Res. 2011;39:8628–8637. doi: 10.1093/nar/gkr452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Walberer BJ, Cheng AC, Frankel AD. Structural diversity and isomorphism of hydrogen-bonded base interactions in nucleic acids. J Mol Biol. 2003;327:767–380. doi: 10.1016/s0022-2836(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 119.Klosterman PS, Hendrix DK, Tamura M, Holbrook SR, Brenner SE. Three-dimensional motifs from the SCOR, structural classification of RNA database: extruded strands, base triples, tetraloops and U-turns. Nucleic Acids Res. 2004;32:2342–2352. doi: 10.1093/nar/gkh537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee JC, Gutell RR. Diversity of base-pair conformations and their occurrence in rRNA structure and RNA structural motifs. J Mol Biol. 2004;344:1225–1249. doi: 10.1016/j.jmb.2004.09.072. [DOI] [PubMed] [Google Scholar]
- 121.Abu Almakarem AS, Petrov AI, Stombaugh J, Zirbel CL, Leontis NB. Comprehensive survey and geometric classification of base triples in RNA structures. Nucleic Acids Res. 2012;40:1407–1423. doi: 10.1093/nar/gkr810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paek KY, Kim CS, Park SM, Kim JH, Jang SK. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol. 2008;82:12082–12093. doi: 10.1128/JVI.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]