

Abstract
Purpose
Muscle paralysis after spinal cord injury (SCI) leads to muscle atrophy, enhanced muscle fatigue, and increased energy demands for functional activities. Phosphorus magnetic resonance spectroscopy (31P-MRS) offers a unique non-invasive alternative of measuring energy metabolism in skeletal muscle and is especially suitable for longitudinal investigations. We determined the impact of spinal cord contusion on in-vivo muscle bioenergetics of the rat hindlimb muscle using 31P-MRS.
Methods
A moderate spinal cord contusion injury (cSCI) was induced at the T8-T10 thoracic spinal segments. 31P-MRS measurements were performed weekly in the rat hindlimb muscles for three weeks. Spectra were acquired in a Bruker 11T/470 MHz spectrometer using a 31P surface coil. The sciatic nerve was electrically stimulated by subcutaneous needle electrodes. Spectra were acquired at rest (5 min), during stimulation (6 min), and recovery (20 min). Phosphocreatine (PCr) depletion rates and the pseudo-first-order rate constant for PCr recovery (kPCr) were determined. The maximal rate of PCr resynthesis, the in-vivo maximum oxidative capacity (Vmax) and oxidative ATP synthesis rate (Qmax), were subsequently calculated.
Results
One week after cSCI, there was a decline in the resting [TCr] of the paralyzed muscle. There was a significant reduction (~24%) in kPCr measures of the paralyzed muscle, maximum in-vivo mitochondrial capacity (Vmax) and the maximum oxidative ATP synthesis rate (Qmax) at 1week post-cSCI.
Conclusions
Using in-vivo MRS assessments, we reveal an acute oxidative metabolic defect in the paralyzed hind limb muscle. These altered muscle bioenergetics might contribute to the host of motor dysfunctions seen after cSCI.
Keywords: 31PMRS, spinal cord contusion, skeletal muscle, oxidative capacity, rat, muscle phosphocreatine
Introduction
Skeletal muscle alterations following spinal cord injury (SCI) have the potential to significantly impact daily functional motor performance and locomotor capabilities; that can ultimately contribute to long-term disability (Yakura, Waters et al. 1990; Gordon and Mao 1994; Wang, Hiatt et al. 1999). Drastic declines in mitochondrial enzyme activity, capillary density and a shift in fiber type composition to type II glycolytic fibers of the paralyzed skeletal muscles is well documented after complete SCI in humans (Kjaer, Mohr et al. 2001) and in spinalized animal models (Jiang, Roy et al. 1991; Durozard, Gabrielle et al. 2000; Gregory, Vandenborne et al. 2003). Skeletal muscle metabolic dysfunction has the potential to decrease oxidative capacity and to negatively impact muscle fatigability (Wang, Hiatt et al. 1999; Bhambhani, Tuchak et al. 2000; McCully, Mulcahy et al. 2011; Erickson, Ryan et al. 2013).
Though metabolic adaptations after SCI appear to be well documented, most investigators have utilized in vitro measurements. These assessments are not only invasive, but suffer from their inability to yield longitudinal follow-up assessments and bio-energetic data from a fully functioning muscle. Alternatively, though maximum oxygen consumption (VO2max) measures are widely used to assess the muscle metabolic oxidative capacity (McCully, Fielding et al. 1993; Wang, Hiatt et al. 1999), these techniques may not necessarily reflect the in-vivo muscle metabolic condition because of their influence from cardiopulmonary functions. Phosphorus magnetic resonance spectroscopy (31P-MRS) has been extensively used in both healthy and diseased muscles to assess the in-vivo metabolic properties of skeletal muscle (Levy, Kushnir et al. 1993; Paganini, Foley et al. 1997; McCully, Mancini et al. 1999; Argov and Arnold 2000; Kent-Braun and Ng 2000; Liu, Walter et al. 2007; McCully, Mulcahy et al. 2011).
The purpose of this study was to assess muscle bioenergetics of hind limb muscles in-vivo after spinal cord contusion injury (cSCI) in rats. We chose a contusion injury model in our study because the majority of new SCIs (~53%) occurring annually are now classified as incomplete and the SCI contusion model is validated and proven to closely correlate with histological, behavioral, electrophysiological evaluations and functional measurements following SCI in the human (Gale, Kerasidis et al. 1985; Noble and Wrathall 1985; Metz, Curt et al. 2000). We hypothesized that the in-vivo bio-energetics of the rat gastrocnemius muscle determined by 31P-MRS, specifically the muscle phosphocreatine (PCr) depletion and re-synthesis rates, along with the maximum mitochondrial capacity and in-vivo maximum oxidative ATP synthesis rate will be drastically impaired one week after cSCI in adult rats.
Methods
Animals
The experimental design for this study is outlined in Figure IA. Twenty-four adult Sprague Dawley female rats (12 week-old, 228–260 g; Charles River, NJ) were housed in a temperature controlled room at 21°C with a 12:12 hours light: dark cycle and provided with rodent chow and water ad libitum. Sixteen rats were moderately injured at the T8-T10 thoracic spinal cord levels and 8 non-injured rats served as controls in providing healthy muscle tissue to run biochemical assays for the quantification of muscle total creatine [TCr] and adenosine triphosphate [ATP]. Of the 16 injured rats, one group of cSCI rats (n=8) was sacrificed at one week after the injury to obtain [TCr] and [ATP]. The remaining animals (n=8) were utilized for 31P-MRS data collected at weeks 1, 2 and 3 after the injury, and subsequently sacrificed. All experimental procedures were performed in accordance with the U.S. Government Principle for the Utilization and Care of Vertebrate Animals by approval of the Institutional Animal Care & Use Committee at the University of Florida.
Fig I.

A) Timeline of experimental procedures and animal groups. After initial handling, animals (n=16) underwent 31P MRS assessments followed by a thoracic spinal cord contusion injury (cSCI). One group of cSCI rats were sacrificed at one week (n=8) to obtain biochemical measurements of [ATP] and [TCr]. The second group of cSCI rats was tested for 31P MRS at 1, 2 and 3 weeks post-cSCI and sacrificed after final testing. Muscle tissue was also harvested for biochemical procedures from one group (n=8) of non-injured animals B) Experimental set-up demonstrating animal posture during 31P-MRS data collection, placement of needle electrodes for stimulation of the sciatic nerve and position of the 31P coil. C) The hindlimb was adequately secured by foam and tape on the MR cradle and the 31P surface coil was secured over the calf muscle.
Spinal Cord Contusion Injury
Spinal cord contusion injury was produced using a NYU (New York University) impactor device. A 10 g weight was dropped from a 2.5 cm height onto the T8 segment of the spinal cord exposed by laminectomy under sterile conditions. Animals received two doses of Ampicillin per day for 5 days, starting at the day of surgery. Procedures were performed under ketamine (100 mg/kg)-xylazine (6.7 mg/kg) anesthesia (Reier, Stokes et al. 1992; Anderson, Howland et al. 1995). Subcutaneous lactated Ringer’s solution (5 ml) and antibiotic spray were administered after completion of the surgery. The animals were kept under vigilant postoperative care, including daily examination for signs of distress, weight loss, dehydration, and bladder dysfunction. Manual expression of bladders was performed 2–3 times daily, as required, and animals were monitored for the possibility of urinary tract infection. All animals were housed individually post surgery. At postoperative day 7, open field locomotion was assessed using the 21 Basso-Beattie-Bresnahan (BBB) locomotor scale (Basso, Beattie et al. 1995) so that if animals did not fall within a preset range (0–7) could be excluded from the study. 31P spectroscopy measurements were performed before injury, and once weekly for a period of three weeks after injury. At the end of the final MR experiments, animals were sacrificed and the gastrocnemius muscle was excised and snap-frozen at −80°C for subsequent biochemical quantification.
31P magnetic resonance spectroscopy experimental set-up
Animals were anesthesized using gaseous isoflurane in oxygen (3% box induction), and maintained at 0.5%–2.5% during the entire MR procedures. Isoflurane at this concentration is commonly used for MRS procedures, without impacting the results obtained (van den Broek, Ciapaite et al. 2010; Torvinen, Silvennoinen et al. 2012). After shaving and cleaning the limb with alcohol the animals were placed horizontally in the prone position on the MR cradle with the hindlimb firmly secured with foam and tape (Fig. IB). MR coils were positioned as described below. Two needle electrodes were placed subcutaneously – one over the region of the third lumbar vertebrae and the other over the greater trochanter (landmarks to stimulate the sciatic nerve). After adequately securing the hindlimb, the sciatic nerve was stimulated to evoke a response from the gastrocnemius muscle. A Grass Stimulator (Quincy, MA) with a Grass Model SIU8T stimulation isolation unit (Grass Instruments, West Warwick, RI) was used to deliver monophasic, rectangular pulse with a 1 ms pulse duration, 1 Hz frequency and of supramaximal intensity (~10V). The stimulus voltage was adjusted to give maximum isometric contraction of the gastrocnemius muscle as confirmed by palpation of the muscle. Electrical stimulation was carried out for four to six minutes to deplete PCr by ~30 to ~40% of the initial resting values. Care was taken to make sure that the PCr content did not deplete by more than 50% to avoid intracellular acidosis (Kemp, Thompson et al. 1994). 31P spectra were acquired at rest, during muscle stimulation, and post-stimulation recovery period for twenty minutes. No attempts were made to synchronize the radiofrequency pulse with the muscle stimulation. Vital signs of the animal were continuously monitored throughout the MR experimental procedure.
31P magnetic resonance spectroscopy data collection
All 31P-MRS data were collected in a Bruker 11Tesla/470 MHz spectrometer. A custom-made 1.5 × 1.7 cm oval 31P (190.5 MHz) tuned surface coil was placed over the belly of the rat gastrocnemius muscle (Fig. IB, C). A 3-cm standard 1H surface coil was placed underneath the hind limb to perform shimming and the animal’s hind limb was extended such that the calf muscles were centered over the surface coil. Resting spectra were acquired with a 50 μs square pulse, a TR of 2 s, spectral width of 10KHz, 150 averages and 8,000 complex data points. Kinetic spectra were collected with a similar sequence but were averaged into 20s bins and acquired at rest (5 min), during electrical muscle stimulation (EMS, 4–6 min), and throughout recovery from EMS (20 min). All data were corrected for T1 saturation by comparing the partially relaxed 31P-MRS spectra at TR of 2 s with fully relaxed spectra acquired at TR of 15 s. The correction factors for PCr, Pi and ATP in our study were 1.40, 1.69 and 1.13, respectively.
31P magnetic resonance spectroscopy data analysis
All free induction decay signals (FIDs) were multiplied by an exponential corresponding to 25 Hz line broadening. Resting spectra were manually phased, and the areas of the -ATP, Pi, and PCr peaks were determined using area integration software (Xwin; Bruker, Billerica, MA, USA). -ATP was used as an internal standard for estimating Pi and PCr concentrations. Absolute concentration of phosphate metabolites was subsequently determined by enzymatically determined ATP concentration in frozen muscle tissue (obtained from one group of animals sacrificed at the end of the experiment as well as from another group of animals that were sacrificed at the end of one week after cSCI) and after accounting for saturation correction.
Intracellular pH was calculated from the chemical shift of the Pi peak relative to PCr using the equation,
| (1) |
where is the chemical shift of the Pi in ppm. The cytosolic resting phosphorylation potential was calculated in reciprocal form since the phosphorylation potential itself is not normally distributed:
| (2) |
Free cytosolic ADP was calculated from the creatine kinase equilibrium reaction as previously described (Mizobata, Prechek et al. 1995; Thompson, Kemp et al. 1995; Pathare, Vandenborne et al. 2008)
| (3) |
where free creatine (Cr) is calculated after subtracting PCr obtained by 31P-MRS from total creatine [TCr] obtained biochemically. Specifically, the PCr content was obtained by 31P-MRS after correction for T1 saturation factors, and the [TCr] content and [ATP] was determined biochemically (see below). [TCr] and [ATP] was first obtained in mmol/kg wet weight assuming a muscle density of 1.06 g/L and converted into mmol/L of intracellular water (mM) assuming a cellular water fraction of 0.73 L/kg as described previously (Pathare, Vandenborne et al. 2008). The intracellular Mg concentration and equilibrium constant (Keq) of the creatine kinase reaction were assumed to be 1 mM and 1.66 × 109 L/mol (M−1) respectively as previously reported (Veech, Lawson et al. 1979). Note that all concentrations are converted to and presented in mM, i.e. mmol/L cell water.
PCr kinetic data
First, the concentrations of PCr were linearly fit for pre and post-cSCI groups and the rate of PCr depletion at the onset of stimulation (Vdep mM/min, a measure of ATP demand) was analyzed using the first five ([PCr] Vdep (0–1.67min) and second set of five ([PCr] Vdep (1.67–3.34min)) data points during stimulation. Second, to measure the dynamic changes in PCr recovery from exercise, we used a custom-designed complex principal component analysis, which accounts for variations in the analysis of multiple spectral peaks of a given data set that are not perfectly phased (Elliott, Walter et al. 1999; Pathare, Vandenborne et al. 2008). Specifically, recovery data were fitted to a single exponential curve, and the pseudo first-order rate constant for PCr recovery (kPCr) was determined (Meyer 1988). The in-vivo mitochondrial oxidative capacity, a measure of Vmax was calculated based on kPCr and baseline PCr values as described previously (Walter, Vandenborne et al. 1997):
| (4) |
Another reliable measure, initial post exercise PCr resynthesis rate (Vmeas), also demonstrated to reflect mitochondrial oxidative capacity and less affected by the end exercise PCr levels and pH (Meyer 1988; Kemp, Thompson et al. 1994; Lodi, Kemp et al. 1997), was determined from the first three to four data points in recovery; depending upon the best linear curve fit
| (5) |
The maximum oxidative ATP synthesis rate (Qmax), which is a function of the intrinsic mitochondrial content and enzyme activity, oxygen and substrate supply to the mitochondria, and the cytosolic redox state (Kemp, Sanderson et al. 1996) was calculated from the known hyperbolic relationship between cytosolic free [ADP] and from the Vmeas as:
| (6) |
where Km is the Michaelis constant and is assumed as 50 M for rat leg muscle (Thompson, Kemp et al. 1995). Measurements of Qmax during recovery in this way, is independent of end exercise pH and PCr (Kemp, Sanderson et al. 1996; Durozard, Gabrielle et al. 2000).
Biochemical Measurements
ATP assay
An ATP assay for gastrocnemius muscle was run for both control and cSCI animals. Tissue was obtained from one group of cSCI rats that were sacrificed at one week after the injury and another group of cSCI animals sacrificed at the end of the experiment. [ATP] was measured as previously described (Hitchins, Cieslar et al. 2001; Gigli and Bussmann 2002; Pathare, Vandenborne et al. 2008). Frozen gastrocnemius muscle was pulverized using a mortar and pestle under dry ice. 100mg of the tissue was homogenized for 30 s with a Mini-bead-beater in a plastic Eppendorf tube containing beads and ice-cold 0.9% perchloric acid (5v/w). The sample was then centrifuged at 9,000 g for 15 min at 4°C (~4,000rpm). The supernatant was extracted and added to 4M KOH (1.125v/w) and centrifuged again for 5 minutes at 4°C and 9,000 g. The supernatant was frozen at −80°C and processed for ATP measurements with an ATP assay kit (Sigma) using a luminometer (Biotek Instruments, CA). Individual [ATP] values were averaged to obtain mean [ATP] concentrations at each time point.
Total creatine assay
Total muscle creatine [TCr] content was determined using the diacetyl/α-napthtol assay as previously described (De Saedeleer and Marechal 1984; Vandenberghe, Gillis et al. 1996; Tarnopolsky and Parise 1999). Approximately 10–15 mg (average 11.55 ± 2.25 mg) of muscle tissue was cut and placed in a vacuum centrifuge (Savant ISS110 SpeedVac™ Concentrator, Thermo Scientific, Milford, MA) to be spun for 18–24 hours. Dried muscle was placed in an ultra-low freezer at −80°C. A porcelain plate and pestle were used to grind the dried muscle samples into powder. The powdered muscle tissue was subsequently extracted in a 0.5 M perchloric acid/1 mM EDTA solution at a relative ratio of 800 μl per 10 mg powdered muscle. The samples were left on ice for 15-minutes, while vortexing periodically. Samples were then spun at 15000 rpm at 4°C for 5 min. The supernatant was neutralized with 2.1 M KHCO3 / 0.3 M MOPS solution at a ratio of 1:5 and then centrifuged again at 15000 rpm for 5 min. In order to determine the muscle [TCr] concentration, 40 μl of the supernatant from the above reaction was combined with 140 μl ddH2O and 20 μl 0.4 N HCl and heated at 65 °C for 10 min to hydrolyze phosphate groups. The solution was then neutralized with 40 μl of 2.0 N NaOH. The reacting solution was allowed to proceed in the dark for 40min and the absorbance of solution eventually read with an excitation wavelength of 460nm. [TCr] values were expressed as mmol/kg dry weight.
Statistical Analysis
Repeated measures ANOVA were used to statistically compare muscle energetic data across time points. We compared the metabolite ratios ([Pi]/[PCr]), phosphorylation ratios - [ADP][Pi]/[ATP], PCr depletion rates (Vdep), PCr rates of recovery (kPCr), maximum mitochondrial capacities (Vmax) and oxidative ATP synthesis rates (Qmax) of the rat hind limb muscle prior to and at 1, 2 and 3 three weeks after cSCI. All hypotheses were tested at an alpha level of 0.05. Post-hoc Bonferroni corrections were used for multiple outcome comparisons. Analyses were performed using SPSS for Windows, Version 13.0.1.
Results
Energy metabolism at rest
The resting phosphate metabolite concentrations measured in the rat gastrocnemius muscles before and after spinal cord contusion injury (cSCI) are summarized in Table I. The resting spectra revealed no change in the inorganic phosphate concentration of the gastrocnemius muscle after cSCI in rats one week post-cSCI. Intracellular pH was slightly higher (by 0.04units) and the [PCr] content significant lower at one week after contusion injury. These parameters resulted in marginal decrease in the Pi-to-PCr ratio (p=0.067) and a significant elevation in the resting phosphorylation ratio (1.5fold, p = 0.002) of the paralyzed gastrocnemius muscles after cSCI. These alterations in resting energy rich phosphate content were completely reversed by 2–3 weeks post-cSCI.
Table I.
Absolute resting phosphate metabolite concentrations determined by 31P-MRS (A) and biochemically (B) before and at various time points (one week, two weeks, three weeks) after spinal cord contusion in rats. C) Kinetic 31P-MRS averaged data at the end of electrical muscle stimulation (End ex) and recovery from exercise. All data are expressed as mean ± standard error.
| A) Resting metabolite concentrations determined by 31P-MRS | ||||
|---|---|---|---|---|
| Pre SCI | 1wk SCI | 2wk SCI | 3wk SCI | |
| [Pi](mM) | 2.46±0.16 | 2.51±0.18 | 2.55±0.11 | 2.21±0.18 |
| [PCr](mM) | 26.34±0.58 | 20.07±0.78* | 28.73±0.56 | 27.73±0.43 |
| [Pi]/[PCr] | 0.09±0.01 | 0.12±0.01 | 0.09±0.004 | 0.08±0.01 |
| pH | 7.14±0.01 | 7.18±0.01* | 7.17±0.01* | 7.17±0.01* |
| [Pi][ADP]/[ATP] | 13.95±1.55 | 21.31±2.0* | 19.26±1.32* | 14.44±1.14 |
| B) Biochemical quantification of absolute metabolite concentrations | ||||
| [ATP](mM) | 6.72±0.64 | 6.03±1.03 | - | 6.85±1.14 |
| [TCr](mM) | 44.9±1.3 | 36.2±3.7* | - | 45.5±4.7 |
| C) Kinetic metabolite concentrations determined by 31P-MRS data | ||||
| kPCr (min−1) | 0.65±0.05 | 0.50±0.06* | 0.67±0.05 | 0.63±0.07 |
| Vmax (mM/min) | 17.52±1.52 | 11.64±1.42* | 16.25±1.28 | 15.97±1.59 |
| End ex pH | 7.02±0.01 | 6.98±0.02* | 6.99±0.02 | 7.00±0.01 |
| Rest – End ex pH (pH units) | 0.12 units | 0.20 units | 0.18 units | 0.17 units |
| End ex [PCr] | 18.98±0.92 | 11.87±0.97* | 19.72±0.9 | 18.84±0.71 |
| End ex [ADP] (μM) | 47.11±4.46 | 55.95±6.32 | 55.14±5.37 | 54.43±2.55 |
| End ex [ADP][Pi]/[ATP] | 67.77±7.18 | 78.27±6.28 | 76.10±13.12 | 77.06±6.25 |
Statistically different from pre injury (<0.05). Abbreviations: [Pi] = inorganic phosphate; [PCr] = phosphocreatine; [ADP] = free cystolic adenosine diphosphate; [ATP] = metabolically determined adenosine triphosphate; [ADP][Pi]/[ATP] = phosphorylation ratio; kPCr = rate constant; 1/k = time constant [TCr] = absolute concentrations of biochemically determined total muscle creatine; Vmax = mitochondrial oxidative capacity based on recovery rate constant; mM= mmol/L cell water.
Energy metabolism during electrical muscle stimulation (EMS) and recovery
Dynamic changes in PCr, Pi and ATP in the rat gastrocnemius muscle measured from 31P spectra during electrical stimulation and the subsequent recovery are shown in Figure IIA, B. As expected, there was a decrease in PCr content with concurrent increases in Pi with the stimulation. However, we noticed some interesting features in PCr kinetics with continued electrical stimulation throughout the recovery period. In control healthy muscles, the rapid loss in PCr during the onset of electrical stimulation dropped off and after approximately four minutes of stimulation a new steady state metabolic condition was reached, with PCr levels equal to ~68% of resting values (Fig. IIIB, approximately 32% drop). In contrast, at one week post-cSCI the rate of PCr depletion remained high throughout the electrical stimulation protocol. As a consequence, electrical stimulation post-cSCI was stopped after around four minutes (instead of 6 minutes), when PCr levels were approximately equal to 58% of resting values (Fig. IIIB, approximately 42% drop). Quantitatively, the rates of PCr depletion during the first 100s ([PCr] Vdep (0–1.67min), − 4.76± 0.37 vs −4.44±0.35mM/min) were not significantly different pre- and post-cSCI. In contrast, the rates of PCr depletion from 100s to 200s into electrical stimulation ([PCr] Vdep (1.67–3.34min)) were statistically significant pre- and one week post-cSCI (Fig. IIIA and IIIB).
Fig II.
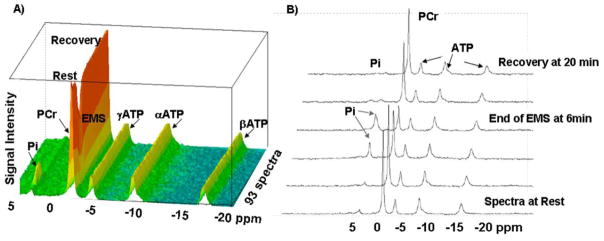
A) Representative three-dimensional stack plot representations of multiple FIDS (93 spectra) showing spectral peaks obtained by 31P-MRS at 11 Tesla from the rat gastrocnemius muscle at rest, during electrical muscle stimulation (EMS) and during recovery from the exercise in a rat before the cSCI. B) Select spectra at rest, at the end of EMS and at 20 min recovery after EMS. Note the depletion of PCr with simultaneous increase in Pi peaks at the end of EMS; and the subsequent recovery of PCr after EMS. The ATP peaks remain unchanged.
Fig III.

A) Graphical representation of the average PCr kinetic data obtained at rest, during and after electrical muscle stimulation (EMS) before (black) and after (red) one week post-cSCI from all animals. Though the time for total stimulation after injury was shorter than before injury; the end stimulation PCr content, when normalized to the baseline PCr content, is lower after than before the cSCI (red and gray dashed lines). B) Expanded view of PCr kinetic data obtained at the onset and upto 6.3 minutes of EMS. PCr depletes earlier and to a greater extent after one week post-cSCI than before injury (red and black dotted lines respectively). C) Histograms depict similar rates of PCr depletion during the first 100seconds (0–1.67min) and slower rates of PCr depletion from 100–200s (1.67–3.34min) at one week post-cSCI as compared to before injury. In B and C, light and dark gray boxes correspond to the first five data points (0–1.67min) and the second five data points (1.67–3.34min) respectively. D) Graphical representation of the average intracellular pH obtained at rest, during and after EMS before and after one week post-cSCI from all animals. * indicates statistically significant differences (p=0.01) between groups. Abbreviations: 2wk cSCI = two weeks post-cSCI; 3wk cSCI = three weeks post-cSCI
The PCr recovery kinetics is shown in Fig. IIIA. Following one week post-cSCI, there was a significant reduction (~24%) in kPCr of the paralyzed gastrocnemius muscle (p=0.001, Table I). Furthermore, Vmax, defined as the maximum mitochondrial oxidative capacity and determined by the rate constant, declined by ~33% at one week post-cSCI (Table I). In addition, we determined the oxidative capacity independent of pH by the maximum rate of mitochondrial ATP production (Qmax). The Qmax was lower after one week of cSCI in all animals (Fig. IVA), but returned to baseline levels by two weeks after injury (Fig. IVB). In fact, almost all measures of phosphate metabolism and kinetics returned to baseline values by two weeks post-cSCI (Table I). As shown in Fig. IVC, a moderate correlation (r=−0.48) was also observed between the rate of ATP production, Qmax and the rate of PCr depletion from 100–200s, Vdep ([PCr] Vdep (1.67–3.34min)).
Fig IV.

A) Maximum oxidative ATP synthesis rate (Qmax) based on the [ADP] model before (pre cSCI) and after one week of spinal cord contusion injury (1wk cSCI) in individual rats (R1–R8). B) Mean maximum oxidative ATP synthesis rate (Qmax) based on the [ADP] model shows significant (*) declines (p=0.01) at 1wk cSCI as compared to pre cSCI and after two and three weeks post-cSCI (2wk cSCI and 3wk cSCI). C) Scatter-plot depicts relationship between the mean maximum oxidative ATP synthesis rate (Qmax) and rate of PCr depletion from 100–200s ([PCr] Vdep(1.67–3.34min)). Data of all animals (pre cSCI, 1wk cSCI, 2wk cSCI and 3wk cSCI) is pooled from each time point.
Note that pH declines below 6.75 units (Taylor, Bore et al. 1983; McCully, Iotti et al. 1994; Boesch 2007) significantly impacts kPCr rates. In our present study, the end stimulation pH in our present work was 6.98 and did not decrease by more than 0.2 units in either the control or SCI groups as compared to the baseline pH (Table I, Fig IIID). Importantly, the fall in cell pH at end exercise was not sufficient to decrease end exercise [ADP] (Table I). Lastly, there was no significant change in [ATP] throughout the experiment.
Biochemical Analysis
Biochemical assay results showed that there were no significant differences in the resting concentrations of [ATP] in the gastrocnemius muscle before, after one week, or following three weeks of cSCI. However, the total muscle creatine values [TCr] were statistically significant before and after one week of injury (Table I).
Discussion
To our knowledge, this is the first in-vivo study that reports acute alterations in energy metabolism in a paralyzed skeletal muscle after a spinal cord contusion injury (cSCI) in rats. Our data reveal a) decrease in the resting phosphocreatine [PCr] content, elevated phosphorylation potential and mild increase in the resting pH of the paralyzed muscle one week after a cSCI b) During recovery from exercise, the rate of PCr recovery in the paralyzed muscle is significantly compromised and associated with a decrease in the mitochondrial oxidative capacity and maximum oxidative ATP synthesis rates c) During exercise, the energy-rich PCr declines to a greater extent and at a faster rate. Our data suggests that there is a significant imbalance in the energy producing and/or consuming states of the paralyzed muscle along with a decrease in the mitochondrial oxidative capacity for oxidative phosphorylation after cSCI.
In-vivo bioenergetics at rest
In our previous studies, we have reported an increase in the resting in-vivo Pi/PCr ratio in response to muscle atrophy following immobilization in both rodents (Pathare, Vandenborne et al. 2008) and humans (Pathare, Stevens et al. 2006); which resulted exclusively from an elevated Pi content in the atrophied muscle. After a cSCI, we do not observe increases in the resting inorganic phosphate content. However, similar to what is seen after a complete spinal cord transection in rats (Durozard, Gabrielle et al. 2000), the trend towards an increase in the resting [Pi]/[PCr] ratios after a cSCI is accompanied with concurrent decreases in the [PCr] content. The exact mechanism of declines in [PCr] content at rest remains unknown, but is suggestive of an imbalance in the resting phosphorylation potential such that the energy available for muscle contraction and other cellular work is decreased (Taylor, Kemp et al. 1993). Additionally, similar to spinal transection in rats (Durozard, Gabrielle et al. 2000), we observed a slight increase in the baseline pH values; possibly indicating a shift to anaerobic metabolism after paralysis.
In-vivo bioenergetics during recovery from exercise
A principal finding of our study is that the rate constant of PCr recovery (kPCr) is significantly decreased after one week of cSCI. Studies have well established that during recovery from exercise, glycolysis ceases and PCr in the muscle cell is replenished at the expense of ATP produced via mitochondrial oxidative phosphorylation (Durozard, Gabrielle et al. 2000). Accordingly, kPCr has been extensively used in both healthy and diseased muscles as estimates of muscle oxidative capacity (Meyer 1988; Paganini, Foley et al. 1997; McCully, Mancini et al. 1999; Argov and Arnold 2000; Kent-Braun and Ng 2000; Pathare, Vandenborne et al. 2007). When measuring oxidative capacity using MRS, it is essential to prevent muscle acidosis. This is because pH declines below 6.75 units (Taylor, Kemp et al. 1993; McCully, Iotti et al. 1994; Boesch 2007) significantly impacts kPCr rates and cannot adequately reflect skeletal muscle oxidative capacity. In the present study, the end stimulation pH was 6.9 and did not decrease by more than 0.2 units as compared to the baseline pH in either the SCI or control groups. We could achieve this by trading against shorter periods of electrical stimulation in our injured group. The lower minimum pH at the end of electrical stimulation in the cSCI rats compared to before injury may suggest reduced cytosolic buffering capacity as well as compensation by anaerobic glycolsis. In fact, similar end-exercise pH (6.96 pH units) is reported after exercise in the vastus lateralis muscle of persons with spinal cord injury without impacting PCr recovery rates (McCully, Mulcahy et al. 2011). Importantly, our control data show similar kPCr values as those reported in literature from healthy rat gastrocnemius muscles (Meyer 1988; Paganini, Foley et al. 1997). Collectively, the decline in kPCr and hence the maximum mitochondrial capacity (Vmax) measures in our present study suggest an overall decrease in mitochondrial oxidative capacity. Additionally, Qmax derived from Vmeas and cytosolic free [ADP] adequately represents the oxidative capacity that is independent of pH (Kemp, Sanderson et al. 1996; Lodi, Kemp et al. 1997) was significantly reduced after cSCI. Physiologically, the in-vivo Qmax is a function of the intrinsic mitochondrial content and enzyme activity, oxygen and substrate supply to the mitochondrion and cytosolic redox (Kemp, Sanderson et al. 1996). In the preset study, a significant decline in the calculated Qmax is accompanied by moderate, but not significant elevations in the end stimulation phosphorylation potential or mitochondrial driving force ([Pi][ADP]/[ATP]), leading us to suggest that the driving force is unable to trigger an adequate mitochondrial response in enhancing oxidative phosphorylation. We believe that this is presumably due to a possible dysregulation between oxygen fluxes in the mitochondria and subsequent ATP synthesis rates. In fact, the finding from our present study that lower ATP synthesis rates (Qmax) accompany faster rates of PCr depletion rates (see below) supports our view (good negative correlation between Qmax and Vdep, r=0.48, Fig. IV). This decrease in oxidative ATP formation after 1 week cSCI was partly compensated by a steeper PCr decrease (see below) as well as anaerobic glycolysis (see pH recovery graph in Fig. IIID).
Oxidative capacity after spinal cord injury
Our finding that the oxidative capacity after cSCI is impaired is in concurrence with in-vivo and in vitro models of complete SCI in both humans (Kjaer, Mohr et al. 2001) and animal models (Gregory, Vandenborne et al. 2003). In-vitro studies show that mixed muscles such as the gastrocnemius and the vastus lateralis have significantly lower mitochondrial enzyme activity following spinalization in both cats and rats (Jiang, Roy et al. 1991; Gregory, Vandenborne et al. 2003). In-vivo studies, on the other hand, report drastic decreases in the kPCr and Qmax measures that suggest a transition in the source of energy supply from oxidative to anaerobic pathways for muscle metabolism following complete paralysis of the rat hind limb muscle (Durozard, Gabrielle et al. 2000). Various mechanisms including skeletal muscle atrophy (Johnston, Finson et al. 2003; Hutchinson, Gomez-Pinilla et al. 2004; Liu, Bose et al. 2008), increase in the number of type II glycolytic fibers of the paralyzed hindlimb muscles (Stevens, Liu et al. 2006), muscle inactivation and muscle deconditioning, as reflected in decreased functional activity and limitation of exercise tolerance to daily activities (Johnston, Betz et al. 2003) have been recognized as precipitating factors in compromising the oxidative capacity of the paralyzed skeletal muscle. Physiologically, this is associated with an overall decrease in the mitochondrial DNA content, capillary density and blood flow of the paralyzed muscle, along with declines in skeletal muscle oxygen uptake following exercise after acute (Gregory, Vandenborne et al. 2003) as well as chronic SCI (Scelsi, Marchetti et al. 1982; Barstow, Scremin et al. 1995; Wang, Hiatt et al. 1999; Bhambhani, Tuchak et al. 2000). These deficits, after complete SCI, are seen acutely (7days) and last chronically (3–6months). However, since the supraspinal input after a spinal cord contusion is partially preserved, our data demonstrate that these muscle adaptations are transient and return to normal two weeks after injury. Lastly, since mitochondrial function can be impacted under low perfusion conditions such as those experienced in peripheral vascular disease (Isbell, Berr et al. 2006; Greiner, Esterhammer et al. 2007) and heart failure (Toussaint, Kwong et al. 1996), deficits in local blood perfusion after cSCI as a potential contributor to mitochondrial oxidative capacity cannot be ruled out.
In-vivo bioenergetics during exercise
An important and novel observation of the present study is a possible impact of the altered oxidative capacity on PCr depletion rates (Vdep) during electrical stimulation in spinal contused rats at one week. Our data demonstrate that during the first 100 sec of electrical muscle stimulation the PCr depletion rate ([PCr] Vdep (0–1.67min)) in the gastrocnemius muscle was similar between animals injured at one-week and controls. However, with continued electrical stimulation for the next 100s the PCr depletion rate ([PCr] Vdep (1.67–3.34min)) was more rapid and to a greater extent after 1 week post-cSCI as compared to the healthy gastrocenemius muscle. Keeping the electrical muscle stimulation parameters constant between animal at different time points, we established that 6 minutes of electrically stimulated contractions were required to deplete the PCr content in our control group (pre-injury) by ~32%. However, in cSCI rats, PCr was depleted by ~42% in only 4 minutes (Fig. IIIB, C). Moreover, while the control group reached a clear steady state by almost three minutes into stimulation, the injured group never reached a steady state with our stimulation protocol. We hypothesize that the faster PCr depletion rates are likely due to increases in ATP demand to perform similar work and decreases in ATP synthesis via oxidative phosphorylation that might be influenced by decreased perfusion secondary to a SCI. We explore these two possibilities below.
Muscle ATP demands and PCr depletion rates
Homeostatic mechanisms within the myocyte couple overall ATP utilization with ATP synthesis; thereby maintaining nearly steady concentrations of ATP during low intensity exercises (Erecinska and Wilson 1982; Kushmerick 1995). Specifically, during muscle contraction, increasing ATP demands in the myocyte are met by PCr breakdown via the creatine kinase equilibrium reaction. This decrease in PCr content indicates the energy buffer role of PCr, and consequently of the phosphorylation ratio ([ADP][Pi]/[ATP]) in response to ATP demands. Accordingly, the magnitude of change in PCr concentration reflects the demand for oxygen and substrates; and PCr depletion rates are purported to measure the ATP needed to meet cellular demands (Kemp, Hands et al. 1995; Toussaint, Kwong et al. 1996). The finding that more amount of PCr is utilized for hydrolysis to ATP implies that the ATP demands for similar intensities of muscle contraction are higher than pre-injury levels. Indeed, sufficient evidence in literature reveals that the paralyzed skeletal muscle is predisposed to increased fatigability, deconditioning, declines in isometric force production over repetitive bouts of contraction and decreases in muscle endurance (Gerrits, De Haan et al. 1999; Shields 2002). Consequently, the paralyzed muscle will function less economically than normal and require more energy to perform a given contraction. Moreover, increases in ATP consumption of paralyzed muscle can also result from ATP consuming events in the tissue such as muscle atrophy (Erkintalo, Bendahan et al. 1998) and the uncoupling of oxidative phosphorylation (Taylor, Kemp et al. 1993; Scheuermann-Freestone, Madsen et al. 2003). However, confirmation from actual measurements of the energy cost of contractions, by the amount of ATP produced for a given power output in the rat model of moderate cSCI are warranted.
Local blood perfusion impact PCr depletion rates and oxidative phosphorylation
Another explanation for the faster Vdep after cSCI is an apparent decrease in the local blood perfusion that interferes with ATP synthesis via oxidative phosphorylation during muscle contraction. During steady state exercise in a healthy muscle, there are increases in oxidative phosphorylation rates; which are supplemented by an increase in the local blood perfusion. Marro et al have shown in the rat hindlimb muscle that during a range of low-level contractile activity, the declines in PCr concentrations are the mirror image of the perfusion increases (Marro, Olive et al. 2007). A correlation exists between energy metabolism and oxygen supply that is fueled by local blood circulation in rat skeletal muscles during exercise (Idstrom, Subramanian et al. 1984; Olive, Dudley et al. 2003). Steady state exercises are met by an initial rapid increase in blood flow till oxygen delivery matches the exercise demands and then plateaus (Van Beekvelt, Shoemaker et al. 2001). Studies have shown that these initial rapid increases in blood flow, in turn, depend upon muscle contractile efficiency and vasomotor tone of the capillary pool (Tschakovsky, Shoemaker et al. 1996; Van Beekvelt, Shoemaker et al. 2001). Noticeably, these data imply that an inadequate blood flow during exercise will result in inadequate delivery of oxygen to the contracting muscle cells and limit the ability of mitochondria to produce ATP via oxidative phosphorylation. Indeed, after SCI in humans, there is an approximate five-fold increase in the time to peak blood flow and a significant delay in the blood flow response at the onset of muscle stimulation in persons with chronic SCI (Olive, Dudley et al. 2003). This increased time of blood delivery, in turn, significantly compromises the ability of the paralyzed muscle to meet the energy demands of muscle contraction secondary to electrical stimulation. Various mechanisms for this deficiency in blood flow have been addressed including alterations in the neural vasomotor tone due to an inadequate vasodilation, decreased contractile ability of the paralyzed muscle to deliver blood, decreased oxidative capacity secondary to shift in muscle fiber type composition towards fast glycolytic fibers and a decreased hyperemic response to muscle contractions (Olive, Dudley et al. 2003). Lastly, similar rapid Vdep rates for a given exercise protocol have been documented after low perfusion states such as peripheral vascular diseases (Kemp, Hands et al. 1995), muscle denervation (Hayashi, Ikata et al. 1997) and chronic heart failure (Toussaint, Kwong et al. 1996). These disease states have shown sizably compromised Vdep rates and local perfusion to the involved muscle; thereby suggesting that the oxygen delivery to the muscle is a likely contributor of the abnormal metabolic response seen.
In view of these findings, we deduce that inherent characteristics of the paralyzed muscle potentially interfere with the local blood perfusion, which in turn hampers the oxygen supply to the exercising muscle. Consequently, ATP production from oxidative phosphorylation during exercise is compromised and is reflected as faster Vdep rates.
Significance of acute metabolic defects after cSCI
In the present study, alterations in energy metabolism observed at rest and during exercise after one week of cSCI reverted to control values by two to three weeks after injury. This rapid adaptation of the paralyzed rat gastrocnemius muscle is remarkable - suggestive of the extreme plasticity of the skeletal muscle in response to neural input - but not surprising. Given that moderate cSCI involves partial loss of descending neural drive, spontaneous motor and physiological recovery after injury is expected. Others and we have previously reported that the partially paralyzed skeletal muscle shows maximum spontaneous reversal of muscle atrophy by three weeks after moderate cSCI (Hutchinson, Gomez-Pinilla et al. 2004; Liu, Bose et al. 2008). Moreover, locomotor impairments too are maximum at one week after the injury and recover gradually by week three, thereby suggesting a possible linkage between alterations in the paralyzed and locomotor deficits (Hutchinson, Gomez-Pinilla et al. 2004; Liu, Bose et al. 2008). Our present work further highlights the potential contribution of metabolic defects associated with acute motor impairments following a cSCI.
Limitations
We did not monitor force in our electrical stimulation protocol. This might constitute a potential limitation as the reproducibility of the work load can not be ensured among animals. However, supramaximal voltage was utilized to give maximum contraction of the gastrocnemius muscle and the stimulation parameters were kept constant throughout the experiment. Though the protocol we used in this study is well documented in the literature (Meyer 1988; Paganini, Foley et al. 1997), we might have stimulated both the gastrocnemius and soleus muscle groups. However, we performed phantom experiments that ascertained that the signal was mainly from the gastrocnemius muscle. In fact, given that the size of the rat plantaris and soleus muscle is small relative to the entire gastrocnemius muscle, we surmise that the soleus muscle is only a minor contributor to the MRS signal measured in this experiment.
Conclusion
In conclusion, the non-invasiveness of 31P-MRS enabled us to longitudinally assess muscle oxidative capacity in real time and with high spectral resolution. We found a decrease in the mitochondrial oxidative phosphorylation rate during steady state exercise along with a drastic elevation in PCr depletion rates (Vdep) at the onset of electrical stimulation after cSCI in rats. Our data uncovers metabolic disturbances in the paralyzed hind limb muscle that might potentially contribute to the host of motor dysfunctions seen after cSCI. Our findings support the potential usefulness of therapeutic interventions aimed at improving aerobic muscle metabolism after this injury.
Abbreviations
- 31PMRS
31 Phosphorus magnetic resonance spectroscopy
- SCI
spinal cord injury
- [ATP]
absolute concentration of biochemically determined free cystolic adenosine triphosphate
- [Pi]
absolute concentration of inorganic phosphate
- [PCr]
absolute concentration of phosphocreatine
- [ADP]
absolute concentration of free cystolic adenosine diphosphate
- [ADP][Pi]/[ATP]
phosphorylation ratio
- Vdep
PCr depletion rate at onset of exercise
- kPCr
rate constant of PCr recovery
- Vmax
mitochondrial oxidative capacity based on recovery rate constant
- Qmax
maximum oxidative ATP synthesis rates based on kPCr and [ADP], independent of pH
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that all experimental procedures were performed in accordance with and comply by the U.S. Government Principle for the Utilization and Care of Vertebrate Animals by approval of the Institutional Animal Care & Use Committee at the University of Florida.
References
- Anderson DK, Howland DR, et al. Fetal neural grafts and repair of the injured spinal cord. Brain Pathol. 1995;5(4):451–57. doi: 10.1111/j.1750-3639.1995.tb00624.x. [DOI] [PubMed] [Google Scholar]
- Argov Z, Arnold DL. MR spectroscopy and imaging in metabolic myopathies. Neurol Clin. 2000;18(1):35–52. doi: 10.1016/s0733-8619(05)70178-x. [DOI] [PubMed] [Google Scholar]
- Barstow TJ, Scremin AM, et al. Gas exchange kinetics during functional electrical stimulation in subjects with spinal cord injury. Med Sci Sports Exerc. 1995;27(9):1284–91. [PubMed] [Google Scholar]
- Basso DM, Beattie MS, et al. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12(1):1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Bhambhani Y, Tuchak C, et al. Quadriceps muscle deoxygenation during functional electrical stimulation in adults with spinal cord injury. Spinal Cord. 2000;38(10):630–8. doi: 10.1038/sj.sc.3101079. [DOI] [PubMed] [Google Scholar]
- Boesch C. Musculoskeletal spectroscopy. J Magn Reson Imaging. 2007;25(2):321–38. doi: 10.1002/jmri.20806. [DOI] [PubMed] [Google Scholar]
- De Saedeleer M, Marechal G. Chemical energy usage during isometric twitches of frog sartorius muscle intoxicated with an isomer of creatine, beta-guanidinopropionate. Pflugers Arch. 1984;402(2):185–9. doi: 10.1007/BF00583333. [DOI] [PubMed] [Google Scholar]
- Durozard D, Gabrielle C, et al. Metabolism of rat skeletal muscle after spinal cord transection. Muscle Nerve. 2000;23(10):1561–8. doi: 10.1002/1097-4598(200010)23:10<1561::aid-mus13>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Elliott MA, Walter GA, et al. Spectral quantitation by principal component analysis using complex singular value decomposition. Magn Reson Med. 1999;41(3):450–5. doi: 10.1002/(sici)1522-2594(199903)41:3<450::aid-mrm4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Wilson DF. Regulation of cellular energy metabolism. J Membr Biol. 1982;70(1):1–14. doi: 10.1007/BF01871584. [DOI] [PubMed] [Google Scholar]
- Erickson ML, Ryan TE, et al. Near-infrared assessments of skeletal muscle oxidative capacity in persons with spinal cord injury. Eur J Appl Physiol. 2013 doi: 10.1007/s00421-013-2657-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkintalo M, Bendahan D, et al. Reduced metabolic efficiency of skeletal muscle energetics in hyperthyroid patients evidenced quantitatively by in vivo phosphorus-31 magnetic resonance spectroscopy. Metabolism. 1998;47(7):769–76. doi: 10.1016/s0026-0495(98)90110-x. [DOI] [PubMed] [Google Scholar]
- Gale K, Kerasidis H, et al. Spinal cord contusion in the rat: behavioral analysis of functional neurologic impairment. Exp Neurol. 1985;88(1):123–34. doi: 10.1016/0014-4886(85)90118-9. [DOI] [PubMed] [Google Scholar]
- Gerrits HL, De Haan A, et al. Contractile properties of the quadriceps muscle in individuals with spinal cord injury. Muscle Nerve. 1999;22(9):1249–56. doi: 10.1002/(sici)1097-4598(199909)22:9<1249::aid-mus13>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Gigli I, Bussmann LE. Effects of exercise on muscle metabolites and sarcoplasmic reticulum function in ovariectomized rats. Physiol Res. 2002;51(3):247–54. [PubMed] [Google Scholar]
- Gordon T, Mao J. Muscle atrophy and procedures for training after spinal cord injury. Phys Ther. 1994;74(1):50–60. doi: 10.1093/ptj/74.1.50. [DOI] [PubMed] [Google Scholar]
- Gregory CM, Vandenborne K, et al. Human and rat skeletal muscle adaptations to spinal cord injury. Can J Appl Physiol. 2003;28(3):491–500. doi: 10.1139/h03-036. [DOI] [PubMed] [Google Scholar]
- Greiner A, Esterhammer R, et al. High-energy phosphate metabolism in the calf muscle of healthy humans during incremental calf exercise with and without moderate cuff stenosis. Eur J Appl Physiol. 2007;99(5):519–31. doi: 10.1007/s00421-006-0379-2. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Ikata T, et al. Time course of recovery from nerve injury in skeletal muscle: energy state and local circulation. J Appl Physiol. 1997;82(3):732–7. doi: 10.1152/jappl.1997.82.3.732. [DOI] [PubMed] [Google Scholar]
- Hitchins S, Cieslar JM, et al. 31P NMR quantitation of phosphorus metabolites in rat heart and skeletal muscle in vivo. Am J Physiol Heart Circ Physiol. 2001;281(2):H882–7. doi: 10.1152/ajpheart.2001.281.2.H882. [DOI] [PubMed] [Google Scholar]
- Hutchinson KJ, Gomez-Pinilla F, et al. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain. 2004;127(Pt 6):1403–1414. doi: 10.1093/brain/awh160. [DOI] [PubMed] [Google Scholar]
- Idstrom JP, V, Subramanian H, et al. Biochemical and 31P-NMR studies of the energy metabolism in relation to oxygen supply in rat skeletal muscle during exercise. Adv Exp Med Biol. 1984;169:489–96. doi: 10.1007/978-1-4684-1188-1_44. [DOI] [PubMed] [Google Scholar]
- Isbell DC, Berr SS, et al. Delayed calf muscle phosphocreatine recovery after exercise identifies peripheral arterial disease. J Am Coll Cardiol. 2006;47(11):2289–95. doi: 10.1016/j.jacc.2005.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BA, Roy RR, et al. Enzymatic responses of cat medial gastrocnemius fibers to chronic inactivity. J Appl Physiol. 1991;70(1):231–9. doi: 10.1152/jappl.1991.70.1.231. [DOI] [PubMed] [Google Scholar]
- Johnston TE, Betz RR, et al. Implanted functional electrical stimulation: an alternative for standing and walking in pediatric spinal cord injury. Spinal Cord. 2003;41(3):144–52. doi: 10.1038/sj.sc.3101392. [DOI] [PubMed] [Google Scholar]
- Johnston TE, Finson RL, et al. Functional electrical stimulation for augmented walking in adolescents with incomplete spinal cord injury. J Spinal Cord Med. 2003;26(4):390–400. doi: 10.1080/10790268.2003.11753711. [DOI] [PubMed] [Google Scholar]
- Kemp GJ, Hands LJ, et al. Calf muscle mitochondrial and glycogenolytic ATP synthesis in patients with claudication due to peripheral vascular disease analysed using 31P magnetic resonance spectroscopy. Clin Sci (Lond) 1995;89(6):581–90. doi: 10.1042/cs0890581. [DOI] [PubMed] [Google Scholar]
- Kemp GJ, Sanderson AL, et al. Regulation of oxidative and glycogenolytic ATP synthesis in exercising rat skeletal muscle studied by 31P magnetic resonance spectroscopy. NMR Biomed. 1996;9(6):261–70. doi: 10.1002/(SICI)1099-1492(199609)9:6<261::AID-NBM430>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Kemp GJ, Thompson CH, et al. pH control in rat skeletal muscle during exercise, recovery from exercise, and acute respiratory acidosis. Magn Reson Med. 1994;31(2):103–9. doi: 10.1002/mrm.1910310203. [DOI] [PubMed] [Google Scholar]
- Kent-Braun JA, Ng AV. Skeletal muscle oxidative capacity in young and older women and men. J Appl Physiol. 2000;89(3):1072–8. doi: 10.1152/jappl.2000.89.3.1072. [DOI] [PubMed] [Google Scholar]
- Kjaer M, Mohr T, et al. Muscle enzyme adaptation to training and tapering-off in spinal-cord-injured humans. Eur J Appl Physiol. 2001;84(5):482–6. doi: 10.1007/s004210100386. [DOI] [PubMed] [Google Scholar]
- Kushmerick MJ. Bioenergetics and muscle cell types. Adv Exp Med Biol. 1995;384:175–84. doi: 10.1007/978-1-4899-1016-5_14. [DOI] [PubMed] [Google Scholar]
- Levy M, Kushnir T, et al. In vivo 31P NMR studies of paraplegics’ muscles activated by functional electrical stimulation. Magn Reson Med. 1993;29(1):53–8. doi: 10.1002/mrm.1910290111. [DOI] [PubMed] [Google Scholar]
- Liu M, Bose P, et al. A longitudinal study of skeletal muscle following spinal cord injury and locomotor training. Spinal Cord. 2008;46(7):488–93. doi: 10.1038/sj.sc.3102169. [DOI] [PubMed] [Google Scholar]
- Liu M, Walter GA, et al. A quantitative study of bioenergetics in skeletal muscle lacking carbonic anhydrase III using 31P magnetic resonance spectroscopy. Proc Natl Acad Sci U S A. 2007;104(1):371–6. doi: 10.1073/pnas.0609870104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi R, Kemp GJ, et al. Influence of cytosolic pH on in vivo assessment of human muscle mitochondrial respiration by phosphorus magnetic resonance spectroscopy. Magma. 1997;5(2):165–71. doi: 10.1007/BF02592248. [DOI] [PubMed] [Google Scholar]
- Marro KI, Olive JL, et al. Time-courses of perfusion and phosphocreatine in rat leg during low-level exercise and recovery. J Magn Reson Imaging. 2007;25(5):1021–7. doi: 10.1002/jmri.20903. [DOI] [PubMed] [Google Scholar]
- McCully K, Mancini D, et al. Nuclear magnetic resonance spectroscopy: its role in providing valuable insight into diverse clinical problems. Chest. 1999;116(5):1434–41. doi: 10.1378/chest.116.5.1434. [DOI] [PubMed] [Google Scholar]
- McCully KK, Fielding RA, et al. Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. J Appl Physiol. 1993;75(2):813–9. doi: 10.1152/jappl.1993.75.2.813. [DOI] [PubMed] [Google Scholar]
- McCully KK, Iotti S, et al. Simultaneous in vivo measurements of HbO2 saturation and PCr kinetics after exercise in normal humans. J Appl Physiol. 1994;77(1):5–10. doi: 10.1152/jappl.1994.77.1.5. [DOI] [PubMed] [Google Scholar]
- McCully KK, Mulcahy TK, et al. Skeletal muscle metabolism in individuals with spinal cord injury. J Appl Physiol. 2011;111(1):143–8. doi: 10.1152/japplphysiol.00094.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz GA, Curt A, et al. Validation of the weight-drop contusion model in rats: a comparative study of human spinal cord injury. J Neurotrauma. 2000;17(1):1–17. doi: 10.1089/neu.2000.17.1. [DOI] [PubMed] [Google Scholar]
- Meyer RA. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am J Physiol. 1988;254(4 Pt 1):C548–53. doi: 10.1152/ajpcell.1988.254.4.C548. [DOI] [PubMed] [Google Scholar]
- Mizobata Y, Prechek D, et al. The duration of infection modifies mitochondrial oxidative capacity in rat skeletal muscle. J Surg Res. 1995;59(1):165–73. doi: 10.1006/jsre.1995.1149. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Wrathall JR. Spinal cord contusion in the rat: morphometric analyses of alterations in the spinal cord. Exp Neurol. 1985;88(1):135–49. doi: 10.1016/0014-4886(85)90119-0. [DOI] [PubMed] [Google Scholar]
- Olive JL, Dudley GA, et al. Vascular remodeling after spinal cord injury. Med Sci Sports Exerc. 2003;35(6):901–7. doi: 10.1249/01.MSS.0000069755.40046.96. [DOI] [PubMed] [Google Scholar]
- Paganini AT, Foley JM, et al. Linear dependence of muscle phosphocreatine kinetics on oxidative capacity. Am J Physiol. 1997;272(2 Pt 1):C501–10. doi: 10.1152/ajpcell.1997.272.2.C501. [DOI] [PubMed] [Google Scholar]
- Pathare N, Vandenborne K, et al. Alterations in inorganic phosphate in mouse hindlimb muscles during limb disuse. NMR Biomed. 2007 doi: 10.1002/nbm.1162. [DOI] [PubMed] [Google Scholar]
- Pathare N, Vandenborne K, et al. Alterations in inorganic phosphate in mouse hindlimb muscles during limb disuse. NMR Biomed. 2008;21(2):101–10. doi: 10.1002/nbm.1162. [DOI] [PubMed] [Google Scholar]
- Pathare NC, Stevens JE, et al. Deficit in human muscle strength with cast immobilization: contribution of inorganic phosphate. Eur J Appl Physiol. 2006;98(1):71–8. doi: 10.1007/s00421-006-0244-3. [DOI] [PubMed] [Google Scholar]
- Reier PJ, Stokes BT, et al. Fetal cell grafts into resection and contusion/compression injuries of the rat and cat spinal cord. Exp Neurol. 1992;115(1):177–88. doi: 10.1016/0014-4886(92)90245-l. [DOI] [PubMed] [Google Scholar]
- Scelsi R, Marchetti C, et al. Muscle fiber type morphology and distribution in paraplegic patients with traumatic cord lesion. Histochemical and ultrastructural aspects of rectus femoris muscle. Acta Neuropathol. 1982;57(4):243–8. doi: 10.1007/BF00692178. [DOI] [PubMed] [Google Scholar]
- Scheuermann-Freestone M, Madsen PL, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107(24):3040–6. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- Shields RK. Muscular, skeletal, and neural adaptations following spinal cord injury. J Orthop Sports Phys Ther. 2002;32(2):65–74. doi: 10.2519/jospt.2002.32.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JE, Liu M, et al. Changes in soleus muscle function and fiber morphology with one week of locomotor training in spinal cord contusion injured rats. J Neurotrauma. 2006;23(11):1671–81. doi: 10.1089/neu.2006.23.1671. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Parise G. Direct measurement of high-energy phosphate compounds in patients with neuromuscular disease. Muscle Nerve. 1999;22(9):1228–33. doi: 10.1002/(sici)1097-4598(199909)22:9<1228::aid-mus9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Kemp GJ, et al. Skeletal muscle bioenergetics in myotonic dystrophy. J Neurol Sci. 1993;116(2):193–200. doi: 10.1016/0022-510x(93)90325-s. [DOI] [PubMed] [Google Scholar]
- Thompson CH, Kemp GJ, et al. Skeletal muscle mitochondrial function studied by kinetic analysis of postexercise phosphocreatine resynthesis. J Appl Physiol. 1995;78(6):2131–9. doi: 10.1152/jappl.1995.78.6.2131. [DOI] [PubMed] [Google Scholar]
- Torvinen S, Silvennoinen M, et al. Rats bred for low aerobic capacity become promptly fatigued and have slow metabolic recovery after stimulated, maximal muscle contractions. PLoS One. 2012;7(11):e48345. doi: 10.1371/journal.pone.0048345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint JF, Kwong KK, et al. Interrelationship of oxidative metabolism and local perfusion demonstrated by NMR in human skeletal muscle. J Appl Physiol. 1996;81(5):2221–8. doi: 10.1152/jappl.1996.81.5.2221. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Shoemaker JK, et al. Vasodilation and muscle pump contribution to immediate exercise hyperemia. Am J Physiol. 1996;271(4 Pt 2):H1697–701. doi: 10.1152/ajpheart.1996.271.4.H1697. [DOI] [PubMed] [Google Scholar]
- Van Beekvelt MC, Shoemaker JK, et al. Blood flow and muscle oxygen uptake at the onset and end of moderate and heavy dynamic forearm exercise. Am J Physiol Regul Integr Comp Physiol. 2001;280(6):R1741–7. doi: 10.1152/ajpregu.2001.280.6.R1741. [DOI] [PubMed] [Google Scholar]
- van den Broek NM, Ciapaite J, et al. Comparison of in vivo postexercise phosphocreatine recovery and resting ATP synthesis flux for the assessment of skeletal muscle mitochondrial function. Am J Physiol Cell Physiol. 2010;299(5):C1136–43. doi: 10.1152/ajpcell.00200.2010. [DOI] [PubMed] [Google Scholar]
- Vandenberghe K, Gillis N, et al. Caffeine counteracts the ergogenic action of muscle creatine loading. J Appl Physiol. 1996;80(2):452–7. doi: 10.1152/jappl.1996.80.2.452. [DOI] [PubMed] [Google Scholar]
- Veech RL, Lawson JW, et al. Cytosolic phosphorylation potential. J Biol Chem. 1979;254(14):6538–47. [PubMed] [Google Scholar]
- Walter G, Vandenborne K, et al. Noninvasive measurement of phosphocreatine recovery kinetics in single human muscles. Am J Physiol. 1997;272(2 Pt 1):C525–34. doi: 10.1152/ajpcell.1997.272.2.C525. [DOI] [PubMed] [Google Scholar]
- Wang H, Hiatt WR, et al. Relationships between muscle mitochondrial DNA content, mitochondrial enzyme activity and oxidative capacity in man: alterations with disease. Eur J Appl Physiol Occup Physiol. 1999;80(1):22–7. doi: 10.1007/s004210050553. [DOI] [PubMed] [Google Scholar]
- Yakura JS, Waters RL, et al. Changes in ambulation parameters in spinal cord injury individuals following rehabilitation. Paraplegia. 1990;28(6):364–70. doi: 10.1038/sc.1990.49. [DOI] [PubMed] [Google Scholar]