

Abstract
In the last few years, there have been many advances in the efforts to cure patients with hepatitis C virus (HCV). The ultimate goal of these efforts is to develop a combination therapy consisting of only direct-antiviral agents (DAA). In this paper, we discuss our efforts that led to the identification of a bicyclic template with potent activity against the NS5B polymerase, a critical enzyme on the life cycle of HCV. Continuing our exploration to improve the stilbene series, the 3,5,6,8-tetrasubstituted quinoline core was identified as replacement of the stilbene moiety. 6-Methoxy-2(1H)-pyridone was identified among several heterocyclic head groups to have the best potency. Solubility of the template was improved by replacing a planar aryl linker with a saturated pyrrolidine. Profiling of the most promising compounds led to the identification of quinoline 41 (RG7109) which was selected for advancement to clinical development.
INTRODUCTION
Hepatitis C virus (HCV), a positive-strand RNA virus member of the Flaviviridae family, chronically infects 150 million people worldwide with an estimated incidence of new cases of 3–4 million each year.1 HCV infection results in mild and acute liver disease, but chronic infections are common and may eventually develop into liver cirrhosis or hepatocellular carcinoma.2
Until 2011, the standard of care (SOC) for treating HCV had been a combination therapy of pegylated interferon-α (Peg-IFN) plus ribavirin (RBV). Response rates for HCV patients having genotypes 2 or 3 on a 24–48 week treatment show a sustained virological response (SVR, defined as the absence of detectable HCV RNA in blood serum for 24 weeks after treatment withdrawal) approaching 80%. Patients infected with HCV genotype 1 (GT-1) do not respond as well to this combination therapy demonstrating SVR rates of < 50% even after treatment therapies of 48 weeks in duration.3
With the recent approval of two HCV protease inhibitors (Telaprevir and Boceprevir) by the FDA, the standard of care for treatment of GT-1 infection is now Peg-IFN/RBV and a protease inhibitor. This triple combination improves the SVR rates up to 75% (Telaprevir) and 68% (Boceprevir) with genotype-1 naive patients,4 and reduces treatment duration. Other protease inhibitors in clinical development are showing improved SVR rates of > 90% in some patients and with a more convenient once daily dosing (q.d.) regimen than the first two compounds approved with three times daily (t.i.d.) and twice daily (b.i.d.) dosing regimens, respectively.5 The use of other direct-acting antivirals with different mechanisms of action (polymerase nucleoside inhibitor (NI) and non-nucleoside inhibitor (NNI) as well as nonstructural 5A (NS5A) inhibitors) have shown SVR > 80% in combination with Peg-IFN/RBV in phase II clinical studies.4
Interferon-associated side effects limit treatment success in a large number of patients.6 Therefore, one of the primary future goals is to develop direct-acting antiviral (DAA) therapy that can achieve high SVR without the use of Peg-IFN. For this objective, there are currently several clinical trials ongoing with combinations of DAAs (protease inhibitor, polymerase NI, polymerase NNI, and NS5A inhibitor) to determine which will deliver the best results with respect to SVR and safety for different patient populations and against the most common genotypes (GT-1,2,3, and 4).6 The development of potent and safe inhibitors of the different viral proteins is crucial to be able to achieve the goal of DAA therapy. The discovery of polymerase inhibitors and the development of a combination therapy that would eventually replace interferon-based treatment has been of interest to us for some time.7,8
The HCV nonstructural 5B (NS5B) protein contains the RNA-dependent RNA polymerase, the catalytic component of the HCV RNA replication machinery. This virus-specific enzyme plays an important role in the viral life cycle and thus represents an attractive therapeutic target. Several classes of inhibitors have been reported for the NS5B polymerase: NIs that bind in the active site, and NNIs that bind in at least 4 different allosteric sites identified so far (palm I, palm II, thumb I and thumb II).9
Different chemotypes have been reported to bind in the palm I allosteric site. From those chemical classes, at least five compounds have entered clinical trials and some of them have shown efficacy. Those are the N-acylpyrrolidine GSK625433 (1),10 benzothiadiazine ANA598 (2),11 benzodiazaphosphinine IDX375 (3),12 and the uracil derivatives ABT-072 (4),13 and ABT-333 (5)14 (Figure 1).
Figure 1.

Known HCV polymerase inhibitors that bind in the palm I allosteric site.
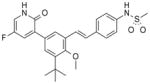
We have been interested in the design of inhibitors that bind to the palm I allosteric site. Recently, we reported the de novo design of fragment 615 with sub-μM potency against the HCV NS5B polymerase and the development of that fragment to stilbene 7, a potent inhibitor of NS5B with single digit nM activity in the HCV subgenomic replicon assay.16 From the start of our efforts, the objective was to find highly potent inhibitors of the replication of HCV with similar activity against both GT-1a and GT-1b. Even though 7 had an excellent biological profile, we continued exploring different scaffolds. It was important to keep the key functionalities (heterocyclic head and the sulfonamide group) in the right position to interact with the critical amino acids in the binding site. Among the different possibilities to achieve this, the option to have a bicyclic core that held both critical groups seemed to fit well based on modeling. Structure 8 was designed by analyzing the cocrystal structure of an analog of stilbene 7 with NS5B16 where it was observed that the vinyl carbon linked to the phenylsulfonamide could be bridged to C-6 of the central core. Several advantages were foreseen by having a bicyclic aromatic core, e.g. restricted rotation into bound conformation for potency, opportunity to explore different linker groups to the sulfonamide, and use of heteroaromatic systems to modulate its drug-like properties. Based on this rationale, it was decided to explore bicyclic aromatic cores.
In this article, we describe our efforts on the exploration of the bicyclic core that led to the discovery of RG7109 (41), a potent inhibitor of the HCV NS5B polymerase that was selected for clinical development.
CHEMISTRY
To determine if a bicyclic core would either maintain similar or improved potency with respect to the stilbene series, four different ring systems were synthesized. Of the four bicyclic cores reported in Table 1, the 3,5,6,8-tetrasubstituted quinoline system was prepared using two similar routes as shown on Schemes 1 and 2.
Table 1.
Bicyclic Core Templates with Replicon (GT-1a and GT-1b) Data.
| compd | Structure | GT-1a EC50 (nM)a |
GT-1b EC50 (nM)a |
|---|---|---|---|
| 7 |
|
8.0 | 3.0 |
| 33 |
|
3.8 | 3.5 |
| 34 |
|
25 | 16 |
| 35 |
|
7.2 | 3.2 |
| 36 |
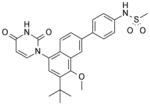
|
4 | 1 |
| 37 |
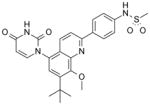
|
83 | 12 |
| 38 |
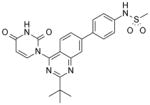
|
17 | NA |
EC50 measured using stable HCV (GT-1a H77, GT-1b Con1) subgenomic replicon assay (n ≥ 2).
Scheme 1. Synthesis of 3,5,6,8-tetrasubstituted quinoline core.

Reagents and conditions: (a) Ph2PON3, Et3N, EtOH, THF, reflux, 81%; (b) aq. KOH, EtOH, reflux, 96%; (c) NBS, DMF, quantitative; (d) Br2, 2-bromoprop-2-enal, AcOH, 100 °C, 45%; (e) 4-(methanesulfonamido)phenylboronic acid, Pd(PPh3)4, Na2CO3, MeOH, toluene, 120 °C, 57%.
Scheme 2. Alternative synthesis of 3,5,6,8-tetrasubstituted quinoline core.
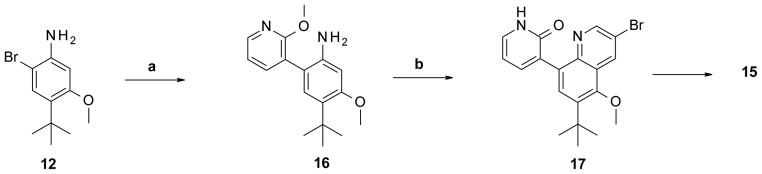
Reagents and conditions: (a) (2-methoxy-3-pyridyl)boronic acid, Pd(PPh3)4, Na2CO3, MeOH, CH2Cl2, 115 °C, 54%; (b) Br2, 2-bromoprop-2-enal, AcOH, 100 °C, 27%.
In Scheme 1, the use of 4-tert-butyl-3-methoxybenzoic acid (9) as starting material allowed us to have two of the four desired substitutions already in place. This starting material was very useful since we planned to maintain these two groups in most of the initial set of bicyclic compounds. The first step of the synthesis was to convert the carboxylic acid 9 to the primary aniline 11 by a Curtius rearrangement via the corresponding carbamate 10. Formation of the desired quinoline ring, using the amine group in 11 as a handle, had the disadvantage of producing two possible regioisomers. To avoid this, selective bromination of 11 para to the methoxy group to afford bromoaniline 12 was carried out prior to the cyclization reaction. Treatment of compound 12 with 2,2,3-tribromopropanal (prepared in situ from 2-bromo acrolein and bromine) achieved the formation of the desired 3,8-dibromoquinoline 13 ring system in moderate yield.17 At this point, compound 13 possessed the two handles needed to add the required substitution on the template to complete the synthesis. Coupling of compound 13 with the corresponding boronic acid using Pd(PPh3)4 occurred in a regioselective manner at the least hindered bromine, affording 3-substituted quinoline 14. Under similar reaction conditions, the heterocycles were attached onto C-8 of the quinoline system to give the desired final compounds. The second route utilized to synthesize derivatives of the quinoline with this specific substitution pattern is depicted in Scheme 2. For this alternative route, attachment of the heterocyclic group was done before the formation of the quinoline system. Scheme 1, being more versatile by virtue of introducing one of the variable elements (heterocyclic head group) after the modest-yielding quinoline-forming cyclization step, was used for most of the examples.
The naphthalene core with the appropriate substitution pattern was built starting from 7-bromotetralin-1-one (18), which was transformed to the corresponding trimethylsilane (TMS) enolate followed by alkylation to yield compound 19 (Scheme 3). Bromination at the α position of the ketone group afforded tetralin-1-one 20, which was subsequently aromatized and the resulting phenol O-alkylated to obtain trisubstituted naphthalene 21. Introduction of the second bromine para to the methoxy group was carried out as described above to obtain compound 22. With the required substitution on the naphthalene core in place, the 4-(methanesulfonamido)phenyl and heterocycle groups were introduced to obtain the final product 24.
Scheme 3. Synthesis of 2,5,7,8-tetrasubstituted naphthalene core.

Reagents and conditions: (a) TMSCl, NaI, Et3N, CH3CN, 69%; (b) 2-chloro-2-methyl-propane, TiCl4, CH2Cl2, −40 °C, 50%; (c) Br2, AcOH, 50 °C, 98%; (d) LiBr, Li2CO3, DMF, 100 °C; (e) MeI, K2CO3, DMF, 96% two steps; (f) Br2, AcOH, 98%; (g) 4-(methanesulfonamido)phenylboronic acid, Pd(PPh3)4, Na2CO3, MeOH, toluene, 115 °C, 54%.
Synthesis of the 2,5,7,8-tetrasubstituted quinoline was achieved by using a three component imino Diels-Alder reaction as reported by Kouznetsov.18 Treatment of aniline 25 with 4-nitrobenzaldehyde followed by addition of the dienophile and Lewis acid gave a mixture of tetrahydroquinolines and desired quinoline 26. The mixture of tetrahydroquinolines was oxidized with ceric ammonium nitrate (CAN) to afford quinoline 26 in 22% overall yield. Compound 26 was converted to the final product by reduction of its nitro group followed by methanesulfonylation of the resultant aniline and addition of the heterocycle head group at the C-5 position via cross coupling (Scheme 4).
Scheme 4. Synthesis of 2,5,7,8-tetrasubstituted quinoline core.
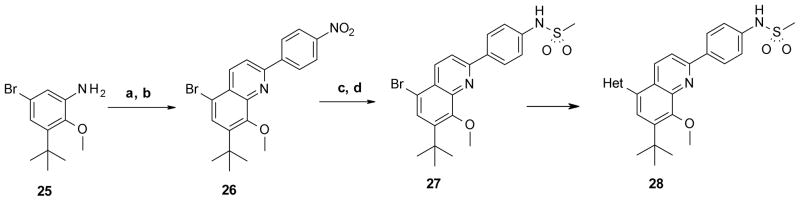
Reagents and conditions: (a) 4-nitrobenzaldehyde, N-vinylpyrrolidin-2-one, BiCl3, CH3CN; (b) CAN, CH3CN, 0 °C, 22% two steps; (c) Fe, NH4Cl, MeOH, water, reflux, 73%; (d) MsCl, pyridine, CH2Cl2, 0 °C, 86%.
The last bicyclic template synthesized was the 2,4,7-trisubstituted quinazoline as described in Scheme 5. 2-Amino-4-bromo-benzamide (29) was reacted with pivaloyl chloride followed by thermal ring closure under alkaline conditions to give corresponding quinazolin-4-one 30. Coupling of compound 30 with 4-(methylsulfonamido)phenylboronic acid followed by reaction with phosphorous oxychloride afforded the chloroquinazoline 31 in good yield. The desired final product 32 was obtained following standard cross coupling conditions for the addition of the heterocycle on the C-4 position.
Scheme 5. Synthesis of 2,4,7-trisubstituted quinazoline core.

Reagents and conditions: (a) Pivaloyl chloride, Et3N, CH2Cl2; (b) aq. NaOH, EtOH, reflux, 52% two steps; (c) 4-(methanesulfonamido)phenylboronic acid, Pd(PPh3)4, Na2CO3, MeOH, toluene, 115 °C, 84%; (d) POCl3, DIPEA, benzene, reflux, 87%.
RESULTS AND DISCUSSION
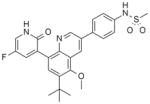
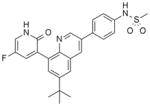
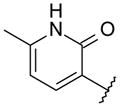
Compound 33, prepared according to Scheme 1, was made first to determine if the bicyclic core could replace the stilbene moiety in our previous series and still maintain potency. Compound 33 has the same 2(1H)-pyridone head as our lead stilbene 7 to allow us to make a close comparison. For 5-fluoropyridone 33, the nitrogen of the quinoline system was expected to face the vicinity of the backbone of Tyr448 and Gly449, where a conserved molecule of water is normally observed in the crystal structures. It was thought that a hydrogen bond acceptor facing that area would be beneficial.7 Compound 33 displayed single-digit nM activity and equipotency against both GT-1a and GT-1b strains in the replicon assay (Table 1). An attractive advantage of 5-fluoropyridone 33 was that it had a slightly better activity against GT-1a than stilbene 7. With this encouraging result, we explored the possibility of removing the methoxy group from the core to determine its effect on potency. A 4-to 6-fold drop in activity was observed with the desmethoxy analog 34 indicating that the bicyclic core required methoxy group for good potency.
After these initial results, we continued exploring different bicyclic cores to determine which would give us the best potency and overall profile. For this exploration, 5-fluoro pyridone head was replaced with the uracil heterocycle while maintaining the methoxy, tert-butyl, and N-phenylmethanesulfonamide substituents. Uracil 35 displayed a decrease in activity against GT-1a, but maintained the same potency against GT-1b with respect to 5-fluoropyridone 33 in the replicon assay. For compounds 36–38, the uracil head group was held constant so the activity of different bicyclic cores could be compared with quinoline 35. Naphthalene 36 showed a 2- to 3- fold increase in potency against both genotypes with respect to quinoline 35. Moving the quinoline nitrogen of 35 to the opposite position (37) caused the potency to decrease significantly. This drop in activity could be partially explained by the expected almost planar orientation of the N-phenylmethanesulfonamide group with respect to the quinoline ring due to the introduction of the quinoline nitrogen adjacent to the phenyl ring. Finally, quinazoline 38 was screened only against GT-1a and also displayed lower activity than quinoline 35.
This initial exploration showed that the naphthalene core (36) had the best potency profile among the four bicyclic cores. Even though the naphthalene core presented the best activity, the 3,5,6,8-tetrasubstituted quinoline core was selected to continue exploring modifications on other parts of the template due to its ease of synthesis, flexibility to make modifications, and excellent potency in the replicon assay.
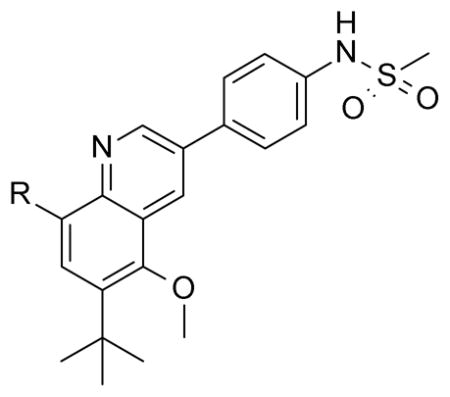
With these initial results, we started our optimization efforts of the 3,5,6,8-tetrasubstituted quinoline core by exploring different heterocyclic head groups. Table 2 shows the compounds made along with their activity in the replicon assay. The first three compounds after compound 35 have a 2(1H)-pyridone as a head group. Of the three, compound 41 stood out because of its excellent potency and equipotency against both GT-1a and GT-1b. The dihydrouracil 42 displayed good potency and selectivity comparable to uracil 35. When the dihydrouracil head of 42 was replaced by a cyclic urea moiety, the potency of the compound (43) decreased by 14-fold against GT-1a and 6-fold against GT-1b. A possible explanation for the decrease in potency is the difference in pKa of the NH of the dihydrouracil with respect to that of the cyclic urea (calculated19 pKa for dihydrouracil 42 8.7 and cyclic urea 43 13.7). The increase in pKa could decrease the strength of the interaction of the NH with the backbone carbonyl of Gln446 resulting in a decrease on potency. C-linked uracil 44 displayed improved potency (~ 2-fold) against both genotypes with respect to N-linked uracil 35.
Table 2.
Different Head Groups on the 3,5,6,8-Tetrasubstituted Quinoline Core with Replicon (GT-1a and GT-1b) Data.
| |||
|---|---|---|---|
| compd | R | GT-1a EC50 (nM)a |
GT-1b EC50 (nM)a |
| 35 |
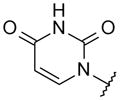
|
7.2 | 3.2 |
| 39 |
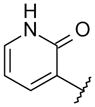
|
5.2 | 3.1 |
| 40 |
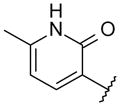
|
4.2 | 2.0 |
| 41 |
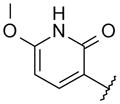
|
1.1 | 1.0 |
| 42 |
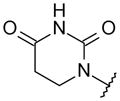
|
4.6 | 3.2 |
| 43 |
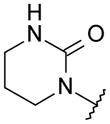
|
64 | 20 |
| 44 |
|
3.0 | 1.5 |
EC50 measured using stable HCV (GT-1a H77, GT-1b Con1) subgenomic replicon assay (n ≥ 2).
The design of the bicyclic cores and head replacements was based on our understanding of the binding mode of the stilbene series in the palm I site.16 The expected binding mode of the bicyclic series was confirmed from the crystal structure of compound 41 bound to NS5B (Figure 3). A careful analysis of the data shows that the 2(1H)-pyridone head is within hydrogen bond distance to the backbone carbonyl of Gln446 and the NH of Tyr448 (2.9 A in both cases). The lower ring of the quinoline template makes edge-to-face interactions with the side chain of Try448, and the tert-butyl group fills up the lower lipophilic pocket. These interactions are the same as those observed previously with the original fragment 6.15 The nitrogen of the quinoline core is within bonding distance to a conserved molecule of water (not shown on Figure 3). The main role for this ring, as replacement of the vinyl group, is to hold the linker of the methanesulfonamide in the right position. The phenyl ring that links the quinoline with the methanesulfonamide makes an edge-to-face interaction with Phe193 and the sulfonamide group is within hydrogen bond distance from the residues: Asp318, Asn 291, and Ser288 (2.7, 2.8, and 3.2 A, respectively). The sulfonamide functional group was essential for the high potency of the template, indicative of the importance of these interactions. The oxygen of the methoxy group on C-6 of the 2(1H)-pyridone is in close proximity (3.3 A) to the carbon of the backbone carbonyl Gly410.
Figure 3.

Cocrystal structure of compound 41 with NS5B polymerase (GT-1b BK) in palm I allosteric site. Crystal structure figures were drawn with PyMOL.20
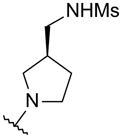
At this point in our efforts to improve activity, the quinoline series had shown excellent potency in the HCV replicon assay against both GT-1a and GT-1b. Also, most of the physicochemical and ADME in vitro properties were within the desirable range (e.g. microsomal stability, CYP inhibition, permeability) except for solubility. Some of the compounds with the best biological activity exhibited low aqueous solubility (< 5 μg/mL). One approach to improve this property was by reducing the planarity of the compounds. With this in mind, we explored different replacements of the phenyl linker between the quinoline and the methanesulfonamide (Table 3). The four carbon alkyl or alkyne linkers (45 and 46, respectively) exhibited activity in the enzyme assay but it was not transferred to the replicon assay. The weak activity observed for these two compounds could be attributed to the different possible conformations of the linker which would prevent the methanesulfonamide group from making favorable interactions with Asp318, Asn 291, and Ser288. In order to minimize the number of possible conformations, we explored saturated ring systems as linkers. Azetidine (47) and piperidine (49) linkers gave similar results as the alkyl chains. To the contrary, the pyrrolidine ring linker (48) displayed the same potency as the corresponding phenyl linker (39). These results were expected since the conformationally-restricted ring systems of 47–49 would place the methanesulfonamide group in slightly different positions resulting in different activities. Compound 48 was cocrystallized with the NS5B polymerase and it demonstrates in the palm I site the same interactions as described for quinoline 41. The pyrrolidine ring is within van der Waals radius contact distance of Phe193 (3.9–4.2 A) and places the methanesulfonamide in the right position to make the same network interactions in the catalytic site as quinoline 41 (Figure 4).
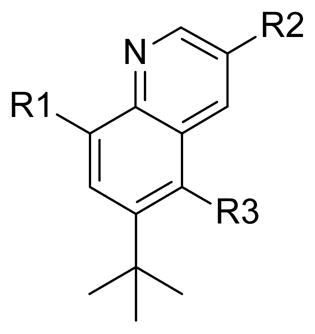
Table 3.
Replacements of the Phenyl Linker with NS5B (GT-1b) and Replicon (GT-1a) data.
| ||||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | GT-1b IC50 (nM)a |
GT-1a EC50 (nM)b |
Aqueous Solubility (μg/mL)c |
| 45 |
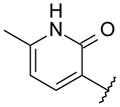
|
|
OMe | 12 | > 1000 | 168 |
| 46 |
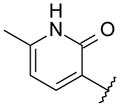
|
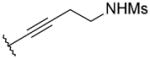
|
OMe | 5 | 341 | < 1 |
| 47 |
|
|
OMe | 3.5 | 532 | NA |
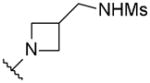
| 48 |
|
|
OMe | 0.2 | 7.9 | 11 |
| 49 |
|
|
H | 625 | > 1000 | 66 |
IC50 against NS5B enzyme were measured using GT-1b Con1 strain and reported as nM.
EC50 measured using stable HCV GT-1a H77 subgenomic replicon assay (n ≥ 2).
Aqueous solubility was measured using a thermodynamic solubility assay.
Figure 4.

Cocrystal structure of compound 48 with NS5B (GT-1b BK) polymerase in palm I allosteric site.
As anticipated, the replacement of the phenyl ring with a saturated linker (in this case the pyrrolidine ring) led to an increase in aqueous solubility (Table 3). The combination of good potency and improved solubility made the pyrrolidine linker an attractive option for further exploration. The phenyl linker of the best compounds described on Tables 1 and 2 was replaced with the pyrrolidine linker. The replacements also led to an increase in aqueous solubility, but potency was not always maintained in comparison to the corresponding phenyl linker compounds (data not shown). From this set of compounds, pyridone 50, the pyrrolidine analog of compound 41, had the best combination of overall properties (Figure 5). Although the potency of pyrrolidine 50 in the replicon assay was weaker than compound 41, it still maintained almost equal potency against both GT-1a and GT-1b and good aqueous solubility.
Figure 5.
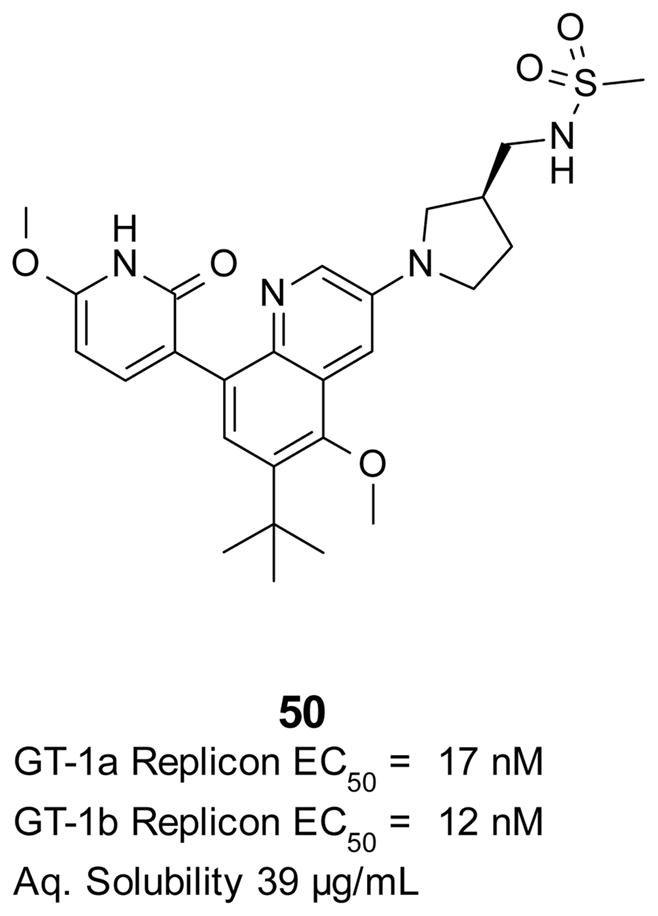
Best pyrrolidine derivative from the bicyclic series.
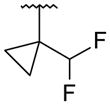
It was desired to select a compound with long half-life in vivo to be able to dose the compound with a q.d. or b.i.d. dosing regimen in the clinic. Even though the majority of the compounds screened in our in vitro hepatocyte assay had medium to high stability (data not shown), a focused effort was carried out to determine if this parameter could be improved. As we showed in the stilbene template,16 oxidation of the tert-butyl group in the bicyclic template was one of the major routes of metabolism (data not shown). Based on this observation, replacements of the tert-butyl were made on the uracil 35 template to determine if it was possible to improve the stability. Replacement of the methyl groups on the tert-butyl by fluorines (trifluoromethyl 51) led to improved stability in human and rat hepatocytes, but a large drop in activity was observed against both GT-1a and GT-1b (Table 4). These results supported our assumption that the metabolism of tert-butyl group was responsible for most of the clearance observed. Two additional modifications were made to determine if cellular activity could be maintained while improving metabolic stability. Extension of the trifluoromethyl group by one carbon (1,1,1,-trifuoroethyl 52) had a similar outcome as with trifluoromethyl 51. The loss of potency seen in compounds 51 and 52 could be due to the less than optimal filling of the lower lipophilic pocket in the palm I site. To address this observation, compound 53 was prepared which contains a cyclopropyl group instead of two of the methyl groups of the tert-butyl and a difluoromethyl in replacement for the third methyl. These two changes maintain similar filling of the pocket as the tert-butyl but with metabolically stable substitutions. Cyclopropyl 53 displayed the expected high metabolic stability with an improvement in potency of 3- to 9-fold with respect to compounds 51 and 52 but still weaker than uracil 35. Similar tert-butyl replacements have been recently reported with similar improvements in in vitro stability.21 Unfortunately, the overall profile of these tert-butyl replacements did not meet our objective and were not pursued further.
Table 4.
Replacements of tert-Butyl Group with Replicon (GT-1a and GT-1b) and in vitro Hepatocytes Data.
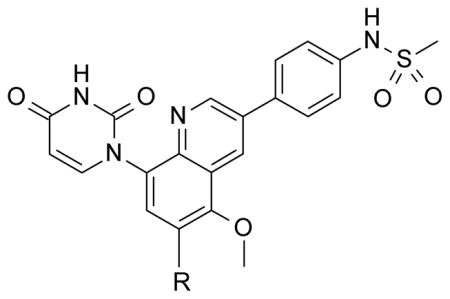
| |||||
|---|---|---|---|---|---|
| compd | R | GT-1a EC50 (nM)a |
GT-1b EC50 (nM)a |
HHb | RHb |
| 35 |
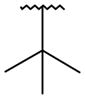
|
7.2 | 3.2 | 7.9 | 20 |
| 51 |
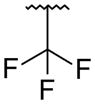
|
821 | 57 | 1.5 | 1.8 |
| 52 |
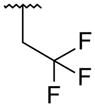
|
839 | 119 | 0.8 | 3.7 |
| 53 |
|
95 | 16 | 0.1 | 1.7 |
EC50 measured using stable HCV (GT-1a H77 and GT-1b Con1) subgenomic replicon assay (n ≥ 2).
Human hepatocytes (HH) and rat hepatocytes (RH) intrinsic clearance is reported in μL/min/106 cells. Human hepatocyte category ranking: high stability < 2.8, low stability > 15; rat hepatocyte category ranking: high stability < 5.4, low stability > 29.
After exploring the four different substitutions of the bicyclic template and understanding their role, five compounds were selected for further profiling in the replicon assay (Table 5). All the compounds selected showed potent inhibition against both GT-1a and GT-1b with almost equipotency against both genotypes (ratio 1a/1b = 1.1–2.2). The compounds were screened in the replicon assay in the presence of 40% human serum (HuS), a more relevant measurement of activity in terms of prediction of clinical efficacy. All the compounds maintained potency at or below 10 nM under the conditions of this assay. The plasma shift observed was relatively small (0.6- to 3.5-fold) for all the compounds except for pyridone 41 (3.8- to 9.4-fold). The EC90 in the presence of 40% HuS was used to determine the plasma trough concentration (Cmin) needed to obtain efficacy in the clinic. We targeted potency at 5-fold coverage above the measured EC90. Since this series generally had a weaker potency against GT-1a in the presence of 40% HuS, we decided to use this value to predict efficacy in human.
Table 5.
Activity in the Replicon Assay for a Selected Set of Compounds in the Bicyclic Series
| compd | GT-1a EC50 (nM)a |
GT-1b EC50 (nM)a |
Ratio 1a/1b | GT-1a 40% HuSb EC50 (nM)a |
GT-1b 40% HuS EC50 (nM)a |
Plasma Shift fold | GT-1a 40% HuS EC90 (nM)a |
GT-1b 40% HuS EC90 (nM)a |
|---|---|---|---|---|---|---|---|---|
| 7 | 8.0 | 3.0 | 2.7 | 66 | 22 | 7.3–8.2 | 531 | 128 |
| 35 | 7.2 | 3.2 | 2.2 | 16 | 5.6 | 1.7–2.2 | 123 | 29 |
| 39 | 5.2 | 3.1 | 1.7 | 4.0 | 11 | 0.8–3.5 | 25 | 47 |
| 41 | 1.1 | 1.0 | 1.1 | 10 | 3.8 | 3.8–9.4 | 63 | 19 |
| 42 | 4.6 | 3.2 | 1.4 | 10 | 3.5 | 1.7–2.1 | 48 | 20 |
| 50 | 17 | 12 | 1.4 | 10 | 7.5 | 0.6 | 61 | 44 |
EC50 and EC90 measured using stable HCV (GT-1a H77, GT-1b Con1) subgenomic replicon assay (n ≥ 2).
Human serum (HS).
From this set of compounds, 41 and 50 were selected for further biological profiling against a panel of GT-1a and GT-1b NS5B clinical isolates from untreated patients in a transient replicon assay.22 Both compounds showed similar or improved potency against the clinical isolates (transient replicon, Table 6) compared to the potencies observed using the stable HCV replicons (Table 5). Differences between the two HCV replicon systems have been previously reported23 and could be due to a better inhibition of the HCV replicase complex in the transient replicon by the non-nucleoside inhibitors than in the stable replicon system. The major loss in potency in comparison with the rest of the GT-1a panel was seen with clinical isolate RO-38. The sequence of this NS5B isolate contains mutation S556G which is located close to the catalytic site where both of our compounds bind. The mean EC50 for compounds 41 and 50 in this set of clinical isolates was shown to be very similar. Against the panel of GT-1b clinical isolates, compound 41 displayed consistent values against the isolates and a mean EC50 similar to the one observed with the stable line. Compound 50 displayed a slight range of activities across the panel, but still with a low single-digit nM mean EC50 value.
Table 6.
Activity of 41 and 50 Against GT-1a and GT-1b Clinical Isolates.
| GT | Clinical Isolate | compd 41 EC50 ± SEM (nM) |
compd 50 EC50 ± SEM (nM) |
|---|---|---|---|
| 1a | H77 | 0.3 ± 0.03 | 0.65 ± 0.12 |
| RO-18 | 0.4 ± 0.05 | 0.5 ± 0.26 | |
| RO-24 | 1.1 ± 0.23 | 2.5 ± 0.41 | |
| RO-28 | 0.9 ± 0.05 | 0.2 ± 0.08 | |
| RO-33 | 0.6 ± 0.09 | 0.7 ± 0.03 | |
| RO-34 | 0.5 ± 0.02 | 0.4 ± 0.08 | |
| RO-35 | 0.3 ± 0.09 | 0.5 ± 0.08 | |
| RO-38 | 4.3 ± 0.07 | 9.5 ± 0.41 | |
| RO-41 | 0.6 ± 0.17 | 0.27 ± 0.07 | |
| mean | 0.72 | 0.8 | |
| 1b | Con1 | 0.32 ± 0.08 | 1.20 ± 0.27 |
| RO-1 | 0.11 ± 0.01 | 0.30 ± 0.06 | |
| RO-2 | 0.19 ± 0.003 | 4.00 ± 1.63 | |
| RO-3 | 0.43 ± 0.02 | 6.1 ± 0.12 | |
| RO-4 | 0.11 ± 0.003 | 0.80 ± 0.16 | |
| RO-7 | 0.22 ± 0.02 | 3.05 ± 0.37 | |
| RO-8 | 0.33 ± 0.10 | 0.33 ± 0.10 | |
| RO-9 | 0.34 ± 0.04 | 3.50 ± 0.41 | |
| RO-10 | 0.34 ± 0.03 | 1.67 ± 0.33 | |
| mean | 0.26 | 1.6 |
EC50 measured using transient HCV subgenomic replicon assay (n ≥ 2). SEM, Standard Error Mean * Geometric mean was calculated.
The pharmacokinetic properties of compounds 41 and 50 were determined in rat and dog. Due to the low solubility of 41 (aqueous solubility < 1 μg/mL), this compound was dosed as a microprecipitated bulk powder (MBP) for oral dosing as described by Shah et al.24,25 Compound 41 showed low clearance in both rat and dog with a t1/2 of 4.3 h, which met our criteria to enable b.i.d. dosing in humans (Table 6). Bioavailability was moderate in both species achieving good levels at Cmax. In addition, concentrations of 0.62 μg/mL (1.22 μM) and 0.99 μg/mL (1.95 μM), in rat and dog respectively, were observed at 8 h after oral dosing. These concentrations cover over 19-fold the EC90 observed for 41 in the GT-1a replicon assay in the presence of 40% human serum. This pharmacokinetic data together with the biological profile described above, lent additional confidence that suppression of the virus in humans could be achieved with compound 41 based on our prediction model.26 It was estimated that 41 will show a viral load reduction of > 1.5 in a 3-days monotherapy with a dose of 49–120 mg b.i.d.
Compound 50 displayed a good profile in both species. When dosed to rats, it showed low clearance and a slightly better t1/2 than 41 with an excellent bioavailability. These characteristics allowed compound 50 to reach a concentration of 1.79 μg/mL (3.5 μM) at 8 h after oral dosing. This concentration covers over 50-fold the EC90 observed for 50 in the GT-1a replicon assay in the presence of 40% human serum. The pharmacokinetic parameters in dog for 50 were similar to those observed in rat.
Compounds 41 and 50 both possess overall characteristics desirable for a clinical candidate. Compound 41 (a.k.a. RO5471354) was ultimately selected for advancement to clinical development.
CONCLUSION
In summary, exploration of a diverse set of bicyclic cores to replace our previous stilbene core led us to the identification of the 3,5,6,8-tetrasubstituted quinolines. This series displayed excellent potency in the HCV replicon assay and a minimal separation in activity between GT-1a and GT-1b. Optimization of the head group steered us to the identification of the 6-methoxypyridone as one of the most promising heads due to its potency and equal activity against both GT-1a and GT-1b. Replacement of the phenyl linker for saturated alkyl and cycloalkyl groups identified 3-substituted pyrrolidine as a promising linker. This linker maintained the potency and improved the solubility of the template. Efforts to replace the tert-butyl for a more metabolically stable group were successful in improving the stability of the template, but the replacements did not maintain the desired potency in the replicon assay. Overall, the optimization efforts led us to select five compounds for further biological profiling. Compounds 41 and 50 were then selected for pharmacokinetic studies in rat and dog to determine their potential as clinical candidates. Both compounds met or exceeded our criteria for clinical advancement based on the results described above. Ultimately, compound 41 was chosen for clinical development.
Our efforts to explore different scaffolds to improve the profile of our lead compound 7 yielded a compound with better biological activity. Compound 41 is equipotent against the two GT and has a 6 fold improvement in the 40% HS replicon assay (Table 5).
The development of fragment 6 to a compound that was selected for clinical development is another successful case where the use of fragments15 together with structure-based design16 led to the selection of a clinical candidate.
EXPERIMENTAL
General information
All reactions were performed in round-bottom flasks. The flasks were fitted with rubber septa and reactions were conducted under inert atmosphere. Plastic syringes and stainless steel needles were used to transfer air- and moisture-sensitive liquids. Most commercial reagents were purchased from Sigma Aldrich, Alfa Aesar, Strem, Lancaster, AstaTech, or TCI, and used as received. Solvents were purchased from Mallinckrodt and anhydrous dimethylformamide (DMF) and tetrahydrofuran (THF) from Sigma-Aldrich in a Sure/Seal system. Purification of crude material was performed using AnaLogix IntelliFlashTM 280 or Teledyne Isco CombiFlash® Companion® systems using silica gel columns RediSep Rf (particle size 40–63 μpowder). Deuterated solvents were purchased from Sigma-Aldrich. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker Avance 300 MHz, 400 MHz, or 500 MHz spectrometers at 25 °C. Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3 δ 7.27, DMSO δ 2.50). Data is represented as follows: chemical shift, multiplicity (br. = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hertz (Hz), and integration. High resolution mass spectrometry (HRMS) data were obtained on a Thermo Finnigan LTQ Orbitrap XL spectrometer operated in ESI mode at a resolution of 30,000. Liquid chromatography-mass spectrometry (LC/MS) analyses were carried out using an Agilent 6140 single quadrupole in ES/APCI dual mode, and Agilent LC with a Zorbax SB-C18 2.1x30mm, 3.5μm column using a 0.02% TFA/water-acetonitrile solvent system. Microwave reactions were carried out in a Biotage Initiator reactor. Purity of tested compounds was ≥ 95% as confirmed by LC/MS.
Ethyl N-(4-tert-butyl-3-methoxy-phenyl)carbamate (10)
To a solution of 4-tert-butyl-3-methoxy-benzoic acid (9) (10.4 g, 50.0 mmol) in THF (200 mL) was added diphenylphosphoryl azide (11.3 mL, 52.4 mmol), Et3N (8.4 mL, 60.3 mmol), and EtOH (29 mL) at rt. The reaction was heated at reflux for 23 h, cooled to rt and concentrated. The residue was dissolved in EtOAc (300 mL), washed with saturated aqueous NaHCO3, dried over MgSO4, and evaporated. The crude material was purified over SiO2 (gradient: 0 to 25% EtOAc in hexane) to give ethyl carbamate 10 (10.12 g, 81%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.6 Hz, 1 H), 6.68 (dd, J = 8.2, 2.9 Hz, 1 H), 6.52 (br s, 1 H), 4.22 (q, J = 7.0 Hz, 2 H), 3.84 (s, 3H), 1.35 (s, 9 H), 1.31 (t, J = 7.3 Hz, 3 H). LC/MS (ES/APCI): 252.1 (M + H)+.
4-tert-Butyl-3-methoxy-aniline (11)
To a solution of ethyl carbamate 10 (11.88 g, 47.3 mmol) in EtOH (86 mL) was added a solution of KOH (29.17 g, 520 mmol) in EtOH (172 mL) at rt. The reaction mixture was heated at reflux for 15 h then cooled to rt and concentrated. The residue was partitioned with EtOAc (400 mL) and brine (400 mL). The organic layer was separated, dried over MgSO4, and concentrated to dryness. The residue was purified over SiO2 (gradient: 0 to 35% EtOAc in hexane) to give aniline 11 (8.16 g, 96%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, J = 7.2, 2.2 Hz, 1 H), 6.42-6.33 (m, 2 H), 3.82 (s, 3H), 1.35 (s, 9 H). LC/MS (ES/APCI): 180.17 (M + H)+.
2-Bromo-4-tert-butyl-5-methoxy-phenylamine (12)
NBS (8.1 g, 45.5 mmol) was added to a solution of aniline 11 (8.16 g, 45.5 mmol) in DMF (150 mL) at rt. The reaction mixture was stirred for 5 h at rt, diluted with EtOAc/hexane (1:1, 300 mL), washed with 1N NaOH (100 mL), brine (300 mL), dried over MgSO4, filtered, and concentrated to afford crude bromoaniline 12 (11.75 g, quantitative) as brownish oil after drying under high vacuum. 1H NMR (400 MHz, CDCl3) δ 7.24 (s, 1 H), 6.40 (br s, 1 H), 3.78 (s, 3H), 1.31 (s, 9 H). LC/MS (ES): 258.04 (M + H)+.
3,8-Dibromo-6-tert-butyl-5-methoxy-quinoline (13)
A solution of Br2 (168 mg, 1.05 mmol) in AcOH (1.3 mL) was added to 2-bromoprop-2-enal (148 mg, 1.1 mmol) in AcOH (1.5 mL) at rt. After stirring for 15 min, a solution of bromoaniline 12 (260 mg, 1.01 mmol) in AcOH (5 mL) was added to the reaction. The reaction mixture was heated at 100 °C for 4 h. The reaction mixture was carefully poured into a cold saturated aqueous NaHCO3 then extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude residue was purified over SiO2 (gradient: 0 to 10% EtOAc in hexane) to afford dibromide 13 (170 mg, 45%) as a brownish solid. 1H NMR (400 MHz, CDCl3) δ 8.94 (d, J = 2.6 Hz, 1 H), 8.53 (d, J = 1.9 Hz, 1 H), 8.08 (s, 1 H), 3.95 (s, 3 H), 1.49 (s, 9 H). LC/MS (ES/APCI): 374.0 (M + H)+.
N-[4-(8-Bromo-6-tert-butyl-5-methoxy-3-quinolyl)-phenyl]methanesulfonamide (14)
A sealed vial containing dibromide 13 (850 mg, 2.27 mmol), 4-(methanesulfonamido)phenylboronic acid (539 mg, 2.5 mmol), Pd(PPh3)4 (263 mg, 0.227 mmol), and Na2CO3 (725 mg, 6.83 mmol) in a mixture of MeOH (8 mL) and toluene (5 mL) was irradiated in a microwave reactor at 120 °C for 1 h. The reaction mixture was cooled to rt and diluted with EtOAc. The organic layer was washed with water, dried over MgSO4, filtered, and concentrated under vacuum. The crude residue was purified over SiO2 (gradient: 0 to 70% EtOAc in hexane) to afford 8-bromoquinoline 14 (600 mg, 57%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 9.20 (d, J = 2.0 Hz, 1 H), 8.50 (d, J = 2.0 Hz, 1 H), 8.08 (s, 1 H), 7.73 (d, J = 8.1 Hz, 2 H), 7.40 (d, J = 7.7 Hz, 2 H), 6.50 (br s, 1 H), 4.00 (s, 3 H), 3.11 (s, 3H), 1.52 (s, 9 H). LC/MS (ES): 462.99 (M + H)+.
4-tert-Butyl-5-methoxy-2-(2-methoxy-3-pyridyl)aniline (16)
A sealed vial containing bromoaniline 12 (700 mg, 2.71 mmol), (2-methoxy-3-pyridyl)boronic acid (612 mg, 4.4 mmol), Pd(PPh3)4 (231 mg, 0.2 mmol), and Na2CO3 (636 mg, 6 mmol) in a mixture of MeOH (1 mL) and CH2Cl2 (9 mL) was irradiated in a microwave reactor at 115 °C for 1 h. The reaction mixture was cooled to rt and diluted with EtOAc. The organic layer was washed with water, dried over MgSO4, filtered, and concentrated. The crude residue was purified over SiO2 (gradient: 0 to 50% EtOAc in hexane) to afford aniline 16 (420 mg, 54%) as a brownish oil. 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 4.7, 1.4 Hz, 1 H), 7.57 (dd, J = 6.8, 1.6 Hz, 1 H), 6.98 (dd, J = 7.1, 4.6 Hz, 1 H), 6.96 (s, 1 H), 6.32 (s, 1 H), 3.99 (s, 3 H), 3.83 (s, 3 H), 3.72-3.64 (br s, 2 H), 1.34 (s, 9 H). LC/MS (ES): 287.13 (M + H)+.
3-(3-Bromo-6-tert-butyl-5-methoxy-8-quinolyl)-1H-pyridin-2-one (17)
A solution of Br2 (191 mg, 1.2 mmol) in AcOH (5 mL) was added to 2-bromoprop-2-enal (230 mg, 1.7 mmol) in AcOH (5 mL) at rt. The solution was titrated to the appearance of a faint reddish color. After stirring at rt for 15 min, a solution of aniline 16 (420 mg, 1.47 mmol) in AcOH (5 mL) was added. The reaction mixture was heated at 100 °C for 2 h. The reaction mixture was carefully poured into a cold saturated aqueous NaHCO3 and then extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated. The crude residue was purified over SiO2 (1:1, EtOAc/hexane) to afford the corresponding 3-bromoquinoline 17 (155 mg, 27%) as a brownish oil. 1H NMR (400 MHz, CDCl3) δ 8.82 (d, J = 2.3 Hz, 1 H), 8.55 (d, J = 1.7 Hz, 1 H), 7.88 (s, 1 H), 7.62 (dd, J = 6.6, 1.5 Hz, 1 H), 7.35 (dd, J = 6.4, 1.8 Hz, 1 H), 6.38 (t, J = 6.9 Hz, 1 H), 3.99 (s, 3 H), 1.52 (s, 9 H). LC/MS (ES): 388.94 (M + H)+.
7-Bromo-2-tert-butyl-tetralin-1-one (19)
To a suspension of 7-bromotetralin-1-one (18) (10.0 g, 44.4 mmol) in CH3CN (10 mL) was added Et3N (7.43 mL, 53.3 mmol) followed by chloro(trimethyl)silane (6.81 mL, 53.6 mmol) at rt under argon. A solution of NaI (7.99 g, 53.3 mmol) in CH3CN (72 mL) was added dropwise to the white suspension via cannula and stirred at rt for 30 min. The reaction mixture was diluted with ice-cold pentane (200 mL) then poured over ice water. The three different layers were separated and the top layer (pentane) dried over MgSO4, filtered, and concentrated to give a yellow oil. The residue was purified over SiO2 (hexane) to give pure (7-bromo-3,4-dihydronaphthalen-1-yl)oxy-trimethyl-silane (9.1 g, 69%). 1H NMR (300 MHz, CDCl3) δ 7.51 (d, J = 1.9 Hz, 1 H), 7.26 (dd, J = 7.9, 2.3 Hz, 1 H), 6.97 (d, J = 7.9 Hz, 1 H), 5.21 (t, J = 4.9 Hz, 1 H), 2.70 (t, J = 7.9 Hz, 2 H), 2.31 (ddd, J = 12.8, 8.7, 4.9 Hz, 2 H), 0.26 (s, 9H).
A solution of (7-bromo-3,4-dihydronaphthalen-1-yl)oxy-trimethyl-silane (6.85 g, 23.0 mmol) in CH2Cl2 (23 mL) was cooled to −40 °C and 2-chloro-2-methyl-propane (1.32 mL, 12.0 mmol) was added under N2. A solution of TiCl4 (1.27 mL, 11.6 mmol) in CH2Cl2 (6 mL) at −40 °C was added to the above solution dropwise while the reaction mixture stirred vigorously to a dark brown/purple suspension. The reaction was allowed to reach rt and stirred over 3 days. The reaction mixture was poured over ice, diluted with EtOAc and water, neutralized with saturated aqueous NaHCO3, and filtered over a plug of Celite® to remove chunky white precipitate. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The residue was purified over SiO2 (5/95, EtOAc/hexane) to yield tert-butyltetralin-1-one 19 (3.26 g, 50%) as yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 2.1 Hz, 1 H), 7.51 (dd, J = 8.2, 2.1 Hz, 1 H), 7.09 (d, J = 8.6 Hz, 1 H), 3.03–2.81 (m, 2 H), 2.34–2.23 (m, 2 H), 2.0–1.83 (m, 1 H), 1.09 (s, 9 H). LC/MS (ES/APCI): 281.1 (M + H)+.
2,7-Dibromo-2-tert-butyl-tetralin-1-one (20)
To a solution of tert-butyltetralin-1-one 19 (3.0 g, 10.7 mmol) in AcOH (40 mL) was added a solution of Br2 (0.6 mL, 11.6 mmol) in AcOH (20 mL) dropwise over 20 min via cannula to give a dark orange suspension. The reaction was stirred at rt for 1 h then warmed to 50 °C and stirred for 1 h. An extra quantity of neat Br2 (0.1 mL, 1.9 mmol) was added and continued heating at 50 °C for another 1.5 h. The reaction was poured over ice, diluted with EtOAc and water, and neutralized with saturated aqueous NaHCO3. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (hexane) to obtain compound 20 (3.8 g, 98%). 1H NMR (300 MHz, CDCl3) δ 8.18 (d, J = 2.1 Hz, 1 H), 7.58 (dd, J = 8.5, 2.1 Hz, 1 H), 7.13 (d, J = 8.1 Hz, 1 H), 3.24 (ddd, J = 17.8, 12.7, 5.1 Hz, 1 H), 2.85 (ddd, J = 17.4, 5.1, 2.5 Hz, 1 H), 2.55 (ddd, J = 14.9, 4.7, 3.0 Hz, 1 H), 2.29 (ddd, J = 17.0, 11.5, 4.7 Hz, 1 H), 1.31 (s, 9 H).
7-Bromo-2-tert-butyl-1-methoxy-naphthalene (21)
A white suspension of compound 20 (3.8 g, 10.6 mmol), LiBr (275 mg, 3.17 mmol), and Li2CO3 (780 mg, 10.6 mmol) in DMF (44.0 mL) was bubbled with argon for 10 min. The mixture was heated at 100 °C under N2 for 1 h then cooled to rt. The reaction was diluted with EtOAc, washed with water, brine, dried over MgSO4, filtered, and concentrated to obtain 7-bromo-2-tert-butyl-naphthalen-1-ol (3.0 g) as a light brown viscous oil in pure form and used in the next reaction without purification. 1H NMR (300 MHz, CDCl3) δ 8.27 (br s, 1 H), 7.64 (d, J = 9.4 Hz, 1 H), 7.49 (dd, J = 8.6, 2.4 Hz, 1 H), 7.23 (d, J = 8.2 Hz, 1 H), 7.37 (d, J = 9.0 Hz, 1 H), 1.52 (s, 9H).
To a suspension of 7-bromo-2-tert-butylnaphthalen-1-ol (2.9 g, 10.4 mmol) and K2CO3 (3.59 g, 26.0 mmol) in DMF (29.7 mL) was added MeI (0.78 mL, 12.5 mmol) and the mixture stirred at 25 °C for 18 h. The mixture was diluted with EtOAc and water and neutralized with 1N HCl. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated to obtain methoxynaphthalene 21 (3.0 g, 96% two steps) as a solid. 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 1.7 Hz, 1 H), 7.67 (d, J = 8.6 Hz, 1 H), 7.53–7.45 (m, 3 H), 3.95 (s, 3 H), 1.49 (s, 9 H).
4,7-Dibromo-2-tert-butyl-1-methoxy-naphthalene (22)
To a solution of methoxynaphthalene 21 (1.6 g, 5.46 mmol) in AcOH (30 mL) was added a solution of Br2 (0.28 mL, 5.46 mmol) in AcOH (20 mL) dropwise via addition funnel under N2. The reaction mixture was stirred at rt for 18 h then diluted with EtOAc and water, and neutralized with saturated aqueous NaHCO3. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated to obtain compound 22 (2.0 g, 98%) in pure form as an oil. 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 1.8 Hz, 1 H), 8.02 (d, J = 9.1 Hz, 1 H), 7.79 (s, 1 H), 7.59 (dd, J = 8.7, 1.8 Hz, 1 H), 3.94 (s, 3 H), 1.47 (s, 9 H).
N-[4-(5-Bromo-7-tert-butyl-8-methoxy-2-naphthyl)phenyl]methanesulfonamide (23)
A sealed vial containing compound 22 (1.0 g, 2.69 mmol), 4-(methanesulfonamido)phenyl boronic acid (578 mg, 2.69 mmol), Pd(PPh3)4 (155 mg, 0.134 mmol), and Na2CO3 (855 mg, 8.06 mmol) in MeOH (7.14 mL), toluene (3.57 mL), and water (1.79 mL) was irradiated in a microwave reactor at 115 °C for 1.5 h. The reaction mixture was diluted with EtOAc and water and was neutralized with 1N HCl. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 20 to 50% EtOAc in hexane) to obtain methanesulfonamide 23 (0.67 g, 54%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.25–8.19 (m, 2H), 7.80 (s, 1 H), 7.78–7.70 (m, 3 H), 7.35 (d, J = 8.5 Hz, 2 H), 6.42 (br s, 1 H), 3.99 (s, 3 H), 3.09 (s, 3 H), 1.50 (s, 9 H).
5-Bromo-7-tert-butyl-8-methoxy-2-(4-nitrophenyl)quinoline (26)
A mixture of 5-bromo-3-tert-butyl-2-methoxy-aniline 25 (2.0 g, 7.75 mmol) and 4-nitrobenzaldehyde (1.4 g, 11.62 mmol) in CH3CN (10 mL) was stirred at rt for 45 min. BiCl3 (0.49 g, 1.55 mmol) was added to this mixture followed by a solution of N-vinylpyrrolidin-2-one (1.72 g, 15.5 mmol) in CH3CN (10 mL) dropwise. The resulting mixture was stirred for 16 h then diluted with EtOAc and water (20 mL). A white solid precipitated and was filtered. The organic layer in the filtrate was dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 15 to 75% EtOAc in hexane) to afford a mixture of tetrahydroquinolines (978 mg) and compound 26 (312 mg).
To a solution of the mixture of tetrahydroquinolines (705 mg, 1.4 mmol) in CH3CN (10 mL) at 0 °C was added dropwise a solution of CAN (1.92 g, 3.51 mmol) in CH3CN (10 mL). The reaction mixture was stirred at 0 °C until all of the starting material was consumed. The reaction was diluted with EtOAc and water. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 1 to 15% EtOAc in hexane) to afford quinoline 26 (302 mg, 622 mg total, 22% overall yield) as a light yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.58 (d, J = 8.9 Hz, 1 H), 8.46 – 8.35 (m, 4 H), 8.0 (d, J = 8.9 Hz, 1 H), 7.88 (s, 1 H), 4.36 (s, 3 H), 1.56 (s, 9 H). LC/MS (ES/APCI): 416.0 [M + H]+.
N-[4-(5-Bromo-7-tert-butyl-8-methoxy-2-quinolyl)phenyl]methanesulfonamide (27)
A solution of quinoline 26 (300 mg, 0.72 mmol), iron powder (121 mg, 2.17 mmol) and NH4Cl (116 mg, 2.17 mmol) in a mixture of MeOH (30 mL) and water (10 mL) was heated at reflux for 1.5 h. The reaction mixture was cooled to rt and filtered through a pad of Celite®. The filtrate was concentrated and the residue was partitioned between EtOAc and water. The organic layer was dried over MgSO4, filtered, and concentrated to afford 4-(5-bromo-7-tert-butyl-8-methoxy-2-quinolyl)aniline (204 mg, 73%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 8.8 Hz, 1 H), 8.11 (d, J = 8.4 Hz, 2 H), 7.86 (d, J = 8.8 Hz, 1 H), 7.73 (s, 1 H), 6.81 (d, J = 8.4 Hz, 2 H), 4.34 (s, 3 H), 3.93 (br s, 1 H), 1.55 (s, 9 H).
To a solution of 4-(5-bromo-7-tert-butyl-8-methoxy-2-quinolyl)aniline (200 mg, 0.52 mmol) in CH2Cl2 (5 mL) at 0 °C was added pyridine (92 μL, 1.14 mmol) followed by MsCl (42 μL, 0.55 mmol). The reaction mixture was stirred at 0 °C for 45 min before it was quenched with water and diluted with EtOAc. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude was purified over SiO2 (gradient: 20 to 70% EtOAc in hexane) to afford methanesulfonamide 27 (208 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 9.2 Hz, 1 H), 8.26 (d, J = 8.5 Hz, 2 H), 7.91 (d, J = 9.1 Hz, 1 H), 7.81 (s, 1 H), 7.37 (d, J = 8.8 Hz, 2 H), 6.53 (br s, 1 H), 4.35 (s, 3 H), 3.09 (s, 3 H), 1.51 (s, 9 H).
7-Bromo-2-tert-butyl-3H-quinazolin-4-one (30)
To a solution of 2-amino-4-bromo-benzamide 29, (3.0 g, 14.0 mmol) and Et3N (1.84 g, 18.2 mmol) in CH2Cl2 (40 mL) at 0 °C was added dropwise pivaloyl chloride (1.68 g, 14.0 mmol). The resulting solution was allowed to reach rt and stirred for 18 h. Another portion of pivaloyl chloride (0.44 g) was added to the reaction at 0 °C and the solution stirred for 2 h at the same temperature then allowed to reach rt and stirred for additional 2 h. The mixture was concentrated under vacuum and the crude material was used in the next reaction without purification. To a suspension of the crude material in EtOH (28 mL) was added a solution of 10 M NaOH (2.8 mL, 28 mmol) and the resulting mixture refluxed under N2 for 1.5 h. The reaction was poured onto ice, neutralized with 1 N HCl, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated to quinazolinone 30 (2.07 g, 53% two steps) as a light yellow solid. 1H NMR (300 MHz, CDCl3) δ 10.21 (br s, 1 H), 8.10 (d, J = 8.4 Hz, 1 H), 7.92 (d, J = 1.7 Hz, 1 H), 7.56 (dd, J = 8.4, 1.5 Hz, 1 H), 1.45 (s, 9 H). LC/MS (ES/APCI): 281.0 [M + H]+.
N-[4-(2-tert-Butyl-4-chloro-quinazolin-7-yl)phenyl]methanesulfonamide (31)
A microwave vial was charged with quinazolinone 30 (500 mg, 1.78 mmol), 4-(methanesulfonamidophenyl)boronic acid (421 mg, 1.96 mmol), Pd(PPh3)4 (206 mg, 0.178 mmol), Na2CO3 (566 mg, 5.34 mmol), MeOH (3 mL), and toluene (1.5 mL). The mixture was purged with argon for 5 min and the vial was sealed. The reaction was heated in a microwave reactor at 115 °C for 30 min. The mixture was concentrated in vacuum and the residue triturated with Et2O to give the crude quinazoline as a brown solid. To a suspension of the crude quinazolinone (445 mg, 1.2 mmol) and DIPEA (358 mg, 0.38 mL, 1.83 mmol) in benzene (3 mL) was added POCl3 (88 μL, 0.97 mmol) and the mixture heated at reflux for 6.5 h. The reaction mixture was cooled to rt and concentrated to dryness. The residue was diluted with EtOAc, washed with water, 1 N HCl, water, brine, dried over MgSO4, and concentrated to give the crude chloroquinazoline 31 (406 mg, 87%) as an orange solid. 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 8.3 Hz, 1 H), 8.18 (d, J = 1.3 Hz, 1 H), 7.85 (dd, J = 8.8, 1.3 Hz, 1 H), 7.76 (d, J = 8.5 Hz, 2 H), 7.39 (d, J = 8.5 Hz, 2 H), 7.12 (br s, 1 H), 3.11 (s, 3 H), 1.51 (s, 9 H). LC/MS (ES/APCI): 388.0 [M - H]−.
N-{4-[6-tert-Butyl-8-(5-fluoro-2-oxo-1,2-dihydro-pyridin-3-yl)-5-methoxy-quinolin-3-yl]-phenyl}-methanesulfonamide (33) hydrobromide
A mixture of the 8-bromoquinoline 14 (150 mg, 0.324 mmol), (5-fluoro-2-methoxy-3-pyridyl)boronic acid (112 mg, 0.657 mmol), Pd(PPh3)4 (40 mg, 0.035 mmol), and Na2CO3 (179 mg, 1.68 mmol) was suspended in MeOH (1.6 mL) and CH2Cl2 (0.4 mL). The reaction mixture was heated in a microwave reactor at 115 °C for 1 h. After cooling the mixture to rt, was poured into saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was separated and washed with saturated aqueous NaHCO3, brine, dried over Na2SO4, filtered, and concentrated under vacuum. The product was purified over SiO2 (gradient: 27 to 50% EtOAc in hexane) to yield N-[4-[6-tert-butyl-8-(5-fluoro-2-methoxy-3-pyridyl)-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (121 mg, 73%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.05 (d, J = 2.3 Hz, 1 H), 8.53 (d, J = 2.3 Hz, 1 H), 8.08 (d, J = 3.0 Hz, 1 H), 7.78 (s, 1H), 7.72 (d, J = 8.7 Hz, 2 H), 7.75 (dd, J = 8.3, 3.0 Hz, 1 H), 7.38 (d, J = 8.3 Hz, 2 H), 6.60 (s, 1 H), 4.05 (s, 3 H), 3.89 (s, 3 H), 3.1 (s, 3 H), 1.54 (s, 9 H).
To a solution of N-[4-[6-tert-butyl-8-(5-fluoro-2-methoxy-3-pyridyl)-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (110 mg, 0.26 mmol) in acetic acid (2.5 mL) was added an aqueous solution of 48% HBr (0.06 mL, 1.1 mmol). The reaction vessel was sealed and heated at 60 °C for 18 h. The reaction was cooled to rt and a yellow powder precipitated. The solid was filtered, washed with Et2O, and dried in the oven under vacuum to give pyridone 33 (71 mg, 55%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.0 (s, 1 H), 9.19 (d, J = 2.0 Hz, 1 H), 8.67 (d, J = 2.0 Hz, 1 H), 7.93 (d, J = 8.1 Hz, 2 H), 7.84 (s, 1 H), 7.77 (dd, J = 8.1, 3.0 Hz, 1 H), 7.71 (br t, J = 3.1 Hz, 1 H), 7.40 (d, J = 8.6 Hz, 2 H), 4.03 (s, 3 H), 3.07 (s, 3 H), 1.50 (s, 9 H). LC/MS (ES): 496.07 (M + H)+.
N-[4-[6-tert-Butyl-8-(2,4-dioxopyrimidin-1-yl)-5-methoxy-3-quinolyl]phenyl] methanesulfonamide (35)
The introduction of the uracil group was done following the procedure reported by Wagner et al.27 A microwave vial was charged with 8-bromoquinoline 14 (200 mg, 0.431 mmol), uracil (72 mg, 0.642 mmol), N-(2-cyanophenyl)pyridine-2-carboxamide (19 mg, 0.085 mmol), CuI (8 mg, 0.042 mmol), K3PO4 (183 mg, 0.86 mmol), CH2Cl2, and MeOH. The suspension was degassed with argon followed by the addition of DMSO (1.5 mL). The vial was sealed and irradiated in a microwave reactor at 150 °C for 5 h. The reaction mixture was cooled to rt and diluted with EtOAc. The organic layer was washed with 1N NaHSO4, dried over MgSO4, filtered, and concentrated. The crude residue was purified over SiO2 (EtOAc) to afford uracil 35 (17 mg, 8%) as a semi solid. 1H NMR (400 MHz, CDCl3) δ 9.31 (br s, 1 H), 8.97 (d, J = 1.7 Hz, 1 H), 8.35 (d, J = 1.7 Hz, 1 H), 7.84 (s, 1 H), 7.63 (br s, 1 H), 7.44 (d, J = 8.7 Hz, 2 H), 7.38 (d, J = 7.9 Hz, 1 H), 7.09 (d, J = 8.4 Hz, 2 H), 5.92 (d, J = 7.9 Hz, 1 H), 4.01 (s, 3 H), 2.95 (s, 3 H), 1.55 (s, 9 H). HRMS (ESI): calcd. for C25H27O5N4S, 495.16967; found, 495.16763 [M + H]+.
N-[4-[7-tert-Butyl-5-(2,4-dioxopyrimidin-1-yl)-8-methoxy-2-naphthyl]phenyl] methanesulfonamide (36)
A flask was charged with N-[4-(5-bromo-7-tert-butyl-8-methoxynaphthyl)phenyl]methanesulfonamide 23 (350 mg, 0.76 mmol), tert-butyl carbamate (124 mg, 1.06 mmol), Pd2(dba)3·CHCl3 (104 mg, 0.1 mmol), 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl (145 mg, 0.34 mmol), sodium tert-butoxide (107 mg, 1.11 mmol), and toluene (10 mL) to give a white suspension. The mixture was flushed with argon for 10 min and stirred at rt over three days. The mixture was diluted with EtOAc and water and was neutralized with 1N HCl. The organic layer was separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 20 to 60% EtOAc in hexane) to give tert-butyl N-[3-tert-butyl-6-[4-(methanesulfonamido)phenyl]-4-methoxy-1-naphthyl]carbamate (196 mg) and used in the next reaction.
A solution of tert-butyl N-[3-tert-butyl-6-[4-(methanesulfonamido)phenyl]-4-methoxy-1-naphthyl]carbamate (0.196 g, 0.2 mmol), 4 M HCl in dioxane (0.49 mL, 1.97 mmol) in CH2Cl2 (1.5 mL) was stirred at rt for 3 h. The reaction was diluted with EtOAc, poured over ice, neutralized with saturated aqueous NaHCO3, washed with brine, dried over MgSO4, and concentrated. The crude product was purified by over SiO2 (gradient: 20 to 50% EtOAc in hexane) to yield N-[4-(5-amino-7-tert-butyl-8-methoxy-2-naphthyl)phenyl]methanesulfonamide (73 mg) as a light brown solid. 1H NMR (300 MHz, CDCl3) δ 8.21 (br s, 1 H), 7.85 (d, J = 8.6 Hz, 1 H), 7.73 (d, J = 8.6 Hz, 2 H), 7.63 (d, J = 9.2 Hz, 1 H), 7.35 (d, J = 8.0 Hz, 2 H), 6.84 (br s, 1 H), 6.74 (br s, 1 H), 3.93 (s, 3 H), 3.07 (s, 3 H), 1.49 (s, 9 H).
In a flask covered with foil, silver cyanate (135 mg, 0.9 mmol) was heated under high vacuum at 50 °C for 18 h. To this dried material was added (E)-3-methoxyacryloyl chloride (65 mg, 0.54 mmol) and dry toluene (1.3 mL) and the slurry heated at 120 °C under N2 for 30 min. The mixture was cooled to rt and the precipitate was allowed to settle in an ice bath. In a separate dry flask, N-[4-(5-amino-7-tert-butyl-8-methoxy-2-naphthyl)phenyl]methanesulfonamide (0.072 g, 0.18 mmol) was taken up in DMF (1 mL) and cooled to 0 °C. To the DMF solution was added dropwise the supernatant from the silver cyanate flask over 10 min. The light brown heterogeneous mixture formed was allowed to stir in an ice bath for 30 min. The mixture was diluted with EtOAc, washed with water, brine, and concentrated. The crude product was taken up in EtOH (1 mL) and added an 11% H2SO4 solution in water (1 mL). The mixture was heated at 120 °C for 1.5 h until all solid was in solution. The mixture was cooled to rt and poured over ice, diluted with EtOAc, washed with water, brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 50 to 100% EtOAc in hexane) to give naphthalene 36 (60 mg, 67%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 11.53 (br s, 1 H), 9.94 (br s, 1 H), 8.27 (br s, 1 H), 7.87 (br d, J = 8.7 Hz, 1 H), 7.82 (d, J = 8.7 Hz, 2 H), 7.72 (d, J = 7.9 Hz, 1 H), 7.67 (d, J = 8.7 Hz, 1 H), 7.60 (s, 1 H), 7.37 (d, J = 8.6 Hz, 2 H), 5.72 (d, J = 7.7 Hz, 1 H), 4.02 (s, 3 H), 3.06 (s, 3 H), 1.48 (s, 9 H). LC/MS (ES/APCI): 491.9 (M - H)−.
N-[4-[7-tert-Butyl-5-(2,4-dioxopyrimidin-1-yl)-8-methoxy-2-quinolyl]phenyl] methanesulfonamide (37)
A microwave vial was charged with bromoquinoline 27 (60 mg, 0.13 mmol), uracil (73 mg, 0.65 mmol), N-(2-cyanophenyl)pyridine-2-carboxamide (14 mg, 0.063 mmol), CuI (6 mg, 0.03 mmol), K3PO4 (137 mg, 0.64 mmol), and DMSO (5 mL). The mixture was irradiated in a microwave reactor at 150 °C for 5 h. The reaction mixture was cooled to rt and the pH adjusted to ca. 2 with 2 N HCl. The mixture was extracted with EtOAc and the organic extract was washed sequentially with water, brine, dried over MgSO4, and concentrated. The crude residue was purified over SiO2 (5/95, MeOH/CH2Cl2) to afford quinoline 37 (9.3 mg, 14%). 1H NMR (300 MHz, DMSO-d6) δ 11.49 (br s, 1 H), 10.09 (br s, 1 H), 8.29 (d, J = 8.8 Hz, 2 H), 8.12 (s, 2 H), 7.72 (d, J = 7.8 Hz, 1 H), 7.58 (s, 1 H), 7.38 (d, J = 8.9 Hz, 2 H), 5.71 (d, J = 7.8 Hz, 1 H), 4.33 (s, 3 H), 3.08 (s, 3 H), 1.47 (s, 9 H). LC/MS (ES/APCI): 495.0 [M + H]+.
N-[4-[2-tert-Butyl-4-(2,4-dioxopyrimidin-1-yl)quinazolin-7-yl]phenyl] methanesulfonamide (38)
A mixture of chloroquinazoline 31 (100 mg, 0.256 mmol), uracil (88 mg, 0.78 mmol), and Cs2CO3 (168 mg, 0.516 mmol) in DMSO (2 mL) was heated in a sealed vial at 125 °C for 2.5 h. The crude reaction was diluted with EtOAc and washed with water, brine, dried over MgSO4, and concentrated. The crude product was purified over SiO2 (gradient: 30 to 50% EtOAc in hexane) to give the desired product quinazoline 38 (54 mg, 45%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.75 (br s, 1 H), 10.06 (br s, 1 H), 8.28 (s, 1 H), 8.11–8.01 (m, 2 H), 8.01–7.91 (m, 3 H), 7.37 (d, J = 8.6 Hz, 2 H), 5.92 (d, J = 7.9 Hz, 1 H), 3.08 (s, 3 H), 1.47 (s, 9 H). LC/MS (ES/APCI): 464.0 [M - H]−.
N-[4-[6-tert-Butyl-5-methoxy-8-(2-oxo-1H-pyridin-3-yl)-3-quinolyl]phenyl] methanesulfonamide (39)
A microwave vial was charged with 3-bromoquinoline 17 (150 mg, 0.387 mmol), 4-(methanesulfonamido)phenylboronic acid (125 mg, 0.581 mmol), Pd(PPh3)4 (45 mg, 0.038 mmol), Na2CO3 (123 mg, 1.16 mmol), MeOH (1.5 mL), and CH2Cl2 (0.2 mL). The vial was sealed and irradiated in a microwave reactor at 115 °C for 1 h. The reaction mixture was cooled to rt and diluted with EtOAc. The organic layer was washed with water, dried over MgSO4, filtered, and concentrated. The crude residue was purified over SiO2 (5/95, MeOH/CH2Cl2) to afford pyridone 39 (40 mg, 21%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 1.7 Hz, 1 H), 8.44 (d, J = 1.7 Hz, 1 H), 7.73 (s, 1 H), 7.67 (dd, J = 6.7, 1.7 Hz, 1 H), 7.50–7.42 (m, 3 H), 7.20 (d, J = 8.8 Hz, 2 H), 6.49 (t, J = 6.7 Hz, 1 H), 4.01 (s, 3 H), 3.0 (s, 3 H), 1.54 (s, 9 H). HRMS (ESI): calcd. for C26H28O4N3S, 478.17950; found, 478.17770 [M + H]+.
N-[4-[6-tert-Butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3-quinolyl]phenyl] methanesulfonamide (41) hydrobromide
Using the two steps procedure for the preparation of 33 from 14 but using (2,6-dimethoxy-3-pyridyl)boronic acid, pyridone 41 (56 mg, 42% two steps) was obtained as yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (br s, 1 H), 9.18 (d, J = 1.7 Hz, 1 H), 8.79 (br s, 1 H), 7.94 (d, J = 8.3 Hz, 2 H), 7.76 (s, 1 H), 7.67 (d, J = 8.1 Hz, 1 H), 7.40 (d, J = 8.3 Hz, 2 H), 6.29 (d, J = 7.5 Hz, 1 H), 4.04 (s, 3 H), 3.89 (s, 3 H), 3.07 (s, 3 H), 1.50 (s, 9 H). HRMS (ESI): calcd. for C27H30O5N3S, 508.19007; found, 508.18848 [M + H]+.
N-[4-[6-tert-Butyl-8-(2,4-dioxohexahydropyrimidin-1-yl)-5-methoxy-3-quinolyl]phenyl] methanesulfonamide (42)
Acrylic acid (0.09 mL, 1.3 mmol) was added in 3 portions (0.02, 0.02, 0.05 mL) to a solution of N-[4-(8-amino-6-tert-butyl-5-methoxy-3-quinolyl)phenyl] methanesulfonamide (prepared with the same procedure as reported for N-[4-(5-amino-7-tert-butyl-8-methoxy-2-naphthyl)phenyl]methanesulfonamide in experimental for 36) (252 mg, 0.631 mmol) in toluene (2.5 mL) at rt. After the first addition, the reaction mixture was stirred at 120 °C for 2 h. After the second addition, the reaction mixture was stirred at 120 °C for 1 h and after the third addition, the reaction mixture was stirred at 120 °C overnight. The reaction mixture was cooled to rt and concentrated. The residue was taken in glacial AcOH (2 mL) and urea (95 mg, 1.58 mmol) was added. The reaction mixture was stirred at 120 °C for 6 h then cooled to rt and the solvent evaporated. The dark brown residue was partitioned between EtOAc and saturated aqueous NaHCO3. The aqueous layer was back extracted twice with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and evaporated. The residue was purified over SiO2 (gradient: 50 to 80% EtOAc in CH2Cl2) to give impure 42 (70 mg) as greenish gray powder. The powder was taken into a minimal amount of CH2Cl2 and the insoluble material was filtered and rinsed with a small amount of CH2Cl2 to give pure dihydrouracil 42 (40 mg, 13%) as a light gray powder. 1H NMR (300 MHz, DMSO-d6) δ 10.33 (br s, 1 H), 9.20 (d, J = 1.9 Hz, 1 H), 8.53 (d, J = 2.0 Hz, 1 H), 7.84 (d, J = 8.6 Hz, 2 H), 7.70 (s, 1 H), 7.31 (d, J = 8.5 Hz, 2 H), 4.00 (s, 3 H), 3.81 (br t, J = 6.5 Hz, 2 H), 2.98 (s, 3 H), 2.80 (t, J = 6.8 Hz, 2 H), 1.47 (s, 9 H). HRMS (ESI): calcd. for C25H29O5N4S, 497.18532; found, 497.18372 [M + H]+.
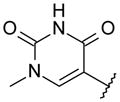
N-[[(3S)-1-[6-tert-Butyl-5-methoxy-8-(2-oxo-1H-pyridin-3-yl)-3-quinolyl]pyrrolidin-3-yl]methyl]methanesulfonamide (48)
A screw-capped vial was charged with bromoquinoline 17 (301 mg, 0.77 mmol), N-(S)-1-pyrrolidin-3-ylmethyl-methanesulfonamide (200 mg, 0.779 mmol), Pd(OAc)2 (18 mg, 0.08 mmol), P(tert-Bu)3 (24 μL, 0.079 mmol), tert-BuONa (298 mg, 3.1 mmol), and toluene (3 mL). The mixture was heated at 100 °C for 18 h. The reaction mixture was cooled to rt and diluted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3, dried over MgSO4, filtered, and concentrated. The crude residue was purified over SiO2 (gradient: 0 to 10% MeOH in CH2Cl2) to obtain product 48 (60 mg, 15%) as a solid. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 2.4 Hz, 1 H), 7.57 (dd, J = 6.8, 1.7 Hz, 1 H), 7.41 (s, 1 H), 7.36 (br d, J = 5.5 Hz, 1 H), 7.08 (d, J = 2.2 Hz, 1 H), 6.88 (br s, 1 H), 6.38 (t, J = 6.6 Hz, 1 H), 3.94 (s, 3 H), 3.47–3.34 (m, 2 H), 3.28 (q, J = 8.2 Hz, 1 H), 3.00–2.91 (m, 2 H), 2.89 (s, 3 H), 2.43 (quintuplet, J = 7.1 Hz, 1 H), 2.17–2.07 (m, 1 H), 1.89–1.77 (br s, 1 H), 1.68–1.58 (m, 1 H), 1.50 (s, 9 H). LC/MS (ES/APCI): 483.1 (M - H)−.
N-[[(3S)-1-[6-tert-butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3-quinolyl]pyrrolidin-3-yl]methyl]methanesulfonamide (50)
A mixture of 3,8-dibromoquinoline 13 (1.876 g, 5.03 mmol), tert-butyl N-[[(3S)-pyrrolidin-3-yl]methyl]carbamate (1.52 g, 7.6 mmol), sodium tert-butoxide (728 mg, 7.49 mmol), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine) (582 mg, 1.01 mmol), and Pd2(dba)3 (460 mg, 0.5 mmol) in degassed toluene (10 mL) was warmed slowly to 100 °C and kept at this temperature for 22 h. The reaction mixture was cooled to rt and partitioned between EtOAc and saturated aqueous NH4Cl. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated. The crude product was purified over SiO2 (gradient: 10 to 50% EtOAc in hexane) to afford tert-butyl N-[[(3R)-1-(8-bromo-6-tert-butyl-5-methoxy-3-quinolyl)pyrrolidin-3-yl]methyl]carbamate (1.77 g, 71%) as a light green foam. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 2.8 Hz, 1 H), 7.66 (s, 1 H), 7.07 (d, J = 2.5 Hz, 1 H), 4.66 (br s, 1 H), 3.85 (s, 3 H), 3.56–3.48 (m, 2 H), 3.46–3.38 (m, 1 H), 3.26–3.11 (m, 3 H), 2.63–2.50 (m, 1 H), 2.21–2.10 (m, 1 H), 1.86–1.74 (m, 1 H), 1.40 (s, 9 H), 1.39 (s, 9 H). LC/MS (ES): 492 (M + H)+.
A solution of tert-butyl N-[[(3R)-1-(8-bromo-6-tert-butyl-5-methoxy-3-quinolyl)pyrrolidin-3-yl]methyl]carbamate (1.76 g, 3.6 mmol) and 4 M HCl in dioxane (9 mL, 36 mmol) in CH2Cl2 (11 mL) was stirred at rt for 16 h. The reaction was concentrated and the residual orange oil was treated with of dry Et2O (150 mL) and stirred at rt for 30 min. The supernatant was removed carefully and residual solid was dried under high vacuum to give [(3R)-1-(8-bromo-6-tert-butyl-5-methoxy-3-quinolyl)pyrrolidin-3-yl]methanamine (1.7 g) as an orange solid. This product was used in the next reaction without purification. To a solution of the primary amine (1.7 g, 3.4 mmol) and Et3N (2.36 mL, 16.9 mmol) in CH2Cl2 (34 mL) was added methanesulfonyl chloride (0.29 mL, 3.8 mmol) at 0 °C. The reaction mixture was stirred at rt for 1 h. The reaction was quenched with water and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated in vacuo to give N-[[(3S)-1-(8-bromo-6-tert-butyl-5-methoxy-3-quinolyl)pyrrolidin-3-yl]methyl]methanesulfonamide (1.44 g, 89%) as an oil. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 2.5 Hz, 1 H), 7.61 (s, 1 H), 7.23 (t, J = 6.2 Hz, 1 H), 7.04 (d, J = 2.7 Hz, 1 H), 3.90 (s, 3 H), 3.64–3.53 (m, 2 H), 3.52–3.43 (m, 1 H), 3.28–3.21 (m, 1 H), 3.07 (t, J = 6.8 Hz, 2 H), 2.93 (s, 3 H), 2.60–2.44 (m, 1 H), 2.24–2.12 (m, 1 H), 1.90–1.78 (m, 1 H), 1.44 (s, 9 H).
A solution of N-[[(3S)-1-(8-bromo-6-tert-butyl-5-methoxy-3-quinolyl)pyrrolidin-3-yl]methyl]methanesulfonamide (6.13 g, 13 mmol), (2,6-dimethoxy-3-pyridyl)boronic acid (2.86 g, 15.6 mmol), [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) chloride 1:1 complex with dichloromethane (412 mg, 0.5 mmol), Cs2CO3 (12.76 g, 39.1 mmol in 13 mL of water) in dioxane (50 mL) was purged with N2 for 10 min and stirred at 80 °C for 1 h. The reaction was quenched with water (200 mL) and extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, and concentrated. The crude product was purified over SiO2 (EtOAc) to obtain N-[[(3S)-1-[6-tert-butyl-8-(2,6-dimethoxy-3-pyridyl)-5-methoxy-3-quinolyl]pyrrolidin-3-yl]methyl]methanesulfonamide (6.26 g, 91%) as a yellow foam. 1H NMR (300 MHz, DMSO-d6) δ 8.42 (d, J = 2.7 Hz, 1 H), 7.58 (d, J = 7.9 Hz, 1 H), 7.25 (s, 1 H), 7.27–7.19 (m, 1 H), 7.06 (d, J = 2.7 Hz, 1 H), 6.44 (d, J = 7.9 Hz, 1 H), 3.93 (s, 3 H), 3.92 (s, 3 H), 3.76 (s, 3 H), 3.61–3.36 (m, 3 H), 3.21 (dd, J = 9.7, 6.6 Hz, 1 H), 3.06 (t, J = 6.4 Hz, 2 H), 2.93 (s, 3 H), 2.60–2.47 (m, 1 H), 2.23–2.09 (m, 1 H), 1.90–1.75 (m, 1 H), 1.45 (s, 9 H). LC/MS (ES/APCI): 529.2 (M + H)+.
To a solution of N-[[(3S)-1-[6-tert-butyl-8-(2,6-dimethoxy-3-pyridyl)-5-methoxy-3-quinolyl]pyrrolidin-3-yl]methyl]methanesulfonamide (120 mg, 0.227 mmol) in AcOH (2 mL) was added aqueous 48% HBr solution (0.13 mL) at rt. The reaction was stirred at 60 °C for 18 h. The reaction mixture was diluted with EtOAc and washed with saturated aqueous NaHCO3. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with saturated aqueous NaHCO3, dried over MgSO4, and concentrated. The crude product was purified over SiO2 (gradient: 0 to 5% MeOH in CH2Cl2) to give pyrrolidine 50 (47 mg, 40%) as a yellow foam. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 2.7 Hz, 1 H), 7.56 (d, J = 8.1 Hz, 1 H), 7.32 (s, 1 H), 7.23 (t, J = 5.9 Hz, 1 H), 7.09 (d, J = 2.6 Hz, 1 H), 6.26 (br s, 1 H), 3.93 (s, 3 H), 3.87 (s, 3 H), 3.63–3.50 (m, 2 H), 3.49–3.41 (m, 1 H), 3.23 (dd, J = 9.6, 6.3 Hz, 1 H), 3.08 (t, J = 6.3 Hz, 2 H), 2.94 (s, 3 H), 2.62–2.51 (m, 1 H), 2.13–2.12 (m, 1 H), 1.90–1.79 (m, 1 H), 1.46 (s, 9 H). HRMS (ESI): calcd. for C26H35O5N4S, 515.23227; found, 515.23059 [M + H]+.
N-[4-[8-(2,4-dioxopyrimidin-1-yl)-5-methoxy-6-(trifluoromethyl)-3-quinolyl]phenyl]methanesulfonamide (51)
A mixture of Cu(I) (10.03 g, 52.6 mmol) and CsF (21.40 g, 140.8 mmol) was finely ground in a mortar while in a glove bag under nitrogen atmosphere to afford a free-flowing powder and transferred to a flask. The flask was then charged with 2-iodo-5-nitroanisole (15.17 g, 54.3 mmol) and sulfolane (30 mL) and stirred rapidly at 45 °C. To this mixture was added trimethyl(trifluoromethyl)silane (19.2 g, 20 mL, 135 mmol) dropwise over 4 h using a syringe pump and the resulting mixture stirred at rt for 18 h. The reaction was diluted with EtOAc (500 mL) and stirred with Celite® for 5 min. The reaction mixture was filtered through a pad of Celite®, diluted with EtOAc (1 L) and washed with 10% NH4OH, 1.0 N HCl, brine, dried over MgSO4, filtered and concentrated. The amber residue was purified over SiO2 (gradient: 0 to 40% CH2Cl2 in hexane) to afford 2-methoxy-4-nitro-1-(trifluoromethyl)benzene (8.61 g, 72%) as a yellow crystalline solid. 1H NMR (300 MHz, CDCl3) δ 7.95–7.83 (m, 2 H), 7.77 (d, J = 8.2 Hz, 1 H), 4.03 (s, 3 H). LC/MS (ES/APCI): 220.9 (M - H)−.
A Parr hydrogenation flask was charged with 2-methoxy-4-nitro-1-(trifluoromethyl)benzene (8.60 g, 38.9 mmol), 10% Pd/C (1.75 g), and EtOH (150 mL). The flask was placed under 57 psi of hydrogen pressure at 55 °C for 18 h. The reaction mixture was cooled and the catalyst filtered and washed with iso-propanol. The solvent was removed and the crude product was purified over SiO2 (gradient: 0 to 40% EtOAc in hexane) to afford 3-methoxy-4-(trifluoromethyl)aniline (7.18 g, 96%) as a waxy off-white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.17 (d, J = 8.2 Hz, 1 H), 6.30 (br s, 1 H), 6.16 (dd, J = 8.2, 1.3 Hz, 1 H), 5.77 (s, 2 H), 3.75 (s, 3 H). LC/MS (ES/APCI): 192.1 (M + H)+.
To a solution of 3-methoxy-4-(trifluoromethyl)aniline (3.01, 15.7 mmol), AcOH (7.5 mL) and anhydrous dioxane (25 mL) was added a solution of Br2 (2.7 g, 16.9 mmol in 20 mL dioxane) dropwise using a syringe pump over 30 min at 0 °C. The reaction mixture stirred at rt for 1 h, poured into a mixture of 1.0 M NaOH (150 mL) and 2.0 M Na2CO3 (150 mL) and extracted with CH2Cl2. The combined extracts were washed sequentially with 0.5 M Na2CO3, brine, dried over Na2SO4, filtered, and concentrated to afford 2-bromo-5-methoxy-4-(trifluoromethyl)aniline (4.10 g, 97%) as a black crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 7.44 (s, 1 H), 6.57 (s, 1 H), 3.77 (s, 3 H).
To a solution of 2-bromoprop-2-enal (1.91 g) in AcOH (50 mL) was added dropwise Br2 (0.72 mL, 13.9 mmol). To this solution was added 2-bromo-5-methoxy-4-(trifluoromethyl)aniline (3.48 g, 12.9 mmol) and stirred at 100 °C for 1 h. The reaction was cooled to rt and poured into a stirred ice cold solution of 2.0 M NaOH (550 mL). To the mixture was slowly added 2.0 M Na2CO3 [vigorous foaming] until the solution reached pH 8. The resulting solution extracted with CH2Cl2 and the organic extracts washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) and re-chromatographed (gradient: 0 to 100% EtOAc in hexane) to afford 3,8-dibromo-5-methoxy-6-(trifluoromethyl)quinoline (938 mg, 19% ) as an off-white solid. 1H NMR (300 MHz, DMSO-d6) δ 9.26 (d, J = 2.2 Hz, 1 H), 8.91 (d, J = 2.2 Hz, 1 H), 8.33 (s, 1 H), 4.06 (s, 3 H).
A vial was charged with 3,8-dibromo-5-methoxy-6-(trifluoromethyl)quinoline (850 mg, 2.2 mmol), N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanesulfonamide (661 mg, 2.22 mmol), an aqueous solution of Cs2CO3 (2.2 g, 6.7 mmol in 2.3 mL of water) and dioxane (10 mL) and the mixture was sparged with N2 for 10 min. Pd(dppf)Cl2·CH2Cl2 (74 mg, 0.09 mmol) was added to the mixture and stirred at 65 °C for 110 min. The reaction was cooled and poured into CH2Cl2 and 0.5 M Na2CO3. The phases were separated and washed sequentially with water, brine, dried over MgSO4, filtered, and concentrated. The product was purified over SiO2 (gradient: 0 to 100% EtOAc in CH2Cl2) to afford N-[4-[8-bromo-5-methoxy-6-(trifluoromethyl)-3-quinolyl]phenyl]methanesulfonamide (538 mg, 51%) as a light amber solid. 1H NMR (300 MHz, DMSO-d6) δ 10.07 (br s, 1 H), 9.53 (d, J = 2.2 Hz, 1 H), 8.73 (d, J = 2.1 Hz, 1 H), 8.27 (s, 1 H), 8.0 (d, J = 8.6 Hz, 2 H), 7.42 (d, J = 8.6 Hz, 2 H), 4.11 (s, 3 H), 3.09 (s, 3 H).
A vial was charged with N-[4-[8-bromo-5-methoxy-6-(trifluoromethyl)-3-quinolyl]phenyl]methanesulfonamide (370 mg, 0.78 mmol), tert-butyl carbamate (110 mg, 0.94 mmol), Pd2(dba)3·CHCl3 (120 mg, 0.116 mmol), 2-di-tert-butylphosphino-2′,4′,6′-tri-iso-propyl-1′,1′-biphenyl (150 mg, 0.353 mmol), sodium tert-butoxide (110 mg, 1.14 mmol), and toluene (3.5 mL) and the resulting mixture was degassed by bubbling argon for 10 min. The reaction mixture was stirred at rt for two days then diluted with EtOAc and washed with saturated aqueous NH4Cl. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified over SiO2 (gradient: 0 to 10% EtOAc in CH2Cl2) to obtain tert-butyl N-[3-[4-(methanesulfonamido)phenyl]-5-methoxy-6-(trifluoromethyl)-8-quinolyl]carbamate (398 mg, 78%) as a light brown foam.
A mixture of tert-butyl N-[3-[4-(methanesulfonamido)phenyl]-5-methoxy-6-(trifluoromethyl)-8-quinolyl]carbamate (390 mg, 0.16 mmol), 4 M HCl in dioxane (4 mL), and CH2Cl2 (4 mL) was stirred at rt for 4 h then concentrated. The residue was partitioned between EtOAc and saturated aqueous NaHCO3. The organic layer was separated, dried over Na2SO4, filtered, and concentrated to give N-[4-[8-amino-5-methoxy-6-(trifluoromethyl)-3-quinolyl]phenyl]methanesulfonamide (300 mg, 73%) as a solid. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (d, J = 2.1 Hz, 1 H), 8.53 (d, J = 2.3 Hz, 1 H), 7.93 (d, J = 8.6 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 6.97 (s, 1 H), 6.17 (br s, 2 H), 3.93 (s, 3 H), 3.08 (s, 3H).
(E)-3-Methoxy-acryloyl chloride (90 mg, 1.12 mmol) was added at rt to a suspension of a previously dried silver cyanate (180 mg, 1.2 mmol) in toluene (2 mL). The resulting slurry was stirred at reflux for 30 min. before being cooled to 0 °C. After the solid had settled, the supernatant was added dropwise to a solution of N-[4-[8-amino-5-methoxy-6-(trifluoromethyl)-3-quinolyl]phenyl]methanesulfonamide (100 mg, 0.243 mmol) in DMF (1.5 mL) at 0 °C and the resulting light orange mixture was stirred for 30 min. The reaction was partitioned between EtOAc and water, the organic layer was dried over Na2SO4, filtered, and concentrated. The residue was taken into EtOH (1.5 mL) and water (1.5 mL) and concentrated H2SO4 (0.2 mL) was added. The resulting sticky slurry was stirred at 110 °C for 1 h. DMF was added and stirred at 110 °C for 2 h before cooling to rt. The clear solution was partitioned between EtOAc and saturated aqueous NaHCO3. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified over SiO2 (gradient: 0 to 40% EtOAc in CH2Cl2) to yield trifluoromethylquinoline 51 (20 mg, 16%). 1H NMR (400 MHz, CDCl3) δ 9.11 (d, J = 2.3 Hz, 1 H), 8.89 (br s, 1 H), 8.59 (br s, 1 H), 8.56 (d, J = 2.3 Hz, 1 H), 7.72 (d, J = 8.7 Hz, 2 H), 7.43 (d, J = 8.6 Hz, 2 H), 6.79 (br s, 1 H), 4.04 (s, 3 H), 3.11 (s, 3 H).
N-[4-[8-(2,4-dioxopyrimidin-1-yl)-5-methoxy-6-(2,2,2-trifluoroethyl)-3-quinolyl]phenyl]methanesulfonamide (52)
In a flask was dissolved 2-methoxy-4-nitro-benzaldehye (9.93 g, 54.8 mmol) in anhydrous DME (100 mL) and stirred to obtain a clear yellow solution. To this solution was added sequentially CF3SiMe3 (9.0 mL, 8.66 g, 60.9 mmol) and CsF (792 mg, 5.2 mmol), sonicated for 20 min, then stirred at rt for 40 min. A 2.0 M HCl solution (100 mL) was added and the resulting mixture stirred at rt for 1 h. EtOAc was added to the mixture, the two layers separated, and the organic phase washed with saturated aqueous NaHCO3, brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) to give 2,2,2-trifluoro-1-(2-methoxy-4-nitro-phenyl) ethanol (13.1 g, 95%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.93 (dd, J = 8.4, 2.0 Hz, 1 H), 7.84 (d, J = 2.0 Hz, 1 H), 7.77 (d, J = 8.6 Hz, 1 H), 7.08 (d, J = 5.8 Hz, 1 H), 5.49 (quintuplet, J = 6.5 Hz, 1 H), 3.97 (s, 3 H).
To a solution of 2,2,2-trifluoro-1-(2-methoxy-4-nitro-phenyl)ethanol (13.01 g, 51.8 mmol) in THF (300 mL) was added NaH (2.25 g, 56.2 mmol, 60% wt dispersion in mineral oil) at rt. The mixture was sonicated for 20 min, then stirred at rt for 10 min. A solution of p-toluenesulfonyl chloride (11.86 g, 62.2 mmol) dissolved in THF (100 mL) was added at rt and the reaction stirred at 50 °C for 1.5 h. The solution was cooled and poured into 0.5 M NaHCO3, diluted with EtOAc, the organic phase separated, washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) to afford [2,2,2-trifluoro-1-(2-methoxy-4-nitro-phenyl)ethyl] 4-methylbenzenesulfonate (20.36 g, 97%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.81–7.73 (m, 2 H), 7.69 (d, J = 8.4 Hz, 2 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.35 (d, J = 8.4 Hz, 2 H), 6.24 (q, J = 6.4 Hz, 1 H), 3.95 (s, 3 H), 2.33 (s, 3 H).
A Parr hydrogenation flask was charged with [2,2,2-trifluoro-1-(2-methoxy-4-nitro-phenyl) ethyl] 4-methylbenzenesulfonate (20.35 g, 50.2 mmol) and dissolved in EtOH (250 mL). To the solution was added 10% Pd/C (4.02 g) and hydrogenated under 55 psi of hydrogen at 50 °C for 1.5 h. The catalyst was filtered through a plug of glass wool and washed with hot EtOH. The filtrate was concentrated and the tosylate salt precipitated as a white solid. The residue was partitioned between 1.0 M NaOH and Et2O. The organic extract was washed with brine, dried over MgSO4, filtered, and concentrated. The residue was purified over SiO2 (CH2Cl2) to obtain 3-methoxy-4-(2,2,2-trifluoroethyl)aniline (9.04 g, 88%) as an off-white solid. 1H NMR (300 MHz, DMSO-d6) δ 6.87 (d, J = 8.1 Hz, 1 H), 6.24 (d, J = 2.0 Hz, 1 H), 6.13 (dd, J = 8.0, 2.0 Hz, 1 H), 5.19 (br s, 2 H), 3.70 (s, 3 H), 3.32 (q, J = 11.7 Hz, 2 H). LC/MS (ES/APCI): 206.1 (M + H)+.
To a solution of 3-methoxy-4-(2,2,2-trifluoroethyl)aniline (8.16 g, 39.8 mmol), AcOH (23 mL), and dioxane (100 mL) was added a solution of Br2 (2.26 mL, 44 mmol) in dioxane (45 mL) dropwise over 30 min at 5 °C. The solution was warmed to rt and stirred for 1 h. The reaction was poured into 1.0 M NaOH and extracted with CH2Cl2. The combined extracts were washed with brine, dried over MgSO4, filtered, and concentrated to afford crude 2-bromo-5-methoxy-4-(2,2,2-trifluoroethyl)aniline (11.42 g) as a pale olive solid. 1H NMR (300 MHz, DMSO-d6) δ 7.21 (br s, 1 H), 6.48 (s, 1 H), 5.41 (br s, 2 H), 3.71 (s, 3 H), 3.38 (q, J = 11.6 Hz, 2 H). LC/MS (ES/APCI): 284.0 (M + H)+.
To a solution of 2-bromoprop-2-enal (2.71 g, 20.1 mmol) in AcOH (25 mL) was added Br2 (3.2 g, 1.0 mL, 20 mmol) at 0 °C. This solution was poured into a stirred solution of 2-bromo-5-methoxy-4-(2,2,2-trifluoroethyl)aniline (5.67 g, 19.9 mmol) and AcOH (25 mL) and the resulting mixture stirred at 100 °C for 2 h. The solution was cooled, diluted with water, and extracted with EtOAc. The extract was washed with 2.0 M NaOH, brine, dried over MgSO4, filtered, and concentrated. The crude product was purified over SiO2 (CH2Cl2) to obtain 3,8-dibromo-5-methoxy-6-(2,2,2-trifluoroethyl)quinoline (2.69 g, 34%) as a light orange solid. 1H NMR (300 MHz, DMSO-d6) δ 9.13 (d, J = 2.3 Hz, 1 H), 8.76 (d, J = 2.3 Hz, 1 H), 8.19 (s, 1 H), 3.95 (s, 3 H), 3.90 (q, J = 11.2 Hz, 2 H). LC/MS (ES/APCI): 399.9 (M + H)+.
A vial was charged with 3,8-dibromo-5-methoxy-6-(2,2,2-trifluoroethyl)quinoline (2.62 g, 6.5 mmol), N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanesulfonamide (1.92 g, 6.5 mmol), an aqueous solution of Cs2CO3 (6.46 g, 19.8 mmol, 6 mL of water) and dioxane (25 mL) and the mixture was sparged with N2 for 10 min. Pd(dppf)Cl2·CH2Cl2 (161 mg, 0.19 mmol) was added to the mixture and stirred at 50 °C for 2 h. The reaction was cooled and poured into CH2Cl2 and 0.5 M Na2CO3. The phases were separated and the organic phase was washed sequentially with water, brine, dried over MgSO4, filtered, and concentrated. The product was purified over SiO2 (gradient: 0 to 60% EtOAc in CH2Cl2) to afford N-[4-[8-bromo-5-methoxy-6-(2,2,2-trifluoroethyl)-3-quinolyl]phenyl]methanesulfonamide (368 mg, 11%) as a solid. 1H NMR (300 MHz, DMSO-d6) δ 10.04 (s, 1 H), 9.40 (d, J = 2.2 Hz, 1 H), 8.62 (d, J = 2.2 Hz, 1 H), 8.13 (s, 1 H), 7.96 (d, J = 8.6 Hz, 2 H), 7.41 (d, J = 8.6 Hz, 2 H), 3.99 (s, 3 H), 3.90 (q, J = 11.2 Hz, 2 H), 3.09 (s, 3 H). LC/MS (ES/APCI): 489.0 (M + H)+.
A flask was charged with N-[4-[8-bromo-5-methoxy-6-(2,2,2-trifluoroethyl)-3-quinolyl]phenyl]methanesulfonamide (1.0 g, 2.04 mmol), tert-butyl carbamate (290 mg, 2.48 mmol), Pd2(dba)3·CHCl3 (106 mg, 0.1 mmol), 2-di-tert-butylphosphino-2′,4′6′-tri-iso-propyl-1′,1′-biphenyl (150 mg, 0.353 mmol), sodium tert-butoxide (287 mg, 3.0 mmol), and toluene (10 mL) and the resulting mixture was degassed by bubbling argon for 10 min. The reaction mixture was stirred at rt for 1.5 h then diluted with EtOAc and washed with saturated aqueous NH4Cl. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified over SiO2 (gradient: 0 to 25% EtOAc in CH2Cl2) to obtain tert-butyl N-[3-[4-(methanesulfonamido)phenyl]-5-methoxy-6-(2,2,2-trifluoroethyl)-8-quinolyl]carbamate (926 mg, 86%) as a foam. 1H NMR (300 MHz, DMSO-d6) δ 10.02 (s, 1 H), 9.24 (d, J = 2.1 Hz, 1 H), 8.88 (s, 1 H), 8.58 (d, J = 2.1 Hz, 1 H), 8.24 (s, 1 H), 7.93 (d, J = 8.7 Hz, 2 H), 7.41 (d, J = 8.7 Hz, 2 H), 3.96–3.79 (m, 2 H), 3.93 (s, 3 H), 3.08 (s, 3 H) 1.54 (s, 9 H). LC/MS (ES/APCI): 526.1 (M + H)+.
A mixture of tert-butyl N-[3-[4-(methanesulfonamido)phenyl]-5-methoxy-6-(2,2,2-trifluoroethyl)-8-quinolyl]carbamate (908 mg, 1.73 mmol), 4 M HCl in dioxane (8 mL), and CH2Cl2 (4 mL) was stirred at rt for 2 h then concentrated. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3. The organic layer was separated, washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified over SiO2 (gradient: 0 to 50% EtOAc in CH2Cl2) to give N-[4-[8-amino-5-methoxy-6-(2,2,2-trifluoroethyl)-3-quinolyl]phenyl]methanesulfonamide (554 mg, 75%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 9.98 (s, 1 H), 9.08 (d, J = 2.2 Hz, 1 H), 8.42 (d, J = 2.1 Hz, 1 H), 7.89 (d, J = 8.7 Hz, 2 H), 7.39 (d, J = 8.5 Hz, 2 H), 6.80 (s, 1 H), 5.87 (s, 2 H), 3.84 (s, 3 H), 3.72 (q, J = 11.4 Hz, 2 H), 3.07 (s, 3 H). LC/MS (ES/APCI): 426.0 (M + H)+.
A suspension of dried silver cyanate (529 mg, 3.5 mmol) and anhydrous toluene (3 mL) was stirred at 90 °C for 20 min. A solution of (E)-3-methoxyprop-2-enoyl chloride (216 mg, 1.8 mmol) in anhydrous toluene (2 mL) was added dropwise to the suspension and stirred at 90 °C for 30 min. The suspension was allowed to stand at rt for 10 min and the supernatant transfer by syringe to a solution of N-[4-[8-amino-5-methoxy-6-(2,2,2-trifluoroethyl)-3-quinolyl]phenyl]methanesulfonamide (249 mg, 0.58 mmol) in DMF (3 mL) at −20 °C. The reaction was allowed to reach rt and stirred for 18 h. The reaction was concentrated and the residue purified over SiO2 (gradient: 0 to 70% EtOAc in CH2Cl2) to obtain a mixture of geometric isomers used in the next reaction (204 mg).
A suspension of the product from previous reaction (178 mg) and H2SO4 (0.5 mL) with EtOH (2 mL) and water (2 mL) was stirred at 110 °C for 2 h. The reaction was cooled to room temperature and let stand overnight. A yellow solid crystallized from the yellow supernatant. The solid was collected by filtration and washed with EtOH/water (1/1). The solid was dissolved in hot EtOH (10 mL) and water (10 mL) added. The resulting solution was allowed to cool to room temperature and a fluffy white crystals formed. The solid was collected by filtration, washed with 1:1 EtOH/water (1/1), and dried under vacuum to obtain the trifluoroethylquinoline 52 (107 mg, 6%) as white crystals. 1H NMR (300 MHz, DMSO-d6) δ 11.51 (d, J = 1.8 Hz, 1 H), 10.03 (s, 1 H), 9.34 (d, J = 2.2 Hz, 1 H), 8.66 (d, J = 2.0 Hz, 1 H), 7.94 (d, J = 8.7 Hz, 2 H), 7.90 (s, 1 H), 7.72 (d, J = 7.9 Hz, 1 H), 7.40 (d, J = 8.7 Hz, 2 H), 5.71 (dd, J = 7.9, 1.9 Hz, 1 H), 4.04 (s, 3 H), 3.96 (d, J = 11.1 Hz, 1 H), 3.88 (d, J = 11.1 Hz, 1 H), 3.08 (s, 3 H). LC/MS (ES/APCI): 521.0 (M + H)+.
N-[4-[6-[1-(Difluoromethyl)cyclopropyl]-8-(2,4-dioxopyrimidin-1-yl)-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (53)
Br2 (4.48 mL, 86.9 mmol) in AcOH (50 mL) was added to a solution of 2-bromoprop-2-enal (11.7 g, 86.7 mmol) in AcOH (100 mL) at rt until the solution showed faint Br2 color. To this solution was added methyl 4-amino-5-bromo-2-methoxy-benzoate (22.6 g, 86.9 mmol) and the resulting solution was gradually heated to 100 °C and stirring continued for 15 min. The reaction was cooled to rt and concentrated. The residue was neutralized with saturated aqueous NaHCO3 and a precipitate formed. The resulting solid was filtered and washed with water and Et2O to afford methyl 3,8-dibromo-5-methoxyquinoline-6-carboxylate (11.04 g, 34%). 1H NMR (300 MHz, CDCl3) δ 9.08 (d, J = 2.3 Hz, 1 H), 8.76 (d, J = 2.4 Hz, 1 H), 8.46 (s, 1 H), 4.08 (s, 3 H), 4.01 (s, 3 H).
To a solution of methyl 3,8-dibromo-5-methoxy-quinoline-6-carboxylate (11.04 g, 29.5 mmol) in CH2Cl2 (550 mL) at 0 °C was added dropwise DIBAL (64.4 mL, 64.4 mmol, 1 M in CH2Cl2). The reaction was quenched with aqueous Rochelle salt and partitioned between water and CH2Cl2. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 0 to 10% MeOH in CH2Cl2) to afford (3,8-dibromo-5-methoxy-6-quinolyl)methanol (9.5 g, 93%). 1H NMR (300 MHz, CDCl3) δ 9.01 (d, J = 2.2 Hz, 1 H), 8.59 (d, J = 2.2 Hz, 1 H), 8.18 (s, 1 H), 4.93 (d, J = 5.7 Hz, 2 H), 3.98 (s, 3 H), 1.98 (t, J = 5.8 Hz, 1 H).
A solution of (3,8-dibromo-5-methoxy-6-quinolyl)methanol (7.82 g, 22.5 mmol), CBr4 (8.97 g, 27.0 mmol), PPh3 (7.09 g, 27.0 mmol) and CH2Cl2 (250 mL) was stirred at rt for 18 h. Additional CBr4 (3.73, 11.2 mmol) and PPh3 (2.95 g, 11.2 mmol) were added to the reaction and stirred for 1 h. The crude reaction mixture was concentrated and the residue purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) to afford 3,8-dibromo-6-(bromomethyl)-5-methoxy-quinoline (7.62 g, 83%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.01 (d, J = 2.3 Hz, 1 H), 8.58 (d, J = 2.2 Hz, 1 H), 8.07 (s, 1 H), 4.67 (s, 2 H), 4.06 (s, 3 H).
A mixture of 3,8-dibromo-6-(bromomethyl)-5-methoxy-quinoline (8.0 g, 19.5 mmol), KCN (12.4 g, 191 mmol), CH2Cl2 (156 mL), and water (140 mL) was heated at reflux for 15 min. The reaction mixture was diluted with water and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated. The residue was purified over SiO2 (gradient: 0 to 5% MeOH in CH2Cl2) to afford 2-(3,8-dibromo-5-methoxy-6-quinolyl)acetonitrile (4.53 g, 65%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.04 (d, J = 2.2 Hz, 1 H), 8.57 (d, J = 2.3 Hz, 1 H), 8.09 (s, 1 H), 4.01 (s, 3 H), 3.93 (s, 2 H).
A mixture of 2-(3,8-dibromo-5-methoxy-6-quinolyl)acetonitrile (5.02 g, 14.1 mmol), 1,2-dibromoethane (1.46 mL, 16.9 mmol), and DMF (60 mL) was cooled to 0 °C and NaH (1.69 g, 42.3 mmol, 60% mineral oil dispersion) was added. The mixture was warmed to rt and stirred for 1 h. The reaction mixture was diluted with water and extracted with CH2Cl2. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) to afford 1-(3,8-dibromo-5-methoxy-6-quinolyl)cyclopropanecarbonitrile (1.91 g, 35%) as a solid. 1H NMR (400 MHz, CDCl3) δ 9.03 (d, J = 2.0 Hz, 1 H), 8.61 (d, J = 2.1 Hz, 1 H), 8.00 (s, 1 H), 4.18 (s, 3 H), 1.83 (dd, J = 7.9, 5.3 Hz, 2 H), 1.49 (dd, J = 7.5, 4.9 Hz, 2 H).
To a solution of 1-(3,8-dibromo-5-methoxy-6-quinolyl)cyclopropanecarbonitrile (280 mg, 0.733 mmol) in CH2Cl2 (14 mL) was added DIBAL (0.87 mL, 0.87 mmol, 1 M in CH2Cl2) dropwise at −78 °C. The reaction mixture was stirred at the same temperature for 1 h. The reaction was quenched with aqueous Rochelle salt and extracted with CH2Cl2. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated. The crude material was purified over SiO2 (gradient: 0 to 100% CH2Cl2 in hexane) to afford 1-(3,8-dibromo-5-methoxy-6-quinolyl)cyclopropanecarbaldehyde (220 mg, 78%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1 H), 9.01 (d, J = 2.2 Hz, 1 H), 8.60 (d, J = 2.1 Hz, 1 H), 7.89 (s, 1 H), 3.92 (s, 3 H), 1.77 (dd, J = 7.6, 4.9 Hz, 2 H), 1.56 (dd, J = 7.0, 4.4 Hz, 2 H).
To a solution of 1-(3,8-dibromo-5-methoxy-6-quinolyl)cyclopropanecarbaldehyde (630 mg, 1.64 mmol) in CH2Cl2 (14 mL) was added diethylaminosulfur trifluoride (DAST) (1.05 g, 6.54 mmol) and the resulting solution was stirred at rt for 72 h. The reaction mixture was diluted with water and extracted with CH2Cl2. The organic layer was washed with water, dried over Na2SO4, filtered, and concentrated. The crude product was purified over SiO2 (gradient: 50 to 100% CH2Cl2 in hexane) to afford 3,8-dibromo-6-[1-(difluoromethyl)cyclopropyl]-5-methoxyquinoline (600 mg, 90%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.0 (d, J = 2.0 Hz, 1 H), 8.57 (d, J = 2.1 Hz, 1 H), 8.07 (s, 1 H), 5.89 (t, J = 57.6 Hz, 1 H), 4.05 (s, 3 H), 1.35 (br t, J = 6.1 Hz, 2 H), 1.18–1.12 (br s, 2 H).
To a microwave vial was added 3,8-dibromo-6-[1-(difluoromethyl)cyclopropyl]-5-methoxyquinoline (600 mg, 1.47 mmol), 4-(methanesulfonamido)phenylboronic acid (317 mg, 1.47 mmol), Pd(PPh3)4 (162 mg, 0.14 mmol), Na2CO3 (469 mg, 4.42 mmol), MeOH (9 mL), and toluene(3 mL). The vial was capped and heated in the microwave reactor at 120 °C for 1 h. The reaction mixture was filtered and the filtrate was concentrated. The residue was diluted with CH2Cl2, washed with water, dried over Na2SO4, and concentrated. The crude material was purified over SiO2 (gradient: 30 to 80% EtOAc in hexanes) to obtain N-[4-[8-bromo-6-[1-(difluoromethyl)cyclopropyl]-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (670 mg, 91%) as a solid. 1H NMR (300 MHz, CDCl3) δ 9.25 (d, J = 2.0 Hz, 1 H), 8.53 (d, J = 2.1 Hz, 1 H), 8.06 (s, 1 H), 7.73 (d, J = 8.4 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 6.69 (s, 1 H), 5.94 (t, J = 57.7 Hz, 1 H), 4.09 (s, 3 H), 3.11 (s, 3 H), 1.40–1.34 (br m, 2 H), 1.21–1.13 (br m, 2 H).
To a flask was added N-[4-[8-bromo-6-[1-(difluoromethyl)cyclopropyl]-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (520 mg, 1.05 mmol), tert-butyl carbamate (147 mg, 1.25 mmol), Pd2(dba)3·CHCl3 (123 mg, 0.12 mmol), 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl (172 mg, 0.4 mmol), sodium tert-butoxide (301 mg, 3.14 mmol), and toluene (8 mL). The mixture was purged with argon for 20 min and stirred at rt for 72 h. The mixture was quenched with NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified over SiO2 (gradient: 30 to 80% EtOAc in hexane) to give tert-butyl N-[6-[1-(difluoromethyl)cyclopropyl]-3-[4-(methanesulfonamido)phenyl]-5-methoxy-8-quinolyl]carbamate (150 mg, 27%) as a solid. 1H NMR (300 MHz, CDCl3) δ 9.01 (d, J = 2.2 Hz, 1 H), 8.82 (br s, 1 H), 8.49 (d, J = 2.3 Hz, 1 H), 8.39 (br s, 1 H), 7.72 (d, J = 8.5 Hz, 2 H), 7.40 (d, J = 8.5 Hz, 2 H), 6.53 (s, 1 H), 6.13 (t, J = 58.4 Hz, 1 H), 4.02 (s, 3 H), 3.10 (s, 3 H), 1.56 (s, 9 H), 1.36 (dd, J = 6.2, 5.0 Hz, 2 H), 1.22–1.14 (br m, 2 H).
To a solution of tert-butyl N-[6-[1-(difluoromethyl)cyclopropyl]-3-[4-(methanesulfonamido)phenyl]-5-methoxy-8-quinolyl]carbamate (150 mg, 0.28 mmol) in CH2Cl2 was added 4 M HCl in dioxane (1.55 mL, 6.2 mmol). The reaction was stirred at rt for 1h then neutralized with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified over SiO2 (gradient: 30 to 80% EtOAc in hexane) to afford N-[4-[8-amino-6-[1-(difluoromethyl)cyclopropyl]-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (110 mg, 90%) as a solid. 1H NMR (400 MHz, CDCl3) δ 8.98 (d, J = 2.2 Hz, 1 H), 8.43 (d, J = 2.2 Hz, 1 H), 7.72 (d, J = 8.4 Hz, 2 H), 7.38 (d, J = 8.4 Hz, 2 H), 6.90 (s, 1 H), 6.49 (br s, 1 H), 6.05 (t, J = 58.4 Hz, 1 H), 4.83 (br s, 2 H), 3.98 (s, 3 H), 3.10 (s, 3 H), 1.31 (dd, J = 6.7, 5.0 Hz, 2 H), 1.14–1.08 (br m, 2 H).
In a flask, (E)-3-methoxyacryloyl chloride (91.8 mg, 0.761 mmol) and dried silver cyanate (190 mg, 1.27 mmol) were combined with toluene (2 mL) and the mixture was heated to 130 °C and stirred for 30 min. The reaction was cooled down to 0 °C and allowed to settle. The supernatant was added dropwise to N-[4-[8-amino-6-[1-(difluoromethyl)cyclopropyl]-5-methoxy-3-quinolyl]phenyl]methanesulfonamide (110 mg, 0.25 mmol) in DMF (1 mL) at 0 °C. The reaction was stirred at 0 °C for 45min then diluted with EtOAc and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude reaction mixture was purified over SiO2 (gradient: 50 to 100% EtOAc in hexane) to obtain a mixture of geometric isomers used in the next reaction (160 mg) as a light yellow solid.
A slurry of the product from previous reaction (160 mg) and concentrated H2SO4 (0.35 mL) with EtOH (4 mL) and water (3 mL) was heated at 110 °C for 1.5 h. Most of EtOH was evaporated and the residue was neutralized with saturated aqueous NaHCO3. The mixture was extracted with EtOAc, the organic layer was dried over Na2SO4, and concentrated. The crude product was concentrated and triturated with Et2O and dried to yield the cyclopropylquinoline 53 (123 mg, 93%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.46 (d, J = 1.5 Hz, 1 H), 10.0 (s, 1 H), 9.31 (d, J = 2.3 Hz, 1 H), 8.65 (d, J = 2.0 Hz, 1 H), 7.93 (d, J = 8.6 Hz, 2 H), 7.87 (s, 1 H), 7.69 (d, J = 8.0 Hz, 1 H), 7.39 (d, J = 8.6 Hz, 2 H), 6.15 (t, J = 57.0 Hz, 1 H), 5.69 (dd, J = 7.8, 1.8 Hz, 1 H), 4.12 (s, 3 H), 3.07 (s, 3 H), 1.32 (br s, 2 H), 1.21–1.15 (m, 2 H). LC/MS (ES/APCI): 529.0 (M + H)+.
HCV NS5B polymerase biochemical assay protocol
The enzymatic activity of HCV polymerase (NS5B570n-Con1) was measured as described previously15 with only two changes as follow: concentration of RNA template was 3 nM and concentration of NS5B570n-Con1 enzyme was 3 nM.
Stable HCV replicon assay
The stable GT-1a H77 and GT-1b Con1 subgenomic replicon cell lines expressing the renilla luciferase as a reporter gene (cell lines 2209-23 and pRLucH771b75S/I, respectively) were used as described by Le Pogam.28
Transient HCV replicon assay using a panel of GT-1 NS5B clinical isolates
The GT-1b Con1 and GT-1a H77 transient replicons in which the wild type NS5B gene has been replaced by NS5B isolates obtained from untreated patients were used as described by Le Pogam.22
HCV NS5B polymerase protein crystallography
HCV NS5B GT1b (BK 1-570) was expressed and purified as previously described.29 Crystals of compound-bound polymerase were grown by sitting drop vapor diffusion set using 7 mg/ml protein plus 5 mM of each respective compound over a reservoir containing 50 mM sodium citrate (pH 4.9), 24% polyethylene glycol 4000, and 7.5% glycerol. Crystals were transferred to a solution of mother liquor plus 20% glycerol for cryoprotection and flash frozen in liquid nitrogen. Data were collected at the Advanced Light Source beamline 5.0.2 and Stanford Synchrotron Radiation Laboratories beam line 7-1. Data were reduced with HKL2000,30 a molecular replacement solution determined with Phaser31 and refined using cycles of Buster32,33 with manual rebuilding using Coot.34
Hepatocyte metabolic stability assay
The compounds were incubated with approximately 1.0 × 106 cells/mL in suspension with Williams’ medium E (supplemented with glutamine, antibiotics, insulin, dexamethasone, and 10% FCS) at 37 °C in an atmosphere of 5% CO2. Hepatocyte incubations were done at different concentrations (0.1, 0.3, 1.0, 3.0, and 10.0 μM) of compounds on a Teleshake 1 Variomag at speed of 900 rpm. Incubations were terminated by the addition of 200 μL of methanol plus internal standard after 2, 10, 30, 60, 120, and 180 min of incubation. Samples were transferred to analytical plates and sealed. The supernatants were analyzed by LC/MS/MS. All incubations were conducted in duplicate, and the percentage of compounds remaining at the end of the incubation was determined from the LC/MS/MS peak area ratio.
Pharmacokinetics
Male Wistar/Han (CRL:WI) rats (Charles River Laboratories, Hollister, CA or Raleigh, NC) used in these studies weighed between 180 and 300 g and were surgically implanted with cannulas in the jugular and/or femoral veins. Male beagle dogs used in these studies. All studies were conducted under IACUC approved protocols, and animals were allowed free access to food and water. Single intravenous bolus doses were administered into the tail vein or into the jugular cannula of dual-cannulated animals using a dosing formulation consisting of 2% dimethyl acetamide/20% PEG400/23.4% HPBCD; the remaining cannulas were used for blood collection. Single oral doses were administered by gavage using a dosing formulation consisting of 2% Klucel LF in water. Blood was collected into tubes containing EDTA at typically 0.08 (iv only), 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. Plasma concentrations were determined by a liquid chromatography/tandem mass spectrometry method (LC/MS/MS) following protein precipitation with acetonitrile.
Preparation of 41 microprecipitated bulk product
Amorphous form of the compound was manufactured by Solvent Controlled Precipitation (SCP) method. The drug and Eudragit L100-55 polymer (copolymers of acrylic and methacrylic acid) were dissolved in the dimethyl acetamide (DMA) by stirring at room temperature. The solution was then added to the temperature controlled anti solvent aqueous media (at dilute HCl, pH 3.0, temperature 2–8 °C) that allows rapid co-precipitation of drug and API. The residual DMA was extracted with frequent washing, followed by filtration and drying. The formulation showed amorphous X-ray diffraction pattern. The yield from SCP process was over 90%. Only Eudragit L100-55 polymer provided stable amorphous formulation which stayed amorphous for more than 60 days in the aqueous media as well as under extreme stress conditions of 40°C/100% RH.
Supplementary Material
Figure 2.

3-Aryl pyridones. Biological data generated with HCV GT-1b strain.
Table 7.
Pharmacokinetic parameters for 41 in rat and dog.
| species |
iv
|
po
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| dosea | Cl (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | dosea,b | Cmax (μg/mL) | C at 8 h (μg/mL) | AUC(inf) (μg•h/mL) | F% | |
| rat | 2.6 | 11.4 | 2.2 | 4.3 | 30 | 2.1 | 0.62 | 13.8 | 26 |
| dog | 0.86 | 7.5 | 2.8 | 4.3 | 30 | 1.5 | 0.99 | 23.7 | 31 |
Dose in mg/kg, 20% PEG400, n = 3.
Amorphous MBP, n = 3.
Table 8.
| species |
iv
|
po
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| dosea | Cl (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | dosea,b | Cmax (μg/mL) | C at 8 h (μg/mL) | AUC(inf) (μg•h/mL) | F% | |
| rat | 3.0 | 13.8 | 2.3 | 5.6 | 30 | 3.8 | 1.79 | 37.0 | 100 |
| dog | 1.0 | 4.4 | 1.5 | 5.1 | 3 | 0.36 | 0.21 | 4.2 | 36 |
Dose in mg/kg, 20% PEG400, n = 3.
Aqueous suspension, n = 3.
Acknowledgments
We gratefully acknowledge all members of the Analytical Department in Roche Palo Alto and the Physical Chemistry Department at Hoffmann-La Roche, Nutley for the characterization of the compounds. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
ABBREVIATIONS USED
- DAA
direct-antiviral agents
- HCV
hepatitis C virus
- MBP
microprecipitated bulk powder
- NI
nucleoside inhibitor
- NNI
non-nucleoside inhibitor
- NS5B
nonstructural 5B
- GT
genotype
- SVR
sustained virological response
Footnotes
Accession Codes
Protein crystal structure coordinates and structure factor files have been deposited in the Protein Data Bank under the accession codes 4MIA (41) and 4MIB (48).
The authors declare no competing financial interest.
Experimental procedure and spectroscopic data for compounds 34, 40, 43–47, and 49, and their corresponding synthetic intermediates. Data collection and refinement statistics for cocrystals of NS5B with 41 and 48. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization. [accessed 05/17/2013]; Available at: http://www.who.int/mediacentre/factsheets/fs164/en/
- 2.McGivern DR, Lemon SM. Virus-Specific Mechanisms of Carcinogenesis in Hepatitis C Virus Associated Liver Cancer. Oncogene. 2011;30:1969–1983. doi: 10.1038/onc.2010.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of Hepatitis C Virus Infection. J Hepatol. 2011;55:245–264. doi: 10.1016/j.jhep.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 4.Asselah T, Marcellin P. Direct Acting Antivirals for the Treatment of Chronic Hepatitis C: One Pill a Day for Tomorrow. Liver International. 2012:88–102. doi: 10.1111/j.1478-3231.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- 5.Tanwar S, Trembling PM, Dusheiko GM. TMC435 for the Treatment of Chronic Hepatitis C. Expert Opin Investig Drugs. 2012;21:1193–1209. doi: 10.1517/13543784.2012.690392. [DOI] [PubMed] [Google Scholar]
- 6.Gane E. Future Prespectives: Towards Interferon-Free Regimens for HCV. Antiviral Therapy. 2012;17:1201–1210. doi: 10.3851/IMP2431. [DOI] [PubMed] [Google Scholar]
- 7.de Vicente J, Hendricks RT, Smith DB, Fell JB, Fischer J, Spencer SR, Stengel PJ, Mohr P, Robinson JE, Blake JF, Hilgenkamp RK, Yee C, Adjabeng G, Elworthy TR, Tracy J, Chin E, Li J, Wang B, Bamberg JT, Stephenson R, Oshiro C, Harris SF, Ghate M, Leveque V, Najera I, Le Pogam S, Rajyaguru S, Ao-Ieong G, Alexandrova L, Larrabee S, Brandl M, Briggs A, Sukhtankar S, Farrell R, Xu B. Non-Nucleoside Inhibitors of HCV Polymerase NS5B. Part 2: Synthesis and Structure–Activity Relationships of Benzothiazine-Substituted Quinolinediones. Bioorg Med Chem Lett. 2009;19:3642–3646. doi: 10.1016/j.bmcl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Gane EJ, Roberts SK, Stedman CAM, Angus PW, Ritchie B, Elston R, Ipe D, Morcos PN, Baher L, Najera I, Chu T, Lopatin U, Berrey MM, Bradford W, Laughlin M, Shulman NS, Smith PF. Oral Combination Therapy with a Nucleoside Polymerase Inhibitor (RG7128) and Danoprevir for Chronic Hepatitis C Genotype 1 Infection (INFORM-1): A Randomised, Double-Blind, Placebo-Controlled, Dose-Escalation Trial. Lancet. 2010;376:1467–1475. doi: 10.1016/S0140-6736(10)61384-0. [DOI] [PubMed] [Google Scholar]
- 9.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J Med Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 10.Haigh D, Amphlett EM, Bravi GS, Bright H, Chung V, Chambers CL, Cheasty AG, Convery MA, Ellis MR, Fenwick R, Gray DF, Hartley CD, Howes PD, Jarvest RL, Medhurst KJ, Mehbob A, Mesogiti D, Mirzai F, Nerozzi F, Parry NR, Roughley N, Skarzynski T, Slater MJ, Smith SA, Stocker R, Theobald CJ, Thomas PJ, Thommes PA, Thorpe JH, Wilkinson CS, Williams E. Identification of GSK625433: A Novel Clinical Candidate for the Treatment of Hepatitis C. Abstract of Papers, 233rd National Meeting of the American Chemical Society; Chicago, IL. March 25–29, 2007; Washington, DC: American Chemical Society; 2007. MEDI 015. [Google Scholar]
- 11.Kapelusznik L, Heil EL, Temesgen Z, Talwani R. Drugs of the Future. 2012;37:725–733. [Google Scholar]
- 12.de Bruijne J, van de Wetering de Rooij J, van Vliet AA, Zhou XJ, Temam MF, Molles J, Chen J, Pietropaolo K, Sullivan-Bolyai JZ, Mayers D, Reesink HW. First-in-Human Study of the Pharmacokinetics and Antiviral Activity of IDX375, a Novel Nonnucloside Hepatitis C Virus Polymerase Inhibitor. Antimicrob Agents Chemother. 2012;56:4525–4528. doi: 10.1128/AAC.00451-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randolph JT, Krueger AC, Donner PL, Motter CE, Pratt JK, Liu D, Rockway TW, Wagner R, Hernandez LE, Beno DW, Lim HB, Beyer JM, Mondal R, Mistry N, Colletti L, Liu Y, Koev G, Kati WM, Longenecker KL, Stewart KD, Molla A, Maring CJ. Discovery of ABT-072, a Potent Inhibitor of HCV NS5B polymerase in Clinical Trials. Abstracts of Papers, 244th National Meeting of the American Chemical Society; Philadelphia, PA. Aug. 19–23, 2012; Washington, DC: American Chemical Society; 2012. MEDI 218. [Google Scholar]
- 14.Pratt JK, Betebenner D, Flentge C, Liu D, Rockway T, Wagner R, Maring C, Liu Y, Koev G, Lim HB, Beyer J, Mondal R, Barnes D, Kotecki B, Beno D, Marsh K, Longenecker K, Kati W, Kempf D, Molla A. Aryl 1-Uracil Inhibitors of HCV Genotype 1 NS5B RNA-dependent RNA Polymerase: Uracil Ring Replacements. Abstracts of Papers, 244th National Meeting of the American Chemical Society; Philadelphia, PA. Aug. 19–23, 2012; Washington, DC: American Chemical Society; 2012. MEDI 146. [Google Scholar]
- 15.Talamas FX, Ao-Ieong G, Brameld KA, Chin E, de Vicente J, Dunn JP, Ghate M, Giannetti AM, Harris SF, Labadie SS, Leveque V, Li J, Lui AS-T, McCaleb KL, Najera I, Schoenfeld RC, Wang B, Wong A. De Novo Fragment Design: A Medicinal Chemistry Approach to Fragment-Based Lead Generation. J Med Chem. 2013;56:3115–3119. doi: 10.1021/jm4002605. [DOI] [PubMed] [Google Scholar]
- 16.Schoenfeld RC, Bourdet DL, Brameld KA, Chin E, de Vicente J, Fung A, Harris SF, Lee EK, Le Pogam S, Leveque V, Li J, Lui AS-T, Najera I, Rajyaguru S, Sangi M, Steiner S, Talamas FX, Taygerly J, Zhao J. Discovery of a Novel Series of Potent Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B. J Med Chem. doi: 10.1021/jm401266k. [Online early access] Published Online: September 27, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boger DL, Boyce CW. Selective Metal Cation Activation of a DNA Alkylating Agent: Synthesis and Evaluation of Methyl 1,2,9,9a-Tetrahydrocyclopropa[c]pyrido[3,2-e]indol-4-one-7-carboxylate (CPyI) J Org Chem. 2000;65:4088–4100. doi: 10.1021/jo000177b. [DOI] [PubMed] [Google Scholar]
- 18.Kouznetsov V, Bohorquez ARR, Saavedra LA, Medina RF. An Efficient Synthesis of New C-2 Aryl Substituted Quinolines Based on Three Component Imino Diels-Alder Reaction. Mol Divers. 2006;10:29–37. doi: 10.1007/s11030-006-2344-8. [DOI] [PubMed] [Google Scholar]
- 19.Milletti F, Storchi L, Sforna G, Cruciani G. New and Original pKa Prediction Method Using Grid Molecular Interaction Fields. J Chem Inf Model. 2007;47:2172–2181. doi: 10.1021/ci700018y. [DOI] [PubMed] [Google Scholar]
- 20.The PyMOL Molecular Graphics System, version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 21.Barnes-Seeman D, Jain M, Bell L, Ferreira S, Cohen S, Chen XH, Amin J, Snodgrass B, Hatsis P. Metabolically Stable tert-Butyl Replacement. ACS Med Chem Lett. 2013;4:514–516. doi: 10.1021/ml400045j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Pogam S, Seshaadri A, Kosaka A, Chiu S, Kang H, Hu S, Rajyaguru S, Symons J, Cammack N, Najera I. Existence of Hepatitis C Virus NS5B Variants Naturally Resistant to Non-nucleoside but not to Nucleoside Polymerase Inhibitors Among Untreated Patients. J Antimicrob Chemother. 2008;61:1205–1216. doi: 10.1093/jac/dkn085. [DOI] [PubMed] [Google Scholar]
- 23.Thompson PA, Patel R, Showalter RE, Li C, Appleman JR, Steffy K. In Vitro Studies Demonstrate That Combinations of Antiviral Agents That Include HCV Polymerase Inhibitor ANA598 Have the Potential to Overcome Viral Resistance. Hepatology; Abstracts of Papers, American Association for the Study of Liver Diseases; San Francisco, CA. Oct. 31–Nov 4, 2008; 2008. p. 1164A. [Google Scholar]
- 24.Shah N, Sandhu H, Phuapradit W, Pinal R, Iyer R, Albano A, Chatterji A, Anand S, Choi DS, Tang K, Tian H, Chokshi H, Singhal D, Malick W. Development of Novel Microprecipitation Bulk Powder (MBP) Technology for Manufacturing Stable Amorphous Formulations of Poorly Soluble Drugs. Int J Pharm. 2012;438:53–60. doi: 10.1016/j.ijpharm.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 25.Shah N, Iyer RM, Mair HJ, Choi DS, Tian H, Diodone R, Fahnrich K, Pabst-Ravot A, TANG K, Scheubel E, Grippo JF, Moreira SA, Go Z, Mouskountakis J, Louie T, Ibrahim PN, Sandhu H, Rubia L, Chokshi H, Singhal D, Malick W. Improved Human Bioavailability of Vemurafenib a Practically Insoluble Drug Using an Amorphous Polymer-Stabilized Solid Dispersion Prepared by a Solvent-Controlled Coprecipitation Process. J Pharm Sci. 2013;102:967–981. doi: 10.1002/jps.23425. [DOI] [PubMed] [Google Scholar]
- 26.Reddy MB, Morcos PN, Le Pogam S, Ou Y, Frank K, Lave T, Smith P. Pharmacokinetic/Pharmacodynamic Predictors of Clinical Potency for Hepatitis C Virus Nonnucleoside Polymerase and Protease Inhibitors. Antimicrob Agents Chemother. 2012;56:3144–3156. doi: 10.1128/AAC.06283-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner R, Tufano MD, Stewart KD, Rockway TW, Randolph JT, Pratt JK, Motter CE, Maring CJ, Longenecker KL, Liu Y, Liu D, Krueger AC, Kati WM, Hutchinson DK, Huang PP, Flentge CA, Donner PL, Degoey DA, Betebenner DA, Barnes DM, Chen S, Franczyk TS, II, Gao Y, Haight AR, Hengeveld JE, Henry RF, Koteki BJ, Lou X, Sarris K, Zhang GGZ. WO2009/039127. Uracil or Thymine Derivative for Treating Hepatitis C. 2009 Mar 26;
- 28.Le Pogam S, Jiang WR, Leveque V, Rajyaguru S, Ma H, Kang H, Jiang S, Singer M, Ali S, Klumpp K, Smith D, Symons J, Cammack N, Najera I. In Vitro Selected Con1 Subgenomic Replicons Resistant to 2′-C-Methyl-Cytidine or to R1479 Show Lack of Cross Resistance. Virology. 2006;351:349–359. doi: 10.1016/j.virol.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 29.Le Pogam S, Kang H, Harris SF, Leveque V, Giannetti AM, Ali S, Jiang WR, Rajyaguru S, Tavares G, Oshiro C, Hendricks T, Klumpp K, Symons J, Browner MF, Cammack N, Najera I. Selection and Characterization of Replicon Variants Dually Resistant to Thumb- and Palm-Binding Nonnucleoside Polymerase Inhibitors of the Hepatitis C Virus. J Virol. 2006;80:6146–6154. doi: 10.1128/JVI.02628-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. In: CW Jr, editor. In Methods in Enzymology Carter. Vol. 276. Academic Press; New York: 1997. pp. 307–326. Ch. 20. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-Enhanced Fast Translation Functions. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, Number 4. The suite collaborative computational project, number 4: programs for protein crystallography. 1994. [DOI] [PubMed] [Google Scholar]
- 33.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.11.4. Cambridge, United Kingdom: Global Phasing Ltd; 2011. [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.