

Abstract
We have previously reported that the 6-aminoquinolone chemotype is a privileged scaffold to obtain antibacterial and antiviral agents. Herein we describe the design, synthesis, enzymatic and cellular characterization of new 6-aminoquinolone derivatives as potent inhibitors of NS5B polymerase, an attractive and viable therapeutic target to develop safe anti-HCV agents. The 6-amino-7-[4-(2-pyridinyl)-1-piperazinyl] quinolone derivative 8 proved to be the best compound of this series, exhibiting IC50 value of 0.069 µM against NS5B polymerase and selective antiviral effect (EC50 = 3.03 µM, EC90 =13.5 µM) coupled with the absence of any cytostatic effect (CC50 >163 µM, SI >54) in Huh-9-13 cells carrying a HCV genotype 1b, as measured by MTS assay. These results indicate that the 6-aminoquinolone scaffold is worthy of further investigation in the context of NS5B-targeted HCV drug discovery programs.
Keywords: Hepatitis C virus, RNA-dependent RNA polymerase, quinolones, NS5B inhibitors, induced-fit docking
Introduction
Hepatitis C virus (HCV), the causative agent of chronic hepatitis C, infects nearly 170 million people worldwide.1 Chronic HCV infection results in major liver disorders such as cirrhosis and hepatocellular carcinoma ultimately necessitating liver transplantation.1,2 Among the six major genotypes, HCV genotype 1b causes the majority of HCV infections in industrialized countries and is the most refractory to therapy.2 To date there is no vaccine available.3 The current combination therapy consists of ribavirin (RBV), pegylated interferon α (pegIFN-α) and for genotype 1 since mid-2011 a NS3/4A protease inhibitor, i.e. telaprevir or boceprevir.4 Although the triple combination therapy induces an improved sustained virological response (SVR), it suffers from long-term toxic effects, high costs, and increased pill-burden.5 Therefore an alternative pegIFN-α/RBV-free treatment, based on a combination of several direct-acting antivirals (DAAs) with different mode of action, is still a need of primary importance.
HCV is a small, enveloped, single-stranded RNA-(+) virus that belongs to the genus Hepacivirus, of the Flaviviridae family.6 Its RNA genome encodes a polyprotein precursor of about 3000 aminoacids, which is processed by cellular and viral proteases to yield four structural (S) and six non-structural (NS) proteins.6 Among the NS proteins, NS5B is a key enzyme for HCV replication with a RNA-dependent RNA polymerase (RdRp) function, thus representing an attractive target for the development of selective antiviral agents.7 NS5B inhibitors are divided into nucleoside inhibitors (NIs) that bind to the active site and non-nucleoside inhibitors (NNIs) that bind to one of the five identified allosteric sites.8 The allosteric sites are currently classified as follows: (i) thumb site I (TSI), (ii) thumb site II (TSII), (iii) palm site I (PSI), (iv) palm site II (PSII), and (v) palm site III (PSIII).8a In the need to identify new anti-HCV drugs targeting NS5B, pharmaceutical companies and academic groups are making tremendous efforts to discover potent and selective inhibitors8b to be combined in a pegIFN-α/RBV-free therapy based on the use of different DAAs. Anyway, although a broad range of NIs and NNIs have been reported in literature, drugs active against NS5B polymerase have not yet been approved by FDA. Regarding the NS5B NIs, sofosbuvir (PSI-7977) and mericitabine (RG7128) are the most advanced drug candidates in clinical trials, whilst filibuvir (PF-00868554) and setrobuvir (ANA598) are the most advanced NNIs.9 In particular, the combination of sofosbuvir with an NS5A inhibitor is the most promising interferon free regimen reaching a SVR >90%.10
During the last few years our research group has been involved in HCV drug discovery, and in particular in the identification of novel chemotypes endowed with anti-NS5B and/or anti-HCV activity.11 As a continuation of this endeavor, we herein report the design and synthesis of new 6-aminoquinolone derivatives as potential anti-HCV compounds. Since many years, we have focused on the 6-aminoquinolone chemotype as a privileged scaffold to obtain novel chemotherapeutic agents. This research work started off by replacing for the first time, the usual fluorine atom at the C-6 position of fluoroquinolone antimicrobial drugs with an amino group.12 This chemical modification produced potent antibacterial agents which were less phototoxic than the corresponding fluorinated compounds.13 Subsequently, we demonstrated that suitable derivatization of the 6-aminoquinolone scaffold resulted in numerous derivatives endowed with a wide range of antiviral activities, such as anti-HIV,14 anti-HCMV15 and anti-HPV.16
Given this background, our attention was caught by a recent publication by Kumar et al.,17 describing the discovery of quinolone benzylesters as a new class of NS5B TSII-NNIs (e.g., compounds 1 and 2, Figure 1A).
Figure 1.

A) Chemical structures of 1 and 2 as representative quinolones TSII-NNIs. B) Schematic representation of the interaction between NS5B polymerase and compound 1 (PDB ID 3PHE). NS5B residues lying within a distance of 4 Å from the bound ligand are shown and color coded as follows: green-hydrophobic; purple-basic; cyan-polar. The TSII pocket is displayed with a line, exhibiting the color of the nearest protein residue. The gap in the line shows the opening of the pocket. Specific interactions between ligand atoms and protein residues are marked with lines: pink, H-bonds to protein backbone; green, π-π stacking interactions. Ligand atoms that are exposed to solvent are marked with gray spheres.
The reported quinolones showed potent NS5B inhibitory activity in the submicromolar range, binding to the NS5B TSII, as confirmed by the co-crystal structure of ligand 1 bound to NS5B polymerase (PDB ID 3PHE)17 (Figure 1B). This structure revealed the formation of two hydrogen bonds between the ligand carbonyl oxygen atoms and the backbone amide NH atoms of Ser476 and Tyr477. The benzyl ester occupied a hydrophobic pocket mainly lined by Leu419, Met423 and Trp528, whereas the N-1 substituted benzyl group filled a pocket defined by Leu419, Val485, Ala486, Leu489 and Leu497, establishing hydrogen-bond interaction with Leu497 through the sulfone moiety. Finally, the methylpiperazine substituent was largely solvent exposed (Figure 1B). The compounds reported by Kumar et al. also demonstrated the ability to inhibit HCV replication in subgenomic replicon system at micromolar concentrations, with derivative 2 showing the best biological profile (IC50 (NS5B) = 0.008 µM; EC50 (replicon) = 0.2 µM; CC50 = 20 µM).17
Based on these observations and given our long-standing experience with the 6-aminoquinolone chemotype, we explored the design of a series of new 6-aminoquinolone derivatives, in an effort to obtain more potent anti-NS5B inhibitors and/or HCV replication inhibitors. In particular, keeping the C-6 amino substituent constant, we designed compounds 3–5 (Scheme 1 and Table 1) in which i) the C-7 position of quinolone scaffold was occupied by a chlorine atom as a suitable replacement of the methoxyl group present in compound 2, and ii) the N-1 and C-3 positions were functionalized with some benzyl substituents already reported as the best fragments in the known anti-NS5B quinolone series. Compounds 6 and 7 were instead designed by keeping the 4-chlorobenzyl moiety constant at both N-1 and C-3 positions, and by replacing the chlorine atom with a piperazine or a methylpiperazine, respectively. These modifications were pursued considering that the latter substituents granted a better solubility in known anti-HCV quinolones such as compound 1. Finally, to better explore the role of the C-7 substituent in this new series of 6-aminoquinolones, we designed derivatives 8–10, where the 1-(2-pyridinyl)piperazine, 2-(1-piperazinyl)-1,3-benzothiazole, and 1-[3-(trifluoromethyl)phenyl]piperazine fragments were placed at the C-7 position, while maintaining the 4-chlorobenzyl substituent at N-1 and C-3 positions (Scheme 1 and Table 1). The three arylpiperazines were used as C-7 substituents in this series of compounds for two main reasons: the C-7 substituents resulted in potent anti-HIV activity,14 and the synthetic pathway to obtain arylpiperazinyl quinolones was well known to us.
Scheme 1.

Synthesis of Target Compounds 3–10a
aReagents and conditions: i) BnNH2, Et2O/EtOH 4:1, rt; ii) K2CO3, dry DMF, 80 °C; iii) 8 N HCl, MW, 120 °C, 15 bar; iv) BnCl, K2CO3, DMF, 60 °C; v) piperazines, Et3N, MeCN, 80 °C; vi) aqueous NaCl 3.5%, Fe-powder, DMF, reflux.
Table 1.
Estimated Ki values, inhibitory activity on NS5B, anti-HCV activity and cytotoxicity of the studied compounds
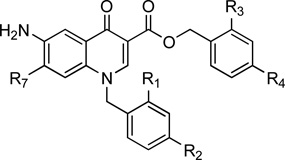 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cpds | R1 | R2 | R3 | R4 | R7 | AutoDock Estimated Ki |
NS5B functional assay |
Replicon assay on Huh5-2 | |||
| Ki (µM)a |
IC50 (µM)b |
EC50 (µM)c | EC90 (µM)d | CC50 (µM)e | SIf | ||||||
| 3 | F | CF3 | CH3 | H | Cl | 0.340 | 1.41±0.2 | 1.5 | NDg | 10.5 | 6.8 |
| 4 | F | CF3 | H | CH3 | Cl | 0.255 | 0.154±0.015 | 72.3 | >192.3 | >192.3 | >2.5 |
| 5 | H | Cl | H | Cl | Cl | 0.249 | NDg | >200 | >200 | >200 | NDg |
| 6 | H | Cl | H | Cl | 0.096 | 0.067±0.02 | 1.7 | NDg | 3.7 | 2.3 | |
| 7 | H | Cl | H | Cl | 0.070 | 0.040±0.004 | 2.7 | NDg | 2.8 | 1.2 | |
| 8 | H | Cl | H | Cl | 0.043 | 0.069±0.002 | 2.2 | 13.5 | >244 | >54 | |
| 9 | H | Cl | H | Cl | 0.093 | 0.138 ± 0.023 | 8.6 | NDg | >149 | >17 | |
| 10 | H | Cl | H | Cl | 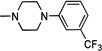 |
0.178 | 6.132 ± 1.6 | 23.8 | 94.6 | >147 | >6 |
| 27 | 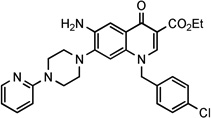 |
2.49 | 3.06± 0.6 | 31.3 | NDg | >193 | 3.06 | ||||
| 28 | 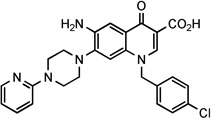 |
NCh | 23.7 ± 6 | 7.3 | NDg | >204 | >28 | ||||
| 2 | 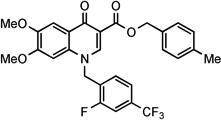 |
0.652 | 0.211± 0.01 (0.008)i |
2.02 | 4.7 | 42.3 | 21 | ||||
AutoDock-predicted inhibition constant (Ki) for each selected IFD complex
IC50 = concentration of compound that inhibits 50% enzyme activity in vitro. The IC50 values of the compounds were determined from dose-response curves employing 8–12 concentrations of the indicated compounds in duplicate in two independent experiments ± SD.
EC50 = the effective concentration required to inhibit virus replication by 50%. The reported values represent the means of data derived at least from three independent experiments.
EC90 = the effective concentration required to inhibit virus replication by 90%. The reported value represents the means of data derived at least from three independent experiments.
CC50 = is the concentration of compound exhibiting 50% antimetabolic effect as evaluated by the MTS assay. The reported value represents the means of data derived at least from three independent experiments.
SI = selectivity index (ratio of CC50 to EC50).
ND = not determined.
NC= not calculated: the IFD top pose of this 6-aminoquinolone derivative showed that the compound was unable to properly occupy TSI.
IC50 value reported by Kumar et al.17
Results and discussion
Induced-fit docking studies
Before initiating chemical synthesis of the described quinolone-based compounds, we performed induced-fit docking (IFD) studies of derivatives 3–10 using Prime and Glide programs.18 Here, receptor flexibility upon ligand binding was taken into account in an attempt to describe the inhibitor binding mode (see Experimental Section). The compounds were prepared using the LigPrep utility19 and docked by the IFD procedure against the crystal structure of NS5B in complex with inhibitor 1 (PDB ID 3PHE).17 In order to validate the IFD performance, test calculations using 1 were carried out, extracting the ligand from the corresponding NS5B complex and then docking it back into the allosteric pocket of the enzyme crystal structure. The top IFD conformation of 1 agreed well with its experimental binding conformation, showing a root-mean square deviation (RMSD) value of 0.7 Å. Moreover, IFD of the known TSII-NNI 2 was carried out as well, since this inhibitor was later used as reference compound in our biological assays, being the quinolone derivative with the highest anti-NS5B potency reported in literature.17 The best IFD structure of 2 showed a ligand binding conformation resembling the conformation observed for compound 1.
IFD of compounds 3–10 into TSII generated a number of NS5B/ligand complexes, and only the best scoring pose for each ligand was retained. This revealed that all the 6-aminoquinolones exhibited similar ligand orientation within the binding pocket, and that they may potentially interact with the NS5B residues of TSII in a comparable fashion to the known TSII-NNIs 1 and 2, as discussed below. For instance, the top-ranked IFD orientation of 6-aminoquinolone 8 is shown in Figure 2, together with the experimental position of compound 1. Beside the ligand-NS5B interactions already highlighted for compound 1 involving the two benzyl groups and the carbonyl group of the quinolone scaffold (Figure 1B), our derivative was able to establish two additional hydrogen-bond interactions; in particular, the C-6 amino group interacted with the backbone carbonyl of Tyr477, whereas the pyridinyl moiety showed hydrogen-bonding to Asn483 (Figure 2). Furthermore, different from 1, derivative 8 was not able to interact with the Ser476 residue through its carbonyl ester group.
Figure 2.

IFD pose of the designed 6-aminoquinolone 8(green) overlaid with the X-ray conformation of the known quinolone TSII-NNI 1 (magenta). Key NS5B residues are represented as sticks, whereas hydrogen bonds are represented as yellow dashed lines.
Subsequently, inhibition constant (Ki) values were estimated for each compound employing the corresponding top IFD complex and the “epdb” function of AutoDock20 (Table 1). The predicted Ki values suggested that the 6-aminoquinolones harbored high binding affinity for NS5B. In particular, the piperazine-based fragments at the C-7 position of the quinolone scaffold were preferred relative to the chlorine atom (compounds 3–5 vs 6–10), with compounds 7 and 8 predicted to be the most potent derivatives within the series (Table 1). Paradoxically though, the AutoDock estimated inhibitory activity of 2, did not agree well with its experimentally reported IC50 value (estimated Ki = 0.652 µM vs experimental IC50 = 0.008 µM). Taken together, the in silico data suggested that, in analogy with the known quinolones, the newly designed compounds 3–10 may act as potent TSII-NNIs, thus providing an incentive to synthesize and test them as potential inhibitors of NS5B polymerase and HCV replication.
Synthesis of the target 6-aminoquinolones
The 6-aminoquinolones 3–10 were prepared through the canonical cycloaracylation procedure, as shown in Scheme 1. In particular, acrylate 1112 was reacted with the appropriate benzylamine in a 4:1 Et2O/EtOH mixture at room temperature (rt) to give the intermediates 12 and 13, which were then cyclized to quinolone esters 14 and 15, using K2CO3 in dry DMF at 80 °C, and subsequently hydrolyzed in aqueous 8 N HCl to the corresponding acids 16 and 17 by using microwave (MW) irradiation at 120 °C for 130 min. The MW irradiation showed some advantages than conventional heating: i) reduction of the reaction time from days to few hours; ii) improvement of yield reaching values close to 90%. Quinolone acids 16 and 17 so obtained were reacted with the appropriate benzylchloride in DMF in presence of K2CO3 at 60 °C, obtaining the key nitro benzylester synthons 18–20 which were converted into their amino target derivatives 3–5 using Fe-powder in a mixture of DMF/aqueous 3.5% NaCl at reflux.21 Although the yields of this reduction were very low (from 10% to 40%), the neutral reaction conditions were necessary to grant the reduction of the amino group avoiding the hydrolysis of the C-3 benzyl ester. The key intermediate 20 was further functionalized with the appropriate piperazine side chains, in dry MeCN in presence of Et3N at 80 °C, to give the derivatives 21–25. This step suffers from low yield (35–50%) when applied to the preparation of intermediates 23–25. Finally the key nitro derivatives 21–25 were reduced to amino target compounds 6–10, using the same conditions as used for the preparation of compounds 3–5.
The low yields obtained for the preparation of intermediates 23–25 prompted us to explore an alternative synthetic procedure. In particular, the planned alternative synthetic pathway reported in Scheme 2 was conveniently applied, selecting compound 8 as synthetic probe. Thus, the ethyl ester intermediate 15 was initially reacted with the 1-(2-pyridinyl)piperazine in dry DMF at 80 °C obtaining intermediate 26 in good yield (85%); it was then hydrogenated using Raney-Ni as catalyst to the corresponding amino derivative 27, which was hydrolyzed under basic conditions into the corresponding acid intermediate 28 and successively benzylated to the target compound 8 by employing the same conditions as previously used. The benzylation reaction of intermediate 28 afforded only the C-3 benzyl ester in fairly good yield, without any trace of C-6 amino benzylation. This alternative synthetic route permitted the avoidance of neutral reduction condition, which suffers from low yields as described above.
Scheme 2.

Alternative Synthesis of Target Compound 8a
aReagents and conditions: i) Arylpiperazines, DMF, 80 °C; ii) H2, Raney-Ni, DMF, rt, atm pressure; iii) aqueous NaOH 5%, EtOH 1:1, reflux; iv) BnCl, K2CO3, DMF, 60 °C.
Biochemical and Biological Studies
The target compounds 3–10 were evaluated by two methods to determine the anti-NS5B and the anti-HCV activities (Table 1). In addition, intermediates 27 and 28 were also evaluated in order to assess the importance of the C-3 benzyl fragment on NS5B inhibition. The cytotoxicity of all ten compounds was also investigated.
NS5B inhibition assay
The anti-NS5B activity was evaluated by the standard primerdependent elongation reaction employing poly rA/U12 template-primer and recombinant HCV NS5BCΔ21 as previously described.22 Compound 2 was re-synthesized23 and included for comparison as a reference, yielding an IC50 value of 0.211 µM under the assay conditions used, in contrast to the reported value of 0.008 µM.17
This discrepancy between the two IC50 values may be due to several reasons such as differences in the type of assay, reaction conditions employed, and the different strains of the HCV NS5B genotype 1b isolate.
For preliminary screening of anti-NS5B candidates, all compounds were first investigated at 25 µM concentration and compounds exhibiting ≥50% inhibition of NS5B RdRp activity at this concentration were subjected to IC50 determination. With the exception of compound 5, all the molecules satisfied this criterion. Thus, compounds 3, 4, 6–10 were further investigated to evaluate their potency, yielding IC50 values ranging from 0.04 to 6.13 µM (Table 1). Compounds 6–8 turned out to be the most potent derivatives within this 6-aminoquinolone series, exhibiting IC50 values of 0.067 µM, 0.040 µM, and 0.069 µM, respectively. These three compounds were characterized by a 4-chlorobenzyl fragment at both N-1 and C-3 positions, and a 1-piperazinyl (compound 6), a 4-methyl-1-piperazinyl (compound 7) or a 4-(2-pyridinyl)-1-piperazinyl (compound 8) group at C-7 position. With the exception of the inactive compound 5, replacement of the C-7 piperazine-based substituents with a chlorine atom, still exhibited NS5B inhibition in the low micromolar range, although there was a 9-fold decrease in activity (compound 3, IC50 = 1.4 µM; compound 4, IC50 = 0.15 µM). It was clearly evident that the chemical nature of the N-4 piperazine substituent significantly influenced the anti-NS5B activity. In particular, insertion of the benzothiazole fragment at N-4 position of the piperazine ring yielded compound 9 (IC50 = 0.138µM), with a slight decrease in its activity by 2 to 3.5-fold compared to derivatives 6–8. By contrast, introduction of a 3-trifluoromethylphenyl group at the same position of the piperazine moiety proved to be highly detrimental as is evident from compound 10 (IC50 = 6.13 µM), which exhibited ~89 to153-fold decrease in its activity relative to compounds 6–8. In line with the SAR reported for known anti-HCV quinolones,17 replacement of the benzyl ester at the C-3 position in the most potent derivative 8 with an ethyl ester group (compound 27) or a free carboxylic acid (compound 28) produced a significant reduction in potency against NS5B by ~45-fold and 345-fold, respectively. These results strongly suggest that the 6-aminoquinolones exert their anti-HCV activity through binding to the TSII-NNIs pocket, and once more highlight the importance of the C-3 benzyl ester in the quinolones series for NS5B inhibition.
In a head to head comparison, compound 7, the most potent NS5B inhibitor among the 6-aminoquinolone series, exhibited 5-fold higher inhibition than the reference compound 2 under our assay conditions. In order to validate the binding of 6-aminoquinolones to NS5B, we next investigated the dissociation constant (KD) for compound 7 as a representative. We reasoned that since KD value is independent of the enzyme and inhibitor amounts used in the assay, it represents the intrinsic affinity of the ligand for its biological target. The KD for compound 7 was evaluated both by Surface Plasmon Resonance (SPR) and fluorescence quenching assays, and yielded values of 14 µM and 1.2 µM, respectively, confirming the binding of our 6-aminoquinolones to NS5B.
We also compared the experimental IC50 values with the predicted Ki values (Table 1) and found that with the exception of compound 10, a high correlation existed between these two data sets (R2 = 0.83). Of note, AutoDock correctly predicted the piperazine-based 6-aminoquinolones 6–9 as the most potent NS5B inhibitors within this series. Interestingly, we also noticed that, even though the inhibition constant predicted by AutoDock for the reference compound 2 (Ki= 0.652 µM) showed low agreement with the inhibitory activity reported in literature (IC50 = 0.008 µM),17 it agreed well with the anti-NS5B potency obtained in our assay (IC50 = 0.211µM).
Replicon system assay
Compounds 3–10 and intermediates 27 and 28 were investigated for their ability to inhibit HCV replication in Huh-5-2 cells carrying a subgenomic genotype 1b replicon. In parallel, we also investigated the cytostatic effect of the compounds on the same cells type. Towards this goal, we evaluated the compound concentration that inhibits virus replication by 50% (EC50) and the concentration that reduces host cell metabolism by 50% (CC50). These values allowed us to calculate the selectivity index (SI = CC50/EC50), as a measure of the therapeutic potential of each compound in the assay system. All values are reported in Table 1. A preliminary analysis showed that most compounds inhibited HCV replication in the micromolar concentration range. 6-Aminoquinolone derivatives 6, 7, and 8, the most potent compounds in the anti-NS5B assay, inhibited HCV replication with EC50 values between 1.7 µM to 2.7 µM. Compound 9 (EC50 = 8.6 µM) exhibited ~3 to 5-fold decrease in anti-HCV activity compared to derivatives 6–8, while quinolones 10 (EC50 = 23.8 µM) and 27 (EC50 = 31.3 µM) displayed ~12 to 18-fold decrease. Compound 3 also displayed a low micromolar anti-HCV activity (EC50 = 1.5 µM), consistent with its low micromolar anti-NS5B activity; moreover, compound 28 (EC50 = 7.3 µM) was more promising than compound 27 (EC50 = 31.3 µM) in the antiviral assay. Finally, the C-7 chloro derivatives 4 and 5 were found to be neither active nor toxic.
It is important to underline that only compounds that produce significant inhibition of viral replication (>70% inhibition) at concentrations that do not elicit an antimetabolic effect on the host cells (cell viability >90%) can be considered selective inhibitors of HCV replication in the replicon system (hit selection criteria). Thus, derivatives 3, 6 and 7 were less promising because of their antimetabolic effects on the cells, as confirmed by their low CC50 values and selectivity index (SI). By contrast, compounds 8 (EC50 = 2.2 µM, CC50 >244 µM), 9 (EC50 = 8.6 µM, EC50 >169 µM) and 28 (EC50 = 7.3 µM, CC50 >204 µM) exhibited reasonable efficacy as deduced from their low EC50 and high SI values. Of these, derivative 8 was the most promising, as it inhibited HCV replication without any cytostatic effects at all concentrations employed. Furthermore, the 6-aminoquinolone 8 achieved a reasonably low EC90 value of 13.5 µM.
As reported in Table 1, the known quinolone 2 was also assayed in parallel in order to compare our molecules with a structurally related reference compound. Compound 2 (EC50 = 2.02 µM) inhibited HCV replication, although with an EC50 different from that reported by Kumar et al. (i.e., EC50 = 0.23 µM),17 thus exhibiting 10-fold less anti-HCV activity under our assay conditions. However, analysis of the dose-response curve (data not shown) unexpectedly underlined that reference 2 was a non-selective anti-HCV agent. In the context of its antimetabolic effect, our data was comparable to that reported previously. Thus, it was gratifying to find that the presence of the C-6 amino group in association with a C-7 4-(2-pyridinyl)-1-piperazinyl substituent as in compound 8 allowed us to obtain a more selective quinolone derivative than the reference compound.
Considering the promising results obtained for derivative 8, it was selected for further studies with the aim to quantitatively validate its anti-HCV activity. In particular, the compound was tested in Huh-9–13 cells by real-time RT-PCR. The data thus obtained were consistent with the data acquired using Huh-5-2 cells. In fact, 6-aminoquinolone 8 showed a selective antiviral effect (EC50 = 3.03 µM) coupled with the absence of any cytostatic effect as measured by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H–tetrazolium (MTS) assay (CC50 >163 µM). However, an in-depth microscopic analysis of the cells highlighted that quinolone 8 had an impact on the cellular morphology as apparent from the observed cell aggregation. The morphological alterations of the treated cells induced by derivative 8 might suggest that its anti-HCV activity could be in part due to potential pleiotropic effects on the cells.
Conclusions
Given our long-standing experience in design, synthesis and biological characterization of 6-aminoquinolone derivatives, and the anti-HCV activity reported for quinolone-based derivatives by Kumar et al., we have herein described new 6-aminoquinolones functionalized at N-1, C-3 and C-7 positions as potential anti-HCV agents. Biochemical and biological data confirmed that the strategy of arylpiperazine moieties insertion at the C-7 position of the quinolone scaffold was successful in developing potent new anti-HCV NS5B quinolones. In particular, compound 8 emerged as one of the most potent NS5B inhibitors and the most selective anti-HCV derivative within this series.
The knowledge acquired from this study will guide the future structural functionalization of the 6-aminoquinolone scaffold, in search of optimized derivatives endowed with enhanced pharmacological profiles.
Experimental section
Induced-fit docking methodology
The crystal structure of NS5B RdRP complexed with inhibitor 1 (PDB ID 3PHE)17 was retrieved from the RCSB Protein Data Bank and used as target structure for our modeling studies. Water molecules were deleted. Schrödinger Protein Preparation Wizard,24 a workflow designed to ensure chemical correctness and to optimize a protein structure for further analysis, was then used to obtain a satisfactory starting structure. In particular, hydrogen atoms were added and bond orders and charges were assigned; the orientation of hydroxyl groups on Ser, Thr, and Tyr, the side chains of Asn and Gln residues, and the protonation state of His residues were optimized. Steric clashes were relieved by performing a small number of minimization steps, not intended to minimize the system completely. In our study, the minimization (OPLS 2005 force field) was stopped when the RMSD of the non-hydrogen atoms reached 0.30 Å.
Prior to IFD experiments, compounds 2–10 were built using the Schrödinger Maestro interface25 and then submitted to the LigPrep utility,19 which rapidly produces low energy 3D structures taking into account ionization states, tautomers, sterochemistries, and ring conformations at the desired pH. For our study, a pH range of 6–8 was set. The ionization state of all the ligands was double checked using MoKa,26 a software able to accurately compute pKa values. In all the compounds, the 6-amino group was considered in the neutral form (average pKa = 2.0). The distal nitrogen atom of the piperazine (pKa = 8.41) and methylpiperazine rings (pKa = 8.0) of compounds 6 and 7 were both modeled as protonated, whereas the arylpiperazines at the C-7 position of the quinolone scaffold in derivatives 8–10 were considered in the neutral form. The methylpiperazine moiety of compound 1 was also considered as protonated (pKa=8.01).
The IFD protocol developed by Schrödinger18 was then employed for accurate prediction of ligand binding modes and concomitant structural changes in the NS5B receptor. Briefly, this methodology was used for the induced-fit docking of compounds 1–10 using the protein prepared as described above and the following steps (the description below is from the IFD manual):
Initial Glide docking of each ligand using a softened potential (van der Waals radii scaling). By default, a maximum 20 poses per ligand are retained, and by default poses to be retained must have a Coulomb-vdW score less than 100 and an H-bond score less than –0.05;
Prime side-chain prediction for each protein-ligand complex, on residues within a given distance of any ligand pose (6 Å in this study);
Prime minimization of the same set of residues and the ligand for each protein/ligand complex pose. The receptor structure in each pose now reflects an induced fit to the ligand structure and conformation.
Glide redocking of each protein-ligand complex structure within a specified energy of the lowest-energy structure (default 30 kcal/mol). The ligand is now rigorously docked, using default Glide settings, into the induced-fit receptor structure.
Estimation of the binding energy (IFD Score) for each output pose.
In our study, all docking calculation were run in the “Standard Precision” mode of Glide, and the grid box was centered on the crystallographic position of TSII-NNI 1. All of the docked structures were automatically ranked according to the IFD score. RMSD value for 1 was computed by all non- hydrogens atoms. The image representing the best IFD pose of 8 (Figure 2) was prepared using PyMOL.27 Figure 1B was instead generated using the Ligand Interaction Diagram tool of Maestro GUI.25
Computation of ligand inhibition constants
AutoDock’s epdb function20 was then used to estimate the inhibition costant (Ki) of derivatives 2–10. The IFD top pose for each ligand was processed by AutodockTools version 1.5.6, that merged nonpolar hydrogens and assigned Gasteiger charges to each of the eight NS5B/ligand complexes. For each of the NS5B proteins, AutoGrid version 4.2.5 was used to generate separate grid maps for all atom types of the corresponding ligand structure, plus one for the electrostatic interactions. The grid was centered on the ligand, and its dimensions were 50Åx50Åx50Å with a spacing of 0.375Å. Finally, AutoDock version 4.2.5.1 was used in non-docking mode to calculate the ligand Ki value; the calculations were performed selecting the “epdb” utility.
Chemistry
All starting materials were commercially available, unless otherwise indicated. Reagents and solvents were purchased from common commercial suppliers and were used as such. Organic solutions were dried over anhydrous Na2SO4 and concentrated with a rotary evaporator at low pressure. All reactions were routinely checked by thin-layer chromatography (TLC) on silica gel 60F254 (Merck) and visualized by using UV or iodine. Column chromatography separations were carried out on Merck silica gel 60 (mesh 70–230), flash chromatography on Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Büchi Electrotermal model 9100) and are uncorrected. Yields were of purified products and were not optimized. The reactions conducted under microwaves irradiation were carried out employing a microwave oven Biotage Initiator™ 2.0 version 2.3 build 6250. 1H NMR spectra were recorded at 200 or 400 MHz (BrukerAvance DRX-200 or 400, respectively) while 13C NMR spectra were recorded at 100 MHz (BrukerAvance DRX-400). Chemical shifts are given in ppm (δ) relative to TMS. Spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR and the spectral data are consistent with the assigned structures. The purity of the compounds was determined by combustion analysis employing a Fisons elemental analyzer, model EA1108CHN, and data for C, H, and N are within 0.4% of the theoretical values (purity ≥95%).
Ethyl 2-(2,4-dichloro-5-nitrobenzoyl)-3-{[2-fluoro-4-(trifluoromethyl)benzyl]amino}acrylate (12)
To a solution of ethyl 2-(2,4-diclhoro-5-nitrobenzoyl)-3-(dimethylamino)acrylate 1112 (2.00 g, 5.5 mmol) in a mixture of Et2O/EtOH 4:1 (30 mL), 2-fluoro-4-trifluoromethylbenzylamine (1.28 mg, 6.6 mmol) was added dropwise and the solution was stirred at room temperature for 5 min. The formed precipitate was filtered under vacuum and washed with Et2O to give the title compound 12 (1.98 g, 70%) as pale yellow solid, pure by TLC in two eluents (CHCl3/MeOH 95:5 and cyclohexane/EtOAc 50:50): mp 134–135 °C. 1H NMR (200 MHz, DMSO-d6): δ 1.00 (t, J = 7.0 Hz, 3H, CH3), 3.90 (q, J = 7.0 Hz, 2H, OCH2), 4.90 (d, J = 6.2 Hz, 2H, NCH2), 7.50–7.80 (m, 3H, benzyl-H), 8.00 (s, 1H, H-3), 8.05 (s, 1H, H-6), 8.40 (d, J = 14.3 Hz, 1H, acrylic-H), 11.00–11.10 (m, 1H, NH).
Ethyl 3-[(4-chlorobenzyl)amino]-2-(2,4-dichloro-5-nitrobenzoyl)acrylate (13)
Following the procedure described to prepare compound 12 and using the 4-chlorobenzylamine, the title compound 13 was obtained as pale yellow solid, pure by TLC in two eluents (CHCl3/MeOH 95:5 and cyclohexane/EtOAc 50:50), in 98% yield: mp 145–146 °C. 1H NMR (200 MHz, DMSO-d6): δ 1.00 (t, J = 7.0 Hz, 3H, CH3), 3.90 (q, J = 7.0 Hz, 2H, OCH2), 4.75 (d, J = 6.2 Hz, 2H, NCH2), 7.35–7.50 (m, 4H, benzyl-H), 8.00 (s, 1H, H-3), 8.05 (s, 1H, H-6), 8.30 (d, J = 14.6 Hz, 1H, acrylic-H), 11.10 (m, 1H, NH).
Ethyl 7-chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (14)
A mixture of compound 12 (1.95 g, 3.8 mmol) in dry DMF (25 mL) and K2CO3 (1.59 g, 11.5 mmol) was heated under stirring at 80 °C for 3 h. The solvent was concentrated and the mixture was poured into ice/water and acidified with 2N HCl (pH~2). The precipitate so obtained was filtered under vacuum to give the title compound 14 (1.68 g, 93%), pure by TLC in two eluents (CHCl3/MeOH 95:5 and CHCl3/acetone 70:30), as pale yellow solid: mp 273–275 °C. 1H NMR (200 MHz, DMSO-d6): δ 1.30 (t, J = 7.0 Hz, 3H, CH3), 4.30 (q, J = 7.0 Hz, 2H, OCH2), 5.90 (s, 2H, NCH2), 7.35–7.45 (m, 1H, benzyl-H-6), 7.60 (d, J = 8.0 Hz, 1H, benzyl-H-5), 7.80 (d, JH-F= 10.0 Hz, 1H, benzyl-H-3), 8.15 (s, 1H, H-8), 8.80 (s, 1H, H-2), 8.95 (s, 1H, H-5).
Ethyl 7-chloro-1-(4-chlorobenzyl)-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (15)
Following the procedure described to prepare compound 14 and starting from intermediate 13, the title compound 15 was obtained as brown solid, pure by TLC in two eluents (CHCl3/MeOH 95:5 and CHCl3/acetone 70:30), in 95% yield: mp 240–242 °C. 1H NMR (200 MHz, DMSO-d6): δ 1.25 (t, J = 7.0 Hz, 3H, CH3), 4.25 (q, J = 7.0 Hz, 2H, OCH2), 5.70 (s, 2H, NCH2), 7.25–7.35 (m, 2H, benzyl-H-2 and -H-6), 7.35–7.45 (m, 2H, benzyl-H-3 and -H-5), 8.00 (s, 1H, H-8), 8.75 (s, 1H, H-2), 8.90 (s, 1H, H-5).
7-Chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid (16)
In a microwave oven tube, compound 14 (0.50 g, 1.07 mmol) was suspended in 8N HCl (10 mL).The mixture was irradiated at 120 °C for 130 min employing the following experimental parameters: pressure 15 bar, cooling on, FHT on, pre-stirring 30 s, very high absorption. The mixture was diluted with water and the solid obtained was filtered under vacuum to give the title compound 16 (0.39 g, 93%), pure by TLC in two eluents (CHCl3/MeOH 90:10 and CHCl3/MeOH/AcOH 85:14.5:0.5), as pale yellow solid: mp 259–260 °C. 1H NMR (200 MHz, DMSO-d6): δ 6.00 (s, 2H, NCH2), 7.35–7.40 (m, 1H, benzyl-H-6), 7.50 (d, J = 8.0 Hz, 1H, benzyl-H-5), 7.80 (d, JH-F = 10.0 Hz, 1H, benzyl-H-3) 8.30 (s, 1H, H-8), 8.10 (s, 1H, H-2), 9.00 (s, 1H, H-5), 14.00 (bs, 1H, COOH).
7-Chloro-1-(4-chlorobenzyl)-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxilic acid (17)
Following the procedure described to prepare the compound 16 and starting from intermediate 15, the title compound 17 was obtained as pale brown solid, pure by TLC in two eluents (CHCl3/MeOH 90:10 and CHCl3/MeOH/AcOH 85:14.5:0.5), in 90% yield: mp 276–278 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.90 (s, 2H, NCH2), 7.30–7.40 (m, 2H, benzyl-H-2 and -H-6), 7.40–7.50 (m, 2H, benzyl-H-3 and -H-5),8.25 (s, 1H, H-8), 8.90 (s, 1H, H-2), 9.30 (s, 1H, H-5), 14.10 (bs, 1H, COOH).
General procedure of esterification (method A)
To a solution of the quinolone 3-carboxylic acids 16 or 17 (1 mmol) in DMF (30 ml), K2CO3 (2 mmol) and the appropriate benzylchloride (1.5 mmol) were added and the mixture was heated at 60 °C for 24 h. The solvent was concentrated, and the mixture was poured into ice/water and acidified with 2N HCl (pH~1–2) to give a precipitated which was filtered under vacuum. The solid obtained was triturated with EtOH affording compounds 18–20.
2-Methylbenzyl 7-chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (18)
Following the general procedure method A and using the 2-methylbenzylchloride, compound 18 was obtained from 16, in 89% yield as yellow solid: mp 183–185 °C. 1H NMR (200 MHz, DMSO-d6): δ 2.30 (s, 3H, CH3), 5.25 (s, 2H, OCH2), 5.80 (s, 2H, NCH2), 7.10–7.25 (m, 3H, O-benzyl-H), 7.30–7.35 (m, 1H, N-benzyl-H-6), 7.40–7.60 (m, 2H, N-benzyl-H-5 and O-benzyl-H), 7.75 (d, JH-F= 10.0 Hz, 1H, N-benzyl-H-3), 8.10 (s, 1H, H-8), 8.75 (s, 1H, H-2), 8.90 (s, 1H, H-5).
4-Methylbenzyl 7-chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (19)
Following the general procedure method A and using the 4-methylbenzylchloride, compound 19 was obtained from 16, in 75% yield as yellow solid: mp 223–224 °C. 1H NMR (200 MHz, DMSO-d6): δ 2.30 (s, 3H, CH3), 5.20 (s, 2H, OCH2), 5.90 (s, 2H, NCH2), 7.10–7.20 (m, 2H, O-benzyl-H-3 and -H-5), 7.30–7.45 (m, 3H, O-benzyl-H-2, -H-6 and N-benzyl-H-6), 7.55 (d, J = 7.0 Hz, 1H, N-benzyl-H-5), 7.75 (d, JH-F = 10.0 Hz, 1H, N-benzyl H-3), 8.10 (s, 1H, H-8), 8.75 (s, 1H, H-2), 8.95 (s, 1H, H-5).
4-Chlorobenzyl 7-chloro-1-(4-chlorobenzyl)-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (20)
Following the general procedure method A and using the 4-chlorobenzylchloride, compound 20 was obtained from 17, in 90% yield as pale brown solid: mp 220–221 °C. 1H NMR (200 MHz, DMSO-d6): δ 5.30 (s, 2H, OCH2), 5.80 (s, 2H, NCH2), 7.30–7.60 (m, 8H, N,O-benzyl-H), 8.10 (s, 1H, H-8), 8.80 (s, 1H, H-2), 9.00 (s, 1H, H-5).
General procedure of coupling with piperazines (method B)
A mixture of compound 20 (1 mmol) in dry CH3CN (20 mL), Et3N (3.0 mmol) and the appropriate piperazine (1.5 mmol) was stirred at reflux for 3–20h. The solvent was then concentrated to half volume and after cooling the precipitated so obtained was collected by filtration under vacuum and purified, if necessary, by the method below reported, affording compounds 21–25.
4-Chlorobenzyl 1-(4-chlorobenzyl)-6-nitro-4-oxo-7-(1-piperazinyl)-1,4-dihydro-3-quinolinecarboxylate (21)
Following the general procedure method B and using piperazine (reaction time 3h), after treatment with Et2O, compound 21 was obtained in 90% yield as a yellow solid: mp 140–143 °C. 1H NMR (200 MHz, DMSO-d6): δ 2.75–3.00 and 3.25–3.50 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.70 (s, 2H, NCH2), 7.10 (s, 1H, H-8), 7.30–7.60 (m, 8H, N,O-benzyl-H), 8.65 (s, 1H, H-2), 9.00 (s, 1H, H-5).
4-Chlorobenzyl 1-(4-chlorobenzyl)-7-(4-methyl-1-piperazinyl)-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (22)
Following the general procedure method B and using 4-methylpiperazine (reaction time 4h), compound 22 was obtained in 71% yield as a yellow crystalline solid: mp 230–232 °C. 1H NMR (200 MHz, CDCl3): δ 2.40 (s, 3H, CH3), 2.45–2.55 and 2.95–3.05 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.40 (s, 2H, NCH2), 6.60 (s, 1H, H-8), 7.10–7.20 (m, 2H, N-benzyl-H-2 and -H-6), 7.35–7.45 (m, 4H, O-benzyl-H), 7.45–7.55 (m, 2H, N-benzyl-H-3 and -H-5), 8.55 (s, 1H, H-2), 8.80 (s, 1H, H-5).
4-Chlorobenzyl 1-(4-chlorobenzyl)-6-nitro-4-oxo-7-[4-(2-pyridinyl)-1-piperazinyl]-1,4-dihydro-3-quinolinecarboxylate (23)
Following the general procedure method B and using 1-(2-pyridinyl)piperazine (reaction time 20h), after purification by flash chromatography eluting with CHCl3/MeOH (97:3), compound 23 was obtained in 50% yield as a yellow solid: mp 199–201 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.10–3.25 and 3.50–3.70 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.75 (s, 2H, NCH2), 6.70 (dd, J = 5.0 and 6.8 Hz, 1H, pyridine-H), 6.85 (d, J = 9.0 Hz, 1H, pyridine-H), 7.10 (s, 1H, H-8), 7.30–7.60 (m, 9H, N,O-benzyl-H and pyridine-H), 8.20 (dd, J = 2.8 and 5.0 Hz, 1H, pyridine-H), 8.60 (s, 1H, H-2), 9.00 (s, 1H, H-5).
4-Chlorobenzyl 7-[4-(1,3-benzothiazol-2-yl)-1-piperazinyl]-1-(4-chlorobenzyl)-6-nitro-4-oxo-1,4-dihydro-3-quinolinecarboxylate (24)
Following the general procedure method B, and using the 2-(1-piperazinyl)-1,3-benzothiazole (reaction time 18h), after purification by flash chromatography eluting with CHCl3/MeOH (97:3), compound 24 was obtained in 35% yield as yellow solid: mp 200–202 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15–3.25 and 3.65–3.75 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.70 (s, 2H, NCH2), 7.10–7.15 (m, 2H, H-8 and benzothiazole-H), 7.25–7.30 (m, 1H, benzothiazole-H), 7.35–7.55 (m, 9H, benzothiazole-H and N,O-benzyl-H), 7.75–7.80 (m, 1H, benzothiazole-H), 8.60 (s, 1H, H-2), 9.00 (s, 1H, H-5).
4-Chlorobenzyl 1-(4-chlorobenzyl)-6-nitro-4-oxo-7-{4-[3-(trifluoromethyl)phenyl]-1-piperazinyl}-1,4-dihydro-3-quinolinecarboxylate (25)
Following the general procedure method B, and using 1-[3-(trifluoromethyl)phenyl]piperazine (reaction time 16 h), after purification by flash chromatography eluting with CHCl3/MeOH (97:3), compound 25 was obtained in 45% yield as yellow solid: mp 213–216 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15–3.25 and 3.30–3.40 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.75 (s, 2H, NCH2), 7.05 (s, 1H, H-8), 7.10 (d, J = 8.0 Hz, 1H, phenyl-H), 7.20–7.25 (m, 2H, phenyl-H), 7.35–7.40 (m, 2H, benzyl-H), 7.35–7.45 (m, 5H, benzyl-H and phenyl-H), 7.55–7.60 (m, 2H, benzyl-H), 8.55 (s, 1H, H-2), 9.00 (s, 1H, H-5).
General procedure for reduction of nitro group (method C)
To a solution of the nitro derivatives 18–25 (1.0 mmol) in DMF (30 mL), aqueous NaCl 3.5% w/w (150 mL) and Fe powder (55 mmol) were added; the mixture was then heated at 150 °C for 24h. The hot mixture was filtered over Celite and the residue washed with warm DMF (50 mL). The filtrate was evaporate to dryness and the residue was further worked-up as follows. For compounds 3–5 the residue was poured into ice/water, acidified with 2N HCl (pH = 4) and the formed precipitated filtered under vacuum, collected and crystallized by EtOH/DMF. In the case of compounds 6–10, the residue was treated with CHCl3 and the inorganic component was removed by filtration. The organic layer was evaporated to dryness and the residue was purified as indicated.
2-Methylbenzyl 6-amino-7-chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-4-oxo-1,4-dihydro-3-quinolinecarboxylate (3)
Following the general procedure method C and starting from intermediate 18, the target compound 3 was obtained in 36% yield as pale yellow solid: mp 259–260 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.30 (s, 3H, CH3), 5.25 (s, 2H, OCH2), 5.75 (s, 2H, NH2), 5.80 (s, 2H, NCH2), 7.20–7.30 (m, 4H, O-benzyl-H), 7.50 (d, J = 7.0 Hz, 1H, N-benzyl-H-6), 7.60 (d, J = 7.0 Hz, 1H, N-benzyl-H-5), 7.65 (s, 1H, H-8), 7.70 (s, 1H, H-5), 7.75 (d, JH-F = 10.0 Hz, 1H, N-benzyl-H-3), 8.75 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 19.00, 50.59, 64.25, 108.82, 110.00, 113.64–113.88 (m, N-benzyl-C-3), 118.30, 122.30 (m, N-benzyl-C-5), 123.65 (q, JC-F = 268.0 Hz, CF3), 124.21, 126.16, 128.38, 128.54 (d, JC-F = 14.0 Hz, N-Benzyl-C-1), 128.96, 129.17, 129.98 (d, JC-F = 5.0 Hz, N-Benzyl-C-6), 130.33, 130.40, 130.80 (dd, JC-F = 9.0 and 33.0 Hz, N-Benzyl-C-4), 135.00, 136.91, 143.47, 149.18, 160.08 (d, JC-F = 250.0 Hz, N-Benzyl-C-2), 165.23, 172.60. Anal. Calcd. for C26H19ClF4N2O3: C, 60.18; H, 3.69; N, 5.40. Found: C, 59.96; H, 3.51; N, 5.80.
4-Methylbenzyl 6-amino-7-chloro-1-[2-fluoro-4-(trifluoromethyl)benzyl]-4-oxo-1,4-dihydro-3-quinolinecarboxylate (4)
Following the general procedure method C and starting from intermediate 19, the target compound 4 was obtainedin 42% yield as pale yellow solid: mp 280–281 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.30 (s, 3H, CH3), 5.20 (s, 2H, OCH2), 5.75 (s, 2H, NH2), 5.80 (s, 2H, NCH2 ), 7.15–7.20 (m, 2H, O-benzyl-H-3 and -H-5), 7.25 (t, J = 7.7 Hz, 1H, N-benzyl-H-6), 7.35–7.40 (m, 2H, O-benzyl-H-2 and -H-6), 7.50 (d, J = 8.1 Hz, 1H, N-benzyl-H-5), 7.60 (s, 1H, H-8), 7.65 (s, 1H, H-5), 7.75 (d, JH-F = 10.0 Hz, 1H, N-benzyl-H-6), 8.75 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 21.19, 50.55, 65.46, 108.85, 109.98, 113.75 (d, JC-F = 21.0 Hz, N-benzyl-C-3), 118.29, 121.5–123.00 (m, N-benzyl-C-5), 123.60 (q, JC-F= 270.0 Hz, CF3), 124.22, 128.21, 128.57 (d, JC-F = 10.0 Hz, N-benzyl-C-1), 129.13, 129.32, 129.89 (d, JC-F = 4.0 Hz, N-benzyl-C-6), 130.33, 130.78 (dd, JC-F = 8.0 and 30.0 Hz, N-benzyl-C-4), 134.13, 137.40, 143.47, 149.16, 160.05 (d, JC-F = 250.0 Hz, N-benzyl-C-2), 165.03, 172.62. Anal. Calcd. for C26H19ClF4N2O3: C, 60.18; H 3.69; N, 5.40. Found:C, 60.30; H, 3.84; N, 5.13.
4-Chlorobenzyl 6-amino-7-chloro-1-(4-chlorobenzyl)-4-oxo-1,4-dihydro-3-quinolinecarboxylate (5)
Following the general procedure method C and starting from intermediate 20, the target compound 5 was obtainedin 34% yield as pale yellow solid: mp 274–275 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.25 (s, 2H, OCH2), 5.50 (s, 2H, NH2), 5.75 (s, 2H, NCH2), 7.20–7.30 (m, 2H, N-benzyl-H-2 and -H-6), 7.30–7.45 (m, 4H, N-benzyl-H-3, and –H-5, O-benzyl-H-2, and -H-6), 7.50–7.60 (m, 3H, H-5, and N-benzyl-H-3, and -H-5), 7.65 (s, 1H, H-8), 8.75 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 55.20, 64.74, 108.26, 109.86, 118.82, 124.07, 128.77, 128.88, 129.36, 129.79, 130.23, 130.70, 132.91, 135.46, 136.29, 143.40, 149.00, 165.19, 172.61. Anal. Calcd. for C24H17Cl3N2O3: C, 59.10; H, 3.51; N, 5.74. Found: C, 59.19; H, 3.64; N, 5.52.
4-Chlorobenzyl 6-amino-1-(4-chlorobenzyl)-4-oxo-7-(1-piperazinyl)-1,4-dihydro-3-quinolinecarboxylate (6)
Following the general procedure method C and starting from intermediate 21, after crystallization by EtOH, the target compound 6 was obtained in 10% yield as yellow solid: mp 221–223 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.75–3.00 and 3.25–3.50 (m, each 4H, piperazine-CH2), 5.30 (s, 2H, OCH2), 5.40 (s, 2H, NH2), 5.75 (s, 2H, NCH2), 7.00 (s, 1H, H-8), 7.20–7.30 (m, 2H, N-benzyl-H-2 and -H-6), 7.40–7.50 (m, 5H, H-5, N-benzyl-H-3, -H-5 and O-benzyl-H-2 and -H-6), 7.50–7.60 (m, 2H, O-benzyl-H-3 and -H-5), 8.75 (s, 1H, H-2), 9.25 (bs, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 49.74, 55.01, 55.54, 64.61, 108.00, 108.16, 109.01, 125.85, 128.76, 129.14, 129.24, 130.02, 131.30, 132.65, 132.80, 135.83, 136.42, 141.04, 143.90, 148.10, 165.54, 173.02. Anal. Calcd. for C28H26Cl2N4O3: C, 62.57; H, 4.88; N, 10.42. Found: C, 62.78; H, 5.08; N, 10.17.
4-Chlorobenzyl 6-amino-1-(4-chlorobenzyl)-7-(4-methyl-1-piperazinyl)-4-oxo-1,4-dihydro-3-quinolinecarboxylate (7)
Following the general procedure method C and starting from intermediate 22, after purification by flash column chromatography eluting with CHCl3/MeOH (95:5),the target compound 7 was obtained in 18% yield as yellow solid: mp 214–217 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.25 (s, 3H, CH3), 2.40–2.60 and 2.70–2.90 (m, each 4H, piperazine-CH2), 5.10 (s, 2H, NH2), 5.30 (s, 2H, OCH2), 5.70 (s, 2H, NCH2), 6.90 (s, 1H, H-8), 7.25–7.35 (m, 2H, N-benzyl-H-2 and -H-6), 7.40–7.50 (m, 5H, H-5, N-benzyl-H-3, -H-5 and O-benzyl-H-2, and -H-6), 7.60–7.70 (m, 2H, O-benzyl-H-3 and -H-5), 8.80 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 45.93, 49.74, 54.91, 55.54, 64.61, 107.94, 108.16, 108.88, 125.85, 128.76, 129.14, 129.24, 129.79, 131.30, 132.65, 132.80, 135.83, 136.42, 141.04, 143.90, 148.10, 165.54, 172.66. Anal. Calcd. for C29H28Cl2N4O3: C, 63.16; H, 5.12; N, 10.16. Found: C, 63.01; H, 4.92; N, 10.41.
4-Chlorobenzyl 6-amino-1-(4-chlorobenzyl)-4-oxo-7-[4-(2-pyridinyl)-1-piperazinyl]-1,4-dihydro-3-quinolinecarboxylate (8)
Following the general procedure method D and starting from intermediate 23, after crystallization by EtOH, the target compound 8 was obtained in 20% yield as pale yellow solid: mp 276–279 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.80–2.90 and 3.60–3.70 (m, each 4H, piperazine-CH2), 5.20 (s, 2H, NH2), 5.25 (s, 2H, OCH2), 5.60 (s, 2H, NCH2), 6.60 (dd, J = 5.0 and 7.0 Hz, 1H, pyridine-H), 6.80 (d, J = 9.0 Hz, 1H, pyridine-H), 7.00 (s, 1H, H-8), 7.25–7.30 (m, 2H, N-benzyl-H-2 and -H-6), 7.30–7.40 (m, 4H, N-benzyl-H-3, -H-5,O-benzyl-H-2, and -H-6), 7.50 (s, 1H, H-5), 7.50–7.60 (m, 3H, O-benzyl-H-3, -H-5 and pyridine-H), 8.15 (dd, J = 1.3 and 5.0 Hz, 1H, pyridine-H), 8.75 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 45.19, 50.00, 55.48, 64.62, 107.58, 108.11, 108.18, 108.90, 113.59, 126.02, 128.76, 129.23, 129.25, 129.79, 131.27, 132.66, 132.85, 135.81, 136.43, 138.03, 141.18, 143.90, 148.04, 159.45, 165.55, 172.67. Anal. Calcd. for C33H29Cl2N5O3: C, 64.50; H, 4.76; N, 11.40. Found: C, 64.70; H, 4.95; N, 11.21.
Compound 8 was advantageously prepared in an alternative way (see scheme 2), starting from intermediate 28, following the general procedure method A and using 4-chlorobenzylchloride. After purification by flash column chromatography eluting with CHCl3/MeOH (97:3), compound 8 was obtained in 70 % yield as white solid: mp, 1H and 13C NMR spectrum are in agreement with the data above reported for this compound.
4-Chlorobenzyl 6-amino-7-[4-(1,3-benzothiazol-2-yl)-1-piperazinyl]-1-(4-chlorobenzyl)-4-oxo-1,4-dihydro-3-quinolinecarboxylate (9)
Following the general procedure method C and starting from intermediate 24, after purification by flash column chromatography eluting CHCl3/MeOH (97:3), the target compound 9 was obtained in 20% yield as pale brown solid: mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.90–3.00 and 3.70–3.80 (m, each 4H, piperazine-CH2), 5.25 (s, 2H, NH2), 5.30 (s, 2H, OCH2), 5.65 (s, 2H, NCH2), 7.05 (s, 1H, H-8), 7.10 (dt, J = 0.9 and 8.1 Hz, 1H, benzothiazole-H), 7.25–7.35 (m, 3H, benzothiazole-H and N-benzyl-H), 7.40–7.50 (m, 6H, H-5, benzothiazole-H, N,O-benzyl-H), 7.60–7.70 (m, 2H, N-benzyl-H), 7.80 (d, J =7.5 Hz, 1H, benzothiazole-H), 8.80 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 48.58, 49.61, 55.36, 64.64, 108.12, 108.47, 108.87, 119.09, 121.71, 121.81, 126.22, 126.50, 128.78, 129.30, 129.81, 130.87, 131.13, 132.66, 132.88, 135.73, 136.41, 141.27, 143.45, 148.10, 152.85, 165.53, 168.59, 172.68. Anal. Calcd. (%) for C35H29Cl2N5O3S: C; 62.69; H, 4.36; N, 10.44. Found: C, 62.99; H, 4.76; N, 10.10.
4-Chlorobenzyl 6-amino-1-(4-chlorobenzyl)-4-oxo-7-{4-[3-(trifluoromethyl)phenyl]-1-piperazinyl}-1,4-dihydro-3-quinolinecarboxylate (10)
Following the general procedure method C and starting from intermediate 25, after purification by flash chromatography eluting CHCl3/MeOH (95:5), the target compound 10 was obtained in 15% yield as white solid: mp 257–259 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.90–3.00 and 3.40–3.50 (m, each 4H, piperazine-CH2), 5.20 (s, 2H, NH2), 5.30 (s, 2H, OCH2), 5.65 (s, 2H, NCH2), 6.95 (s, 1H, H-8), 7.15 (d, J = 7.5 Hz, 1H, phenyl-H), 7.25 (s, 1H, H-5), 7.30–7.35 (m, 3H, N-benzyl-H-2, -H-6 and phenyl-H), 7.40–7.50 (m, 6H, N-benzyl-H-3, -H-5, O-benzyl-H-2,-H-6, and phenyl-H), 7.55–7.60 (m, 2H, O-benzyl-H-3 and -H-5), 8.80 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 48.17, 49.88, 55.16, 64.66, 108.03, 108.21, 108.86, 111.40–111.43 (m, phenyl-C-2), 115.20–115.25 (m, phenyl-C-4), 119.30, 124.15 (q, JC-F = 270.0 Hz, CF3), 125.95, 128.76, 129.20, 129.25, 130.21, 130.50 (bs, phenyl-C-5), 131.34, 132.70 (q, JC-F = 30.0 Hz, phenyl-C-3), 132.88, 135.70, 136.36, 141.02, 143.77, 148.08, 151.65, 165.50, 172.77. Anal. Calcd. (%) for C35H29Cl2F3N4O3: C, 61.68; H, 4.29; N, 8.22. Found: C, 61.79; H, 4.42; N, 8.10.
Ethyl 1-(4-chlorobenzyl)-6-nitro-4-oxo-7-[4-(2-pyridinyl)-1-piperazinyl]-1,4-dihydro-3-quinolinecarboxylate (26)
To a warmed solution of compound 15 (1.0 g, 2.4mmol) in dry DMF (60 mL), 1-(2-pyridinyl)piperazine (1.2 g, 7.2 mmol) was added. The solution was heated at 80 °C for 8h and then concentrated to one-third of the volume under reduced pressure. After cooling the precipitate formed was collected by filtration under vacuum and crystallized by EtOH/DMF (1:2) to give compound 26 (1.1 g) in 85% yield as yellow solid: mp 213–215 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.25 (t, J = 7.1 Hz, 3H, CH3), 3.15–3.25 and 3.65–3.75 (m, each 4H, piperazine-CH2), 4.25 (q, J = 7.1 Hz, 2H, OCH2), 5.75 (s, 2H, NCH2), 6.75–6.80 (m, 1H, pyridine-H), 6.90 (d, J = 8.4 Hz, 1H, pyridine-H), 7.05 (s, 1H, H-8), 7.30–7.55 (m, 4H, benzyl-H), 7.65–7.75 (m, 1H, pyridine-H), 8.15 (dd, J = 1.4 and 5.1 Hz, 1H, pyridine-H), 8.55 (s, 1H, H-2), 8.90 (s, 1H, H-5).
Ethyl 6-amino-1-(4-chlorobenzyl)-4-oxo-7-[4-(2-pyridinyl)-1-piperazinyl]-1,4-dihydro-3-quinolinecarboxylate (27)
A stirred solution of nitro derivative 26 (1 g, 1.8 mmol) in DMF (60 mL) was hydrogenated over a catalytic amount of Raney nickel at room temperature and atmospheric pressure for 6 h. The mixture was then filtered over Celite, the filtrate was evaporated to dryness, and the residue was crystallized by EtOH/DMF to give amino derivative 27 (0.7 g) in 70 % yield as pale brown solid: mp >300 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.25 (t, J = 7.1 Hz, 3H, CH3), 2.75–2.85 and 3.65–3.75 (m, each 4H, piperazine-CH2), 4.25 (q, J = 7.1 Hz, 2H, OCH2), 5.25 (s, 2H, NH2), 5.65 (s, 2H, NCH2), 6.70 (dd, J = 5.0 and 7.0 Hz, 1H, pyridine-H), 6.80 (d, J = 8.6 Hz, 1H, pyridine-H), 6.95 (s, 1H, H-8), 7.25–7.30 (m, 2H, benzyl-H-2 and -H-3), 7.35–7.40 (m, 2H, benzyl-H-3 and –H-5), 7.45 (s, 1H, H-5), 7.55 (dt, J = 2.0 and 7.2 Hz, 1H, pyridine-H), 8.15 (dd, J = 1.3 and 5.0 Hz, 1H, pyridine-H), 8.75 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 14.76, 45.08, 49.90, 55.33, 59.85, 107.53, 107.95, 108.57, 108.71, 113.52, 125.87, 129.08, 129.96, 131.47, 132.71, 135.82, 138.01, 140.98, 143.78, 147.77, 147.92, 159.33, 165.47, 172.58. Anal. Calcd. (%) for C28H28ClN5O3: C, 64.92; H, 5.45; N, 13.52. Found: C, 65.22; H, 5.74; N, 13.17.
6-amino-1-(4-chlorobenzyl)-4-oxo-7-[4-(2-pyridinyl)-1-piperazinyl]-1,4-dihydro-3-quinolinecarboxylic acid (28)
A stirred mixture of ethyl ester 27 (0.6 g, 1.16mmol) in aqueous 4% NaOH (10 ml) and EtOH (10 ml) was refluxed for 2 h and then concentrated to half volume under reduced pressure. The formed precipitated was filtered under vacuum and re-suspended in ice/water and acidified with 2N HCl (pH = 2). The solid so obtained was filtered to give compound 28 (0.46 g) in 81 % yield as yellow solid, pure by TLC in two eluents (CHCl3/MeOH 95:5 and CHCl3/MeOH/AcOH 85:14.5:0.5): mp> 300 °C. 1H NMR (400, DMSO-d6): δ 3.20–3.30 and 3.90–4.00 (m, each 4H, piperazine-CH2), 5.85 (s, 2H, NCH2), 6.95–7.00 (m, 1H, pyridine-H), 7.20 (s, 1H, H-8), 7.25–7.50 (m, 5H, pyridine-H and benzyl-H), 7.55 (s, 1H, H-5), 7.95–8.00 (bs, 1H, pyridine-H), 8.05 (dd, J = 1.0 and 6.0 Hz, 1H, pyridine-H), 9.05 (s, 1H, H-2). 13C NMR (100 MHz, DMSO-d6): δ 46.12, 48.90, 56.25, 107.48, 107.89, 108.55, 108.72, 114.00, 125.80, 129.18, 129.95, 131.50, 132.88, 135.82, 137.99, 141.01, 144.54, 147.56, 147.98, 161.35, 167.07, 174.39. Anal. Calcd. (%) for C26H24ClN5O3: C, 63.74; H, 4.94; N, 14.29. Found: C, 63.92; H, 5.19; N, 12.97.
Biochemical and biological assays
NS5B polymerase assay
Recombinant NS5B 1b (HC-J4) carrying an N-terminal histidine-tag and 21-amino acid truncation at its C-terminus was purified by Ni-NTA chromatography as previously described.22 The compounds were dissolved in DMSO as 10 mM stocks and serially diluted in DMSO immediately prior to the assay. Anti-HCV NS5B activity of the compounds was investigated by the standard primer dependent elongation assay as previously described.28 Preliminary screening was conducted at 25 µM compound concentration to identify candidate NS5B inhibitors. The assays were carried out at 30°C for 1 h and reactions stopped with chilled 5% (v/v) trichloroacetic acid (TCA) containing 0.5 mM pyrophosphate. Nascent RNA products were precipitated on GF-B filters and quantified by liquid scintillation counting. NS5B activity in the presence of DMSO control was set at 100% and that in the presence of the compounds was determined relative to this control. Compounds exhibiting ≥50% inhibition at 25 µM concentration were further investigated for their IC50 values. Dose-response curves of 8–12 concentrations of the compounds in duplicate in two independent experiments were plotted using nonlinear regression analysis and IC50 values were determined using CalcuSyn V2 software.
Fluorescence quenching assay (FQ)
FQ assay was performed as described previously.29 In Brief, HCV NS5B (CΔ21) was diluted to a final concentration of 1 µM in a buffer containing 50 mM Tris HCl (pH 7.8), 50 mM NaCl, 10% glycerol, 10% DMSO and 0.5 mM MnCl2 in a final volume of 100 µl per well in a 96 well Fluotrac 200 plate. Serially diluted compound 7 in DMSO or DMSO alone (control) was incubated for 5 min at room temperature. Excitation and emission intensities, 280 nm and 330 nm, respectively, were used to determine the dissociation constant (KD) value for the compound. The excitation and emission wavelengths were set at monochromator bandwidths of 20 and 5 nm, respectively. The background emission was eliminated by buffer containing equal amount of test compound or DMSO. The KD value was determined using the equation, ΔF/ΔFmax = [compound] total / (KD + [compound] total), where ΔF is the difference in emission fluorescence intensity of NS5B in the absence and presence of compound. ΔFmax is the difference in emission fluorescence intensity of NS5B in the absence of compound and in the presence of infinite concentration of compound and KD is the dissociation constant.
SPR Interaction analysis
The ectodomain of HCV NS5B (Con 1 strain isolate), with a C-terminal hexa histidine fusion tag, was immobilized by standard amine coupling to a CM5 biosensor chip in a Biacore S51 instrument (GE Healthcare). The enzyme was transferred to a 10 mM MES buffer (pH 6.0) and injected until an immobilization level of 4–5 kRU was reached. HSB-P (20 mM HEPES pH 7.4, 150 mM NaCl, 0.05 % (v/v) Tween20) was used as system running buffer during the immobilization, and TBS-P (20 mM Tris pH 7.4 at room temperature, 130 mM NaCl, 0.05 % (v/v) Tween20, 5 % (v/v) DMSO) was used during the subsequent characterization of the synthesized compound. Quinolone derivative 7 was injected over the protein surface for 60 s at 30 µL/min using two-fold dilution concentration series between 6 and 200 µM. Filibuvir (PF-00868554)30 was used as a positive control substance to assess the stability of the immobilized enzyme over time.
Raw data was processed according to standard procedures, including reference and blank subtraction along with solvent correction to eliminate any artifacts such as non-specific binding and discrepancies in buffer composition in the Biacore T100 Evaluation software. Global nonlinear regression with the 1:1 Langmuir binding model was used to determine the dissociation equilibrium constant (KD) and estimate the maximal signal (Rmax) as described previously.11c
Cells and viruses
The Huh 5-2 and Huh 9–13 HCV subgenomic replicon-containing cells were provided by Prof R Bartenschlager (University of Heidelberg, Heidelberg, Germany).
Antiviral assays
Huh 5.2 cells, containing the hepatitis C virus genotype 1b I389luc-ubineo/NS3-3’/5.1 replicon31 were sub-cultured in DMEM supplemented with 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin and 2% Geneticin at a ratio of 1:3 to 1:4, and grown for 3–4 days in 75 cm² tissue culture flasks. One day before addition of the compound, cells were harvested and seeded in assay medium (DMEM, 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin) at a density of 6 500 cells/well (100 µL/well) in 96-well tissue culture microtiter plates for evaluation of anti-metabolic effect and CulturPlate (Perkin Elmer) for evaluation of the antiviral effect. The microtiter plates were incubated overnight (37 °C, 5% CO2, 95–99% relative humidity), yielding a non-confluent cell monolayer.
The evaluation of the anti-metabolic as well as antiviral effect of each compound was performed in parallel. Four-step, 1-to-5 compound dilution series were prepared for the first screen, to collect data for a more detailed dose-response curve, an eight-step, 1-to-2 dilution series was used. Following assay setup, the microtiter plates were incubated for 72 hours (37 °C, 5% CO2, 95–99% relative humidity). For the evaluation of anti-metabolic effects, the assay medium was aspirated, replaced with 75 µL of a 5% MTS solution in phenol red-free medium and incubated for 1.5 hours (37 °C, 5% CO2, 95–99% relative humidity). Absorbance was measured at a wavelength of 498 nm (Safire², Tecan), and optical densities (OD values) were converted to percentage of untreated controls. For the evaluation of antiviral effects, assay medium was aspirated and the cell monolayers were washed with PBS. The wash buffer was aspirated, and 25 µL of Glo Lysis Buffer (Promega) was added allowing for cell lysis to proceed for 5 min at room temperature. Subsequently, 50 µL of Luciferase Assay System (Promega) was added, and the luciferase luminescence signal was quantified immediately (1000 ms integration time/well, Safire², Tecan). Relative luminescence units were converted into percentage of untreated controls.
The EC50 and EC90 (values calculated from the dose-response curve) represent the concentrations at which 50% and 90% inhibition, respectively, of viral replication is achieved. The CC50 (value calculated from the dose-response curve) represents the concentration at which the metabolic activity of the cells is reduced by 50 % as compared to untreated cells.
A concentration of compound is considered to elicit a genuine antiviral effect in the HCV replicon system when the anti-replicon effect is well above the 70% threshold at concentrations where no significant anti-metabolic activity is observed.
Compounds that reproducibly matched the above-outlined selection criteria were evaluated for selective antiviral activity in the Huh 9–13 replicon system. A similar assay setup was used as described above; the antiviral and anti-metabolic effect of the compounds was evaluated in parallel. The anti-metabolic activity of the compounds was quantified as outlined above. For the evaluation of the antiviral effect, assay medium was aspirated and the plates with dry monolayer were stored at −80°C awaiting extraction. Following thawing of the plates at room temperature, the cell monolayer was lysed with 100 µL of cell-to-cDNA lysis buffer (Invitrogen). Lysis of the cells was allowed to proceed for 10 min at room temperature after which all liquid was transferred to a PCR plate (Axygen). The PCR plate was incubated for 15min at 75 °C (T3, Biometra). The lysate was diluted 1:2 with RNase/DNase-free water, after which 5 µL was transferred to a real-time PCR plate (Applied Biosystems). Replicon RNA content was quantified using a real-time quantitative one-step RT-PCR method (RT-qPCR). Per sample, 20 µL master mix was added containing 12.5 µL 2x RT-qPCR mix (Low Rox One-Step RT-qPCR master mix, Abgene), 0.125 µL of a 60 µM forward primer solution (5’-CCA GAT CAT CCT GAT CGA CCA G-3’, final [ ] of 300 nM), 0.125 µL of a 60 µM reversed primer solution (5’-CCG GCT ACC TGC CCA TTC-3’, final [ ] of 300 nM), 0.3µL of a 5 µM probe solution (5’-ACA TCG CAT CGA GCG AGC ACG TAC-3’, final [ ] of 60 nM) and 6.825 µL of DNase/RNase-free water (ACROS). The samples were analyzed using a SDS7500F (Applied Biosystems, standard thermocycling profile: 30 min at 48 °C, 10 min at 95 °C, 40 cycles of 15 sec at 95 °C and 1 min at 60 °C). Replicon RNA quantities were converted to percentage of untreated controls, allowing the calculation of EC50 and EC90 values. Similar to the Huh 5-2 assay, a compound is only considered to be a selective inhibitor of HCV replication when clear inhibition of virus replication is observed at concentrations that do not elicit a significant anti-metabolic effect on the host cells.
ACKNOWLEDGMENT
We thank Roberto Bianconi, Stijn Delmotte, Katrien Geerts and Inge Vliegen for excellent technical assistance. This work was supported by the UMDNJ Foundation and National Institutes of Health Research Grant CA153147 to N.K.-B.
ABBREVIATIONS
- RBV
ribavirin
- pegIFN-α
pegylated interferon-α
- SVR
sustained virological response
- DAAs
direct-aging antivirals
- NS5B
nonstructural 5B
- RdRp
RNA-dependent RNA polymerase
- NIs
nucleoside inhibitors
- NNIs
non-nucleoside inhibitors
- TSI
thumb site I
- TSII
thumb site II
- PSI
palm site I
- PSII
palm site II
- PSIII
palm site III
- HCMV
human cytomegalovirus
- IFD
induced fit docking
- MW
microwaves
- SI
selectivity index
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H–tetrazolium
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Lavanchy D. Evolving Epidemiology of Epatitis C Virus. Clin. Microbiol. Infect. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. Natural History of Chronic Hepatitis C. Hepatology. 2002;36:S35–S46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 3.Fauvelle C, Lepiller Q, Felmlee DJ, Fofana I, Habersetzer F, Stoll-Keller F, Baumert TF, Fafi-Kremer S. Hepatitis C virus vaccines - Progress and perspectives. Microbial Pathogenesis. 2013;58:66–72. doi: 10.1016/j.micpath.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Scheridan C. New Merck and Vertex Drugs Raise Standard of Care in Hepatitis C. Nat. Biotechnol. 2011;29:553–554. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- 5.Ferenci P, Reddy KR. Impact of HCV Protease-Inhibitor-BasedTriple Therapy for Chronic HCV Genotype 1 Infection. Antivir. Ther. 2011;16:1187–1201. doi: 10.3851/IMP1934. [DOI] [PubMed] [Google Scholar]
- 6.Pawlotsky JM, Chevaliez S, McHutchison JG. The Hepatitis C Virus Life Cycle as a Target for New Antiviral Therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- 7.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA Polymerase of Hepatitis C Virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 8.a) Barreca ML, Iraci N, Manfroni G, Cecchetti V. Allosteric Inhibition of the Hepatitis C Virus NS5B Polymerase: in Silico Strategies for Drug Discovery and Development. Future Med. Chem. 2011;3:1027–1055. doi: 10.4155/fmc.11.53. [DOI] [PubMed] [Google Scholar]; b) Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, Nucleotide and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 9.Garber L, Welzel TM, Zeuzem S. New Therapeutic Strategies in HCV: Polymerase Inhibitors. Liver Int. 2013;33:85–92. doi: 10.1111/liv.12068. [DOI] [PubMed] [Google Scholar]
- 10.Asselah T. Sofosbuvir-based interferon-free therapy for patients with HCV infection. J. Hepatol. 2013 doi: 10.1016/j.jhep.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 11.a) Manfroni G, Paeshuyse J, Massari S, Zanoli S, Gatto B, Maga G, Tabarrini O, Cecchetti V, Fravolini A, Neyts J. Inhibition of Subgenomic Hepatitis C Virus RNA Replication by Acridone Derivatives: Identification of an NS3 Helicase Inhibitor. J. Med. Chem. 2009;52:3354–3365. doi: 10.1021/jm801608u. [DOI] [PubMed] [Google Scholar]; b) Manfroni G, Meschini F, Barreca ML, Leyssen P, Samuele A, Iraci N, Sabatini S, Massari S, Maga G, Neyts J, Cecchetti V. Pyridobenzothiazole Derivatives as New Chemotype Targeting the HCV NS5B Polymerase. Bioorg. Med. Chem. 2012;20:866–876. doi: 10.1016/j.bmc.2011.11.061. [DOI] [PubMed] [Google Scholar]; c) Barreca ML, Manfroni G, Leyssen P, Winquist J, Kaushik-Basu N, Paeshuyse J, Krishnan R, Iraci N, Sabatini S, Tabarrini O, Basu A, Danielson UH, Neyts J, Cecchetti V. Structure-based Discovery of Pyrazolobenzothiazine Derivatives as Inhibitors of Hepatitis C Virus Replication. J. Med. Chem. 2013;56:2270–2282. doi: 10.1021/jm301643a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cecchetti V, Clementi S, Cruciani G, Fravolini A, Pagella PG, Savino A, Tabarrini O. 6-Aminoquinolones: A New Class of Quinolone Antibacterials? J. Med. Chem. 1995;38:973–982. doi: 10.1021/jm00006a017. [DOI] [PubMed] [Google Scholar]
- 13.a) Wise R, Pagella PG, Cecchetti V, Fravolini A, Tabarrini O. In Vitro Activity of MF 5137, a New Potent 6-aminoquinolone. Drugs. 1995;49:272–273. doi: 10.2165/00003495-199500492-00067. [DOI] [PubMed] [Google Scholar]; b) Miolo G, Viola G, Vedaldi D, Dall’Acqua F, Fravolini A, Tabarrini O, Cecchetti V. In Vitro Phototoxic Properties of New 6-Desfluoro- and 6-Fluoro-8-methylquinolones. Toxicol. In Vitro. 2002;16:683–693. doi: 10.1016/s0887-2333(02)00093-0. [DOI] [PubMed] [Google Scholar]
- 14.a) Cecchetti V, Parolin C, Moro S, Pecere T, Filipponi E, Calistri A, Tabarrini O, Gatto B, Palumbo M, Fravolini A, Palu’ G. 6-Aminoquinolones as New Potential anti-HIV Agents. J. Med. Chem. 2000;43:3799–3802. doi: 10.1021/jm9903390. [DOI] [PubMed] [Google Scholar]; b) Tabarrini O, Stevens M, Cecchetti V, Sabatini S, Dell’Uomo M, Manfroni G, Palumbo M, Pannecouque C, De Clercq E, Fravolini A. Structure Modifications of 6-Aminoquinolones with Potent Anti-HIV Activity. J. Med. Chem. 2004;47:5567–5578. doi: 10.1021/jm049721p. [DOI] [PubMed] [Google Scholar]; c) Tabarrini O, Massari S, Daelemans D, Stevens M, Manfroni G, Sabatini S, Balzarini J, Cecchetti V, Pannecouque C, Fravolini A. Structure-activity Relationship Study on Anti-HIV 6-Desfluoroquinolones. J. Med. Chem. 2008;11:5454–5458. doi: 10.1021/jm701585h. [DOI] [PubMed] [Google Scholar]; d) Tabarrini O, Massari S, Cecchetti V. 6-Desfluoroquinolones as HIV-1 Tat-mediated Transcription Inhibitors. Future Med. Chem. 2010;2:1161–1180. doi: 10.4155/fmc.10.208. [DOI] [PubMed] [Google Scholar]; e) Massari S, Daelemans D, Manfroni G, Sabatini S, Tabarrini O, Pannecouque C, Cecchetti V. Studies on anti-HIV Quinolones: new Insights on the C-6 Position. Bioorg. Med. Chem. 2009;17:667–674. doi: 10.1016/j.bmc.2008.11.056. [DOI] [PubMed] [Google Scholar]
- 15.a) Loregian A, Mercorelli B, Muratore G, Sinigalia E, Pagni S, Massari S, Gribaudo G, Gatto B, Palumbo M, Tabarrini O, Cecchetti V, Palù G. The 6-aminoquinolone WC5 Inhibits Human Cytomegalovirus Replication at an Early Stage by Interfering with the Transactivating Activity of Viral Immediate-Early 2 Protein. Antimicrob. Agents Chemother. 2010;54:1930–1940. doi: 10.1128/AAC.01730-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Massari S, Mercorelli B, Sancineto L, Sabatini S, Cecchetti V, Gribaudo G, Palù G, Pannecouque C, Loregian A, Tabarrini O. Design, Synthesis, and Evaluation of WC5 Analogues as Inhibitors of Human Cytomegalovirus Immediate-Early 2 Protein, a Promising Target for Anti-HCMV Treatment. Chem. Med. Chem. 2013;8:1403–1414. doi: 10.1002/cmdc.201300106. [DOI] [PubMed] [Google Scholar]
- 16.Massari S, Donaliso M, Ansideri F, Sancineto L, Sabatini S, Manfroni G, Barreca ML, Cecchetti V, Lembo D, Tabarrini O. Small Molecules Targeting HPV E6 and E7 Oncoprotreins Expression. XXII National Meeting on Medicinal Chemistry, Rome, Italy, 10–13. 2013 Accepted abstract. [Google Scholar]
- 17.Kumar DV, Rai R, Brameld KA, Somoza JR, Rajagopalan R, Janc JW, Xia YM, Ton TL, Shaghafi MB, Hu H, Lehoux I, To N, Young WB, Green MJ. Quinolones as HCV NS5B Polymerase Inhibitors. Bioorg. Med. Chem. Lett. 2011;21:82–87. doi: 10.1016/j.bmcl.2010.11.068. [DOI] [PubMed] [Google Scholar]
- 18.a) Schrödinger Suite 2012 Induced Fit Docking protocol. New York, NY: Glide version 5.8, Schrödinger, LLC; Prime version 3.1, Schrödinger, LLC, New York, NY, 2012. [Google Scholar]; b) Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. “Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects”. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]; c) Sherman W, Beard HS, Farid R. “Use of an Induced Fit Receptor Structure in Virtual Screening,”. Chem. Biol. Drug Des. 2006;67:83–84. doi: 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 19.LigPrep, version 2.5. New York: Schrödinger, LLC; [Google Scholar]
- 20.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agraval A, Tratneyek PG. Reduction of Nitro Compounds by Zero-valent Iron Metal. Environ. Sci. Technol. 1996;30:153–160. [Google Scholar]
- 22.a) Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Costa PR, da Silva AJ, Sarafianos SG, Noel F. Identification and Characterization of Coumestans as Novel HCV NS5B Polymerase Inhibitors. Nucleic Acids Res. 2008;36:1482–1496. doi: 10.1093/nar/gkm1178. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, Talele TT, Arora P, Kaushik-Basu N. Characterization of Aurintricarboxylic Acid as a Potent Hepatitis C Virus Replicase Inhibitor. Antivir. Chem. Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- 23.Compound 2 was synthesized by us following the experimental procedure reported in Kumar, D., Rai. Antiviral agents. WO2007/005779A2, 2007. Its identity was verified by 1H NMR; the purity was determined by HPLC analysis and was found to be 95%. Compound 2: 1H NMR (400 MHz, DMSO-d6): δ 2.25 (s, 3H, CH3), 3.75 and 3.80 (s, each 3H, OCH3), 5.20 (s, 2H, OCH2), 5.80 (s, 2H, NCH2), 6.95 (s, 1H, H-8), 7.15–7.20 (m, 2H, O-benzyl-H-3 and –H-5), 7.30–7.40 (m, 3H, N-benzyl-H-6, O-benzyl-H-2, and –H-6), 7.60 (d, J = 8.0 Hz, 1H, N-benzyl-H-5), 7.65 (s, 1H, H-5), 7.75 (d, JH-F = 11.0 Hz, 1H, N-benzyl-H-3), 8.80 (s, 1H, H-2). The analytical HPLC measurements were made on a Shimadzu (Kyoto, Japan) LC-20A Prominence, equipped with a CBM-20A communication bus module, two LC-20AD dual piston pumps, a SPD-M20A photodiode array detector (DAD, SPD20A) and a Rheodyne 7725i injector (Rheodyne Inc., Cotati, CA, USA) with a 20 µL stainless steel loop. A GraceSmart RP18 column (Grace, Sedriano, Italy) 250 × 4.6 mm i.d., 5 mm, 100 A was used as the analytical column. The column temperature was controlled through a Grace (Sedriano, Italy) heather/chiller (Model 7956R) thermostat. The mobile phase was prepared as a mixture of H2O/MeCN—50/50 (v/v). The analyses were carried out at 1.0 mL min−1 eluent flow rate after previous conditioning by passing through the column the selected mobile phase for at least 30 min at the same eluent velocity. All the analyses were conducted at a 25 °C column temperature. The detection was performed by a photodiode array detector (190-400nm).
- 24.Schrödinger Suite 2012 Protein Preparation Wizard; Epik version 2.3. New York, NY: Schrödinger, LLC; 2012. Impact version 5.8, Schrödinger, LLC, New York, NY, 2012; Prime version 3.1, Schrödinger, LLC, New York, NY. [Google Scholar]
- 25.Maestro, version 9.3. New York, NY: Schrödinger, LLC; 2012. [Google Scholar]
- 26.a) Milletti F, Storchi L, Sforna G, Cruciani G. New and Original pKa prediction method using Grid Molecular Interaction Fields. J. Chem. Inf. Mod. 2007;47:2172–2181. doi: 10.1021/ci700018y. [DOI] [PubMed] [Google Scholar]; b) Milletti F, Storchi L, Sforna G, Cross S, Cruciani G. Tautomer Enumeration and Stability Prediction for Virtual Screening on Large Chemical Databases. J. Chem. Inf. Mod. 2009;49:68–75. doi: 10.1021/ci800340j. [DOI] [PubMed] [Google Scholar]
- 27.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 28.Nichols DB, Fournet G, Gurukumar KR, Basu A, Lee JC, Sakamoto N, Kozielskie F, Musmuca I, Joseph B, Ragno R, Kaushik-Basu N. Inhibition of Hepatitis C Virus NS5B Polymerase by S-Trityl-L-Cysteine Derivatives. Eur. J. Med. Chem. 2012;49:191–199. doi: 10.1016/j.ejmech.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manvar D, Mishra M, Kumar S, Pandey VN. Identification and Evaluation of anti-Hepatitis C virus Phytochemicals from Eclipta alba. J. Ethnopharmacol. 2012;144:545–554. doi: 10.1016/j.jep.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Tatlock J, Linton A, Gonzalez J, Jewell T, Patel L, Ludlum S, Drowns M, Rahavendran SV, Skor H, Hunter R, Shi ST, Herlihy KJ, Parge H, Hickey M, Yu X, Chau F, Nonomiya J, Lewis C. Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5, 6-dihydropyran-2-one (PF-00868554) as a Potent and Orally Available Hepatitis C Virus Polymerase Inhibitor. J. Med. Chem. 2009;52:1255–1258. doi: 10.1021/jm8014537. [DOI] [PubMed] [Google Scholar]
- 31.Vrolijk JM, Kaul A, Hansen BE, Lohmann V, Haagmans BL, Schalm SW, Bartenschlager R. A Replicon-Based Bioassay for the Measurement of Interferons in Patients with Chronic Hepatitis C. J. Virol. Methods. 2003;110:201–209. doi: 10.1016/s0166-0934(03)00134-4. [DOI] [PubMed] [Google Scholar]